

Abstract
Baker's yeast (Saccharomyces cerevisiae) has been genetically engineered to ferment the pentose sugar xylose present in lignocellulose biomass. One of the reactions controlling the rate of xylose utilization is catalyzed by xylose reductase (XR). In particular, the cofactor specificity of XR is not optimized with respect to the downstream pathway, and the reaction rate is insufficient for high xylose utilization in S. cerevisiae. The current study describes a novel approach to improve XR for ethanol production in S. cerevisiae. The cofactor binding region of XR was mutated by error-prone PCR, and the resulting library was expressed in S. cerevisiae. The S. cerevisiae library expressing the mutant XR was selected in sequential anaerobic batch cultivation. At the end of the selection process, a strain (TMB 3420) harboring the XR mutations N272D and P275Q was enriched from the library. The Vmax of the mutated enzyme was increased by an order of magnitude compared to that of the native enzyme, and the NADH/NADPH utilization ratio was increased significantly. The ethanol productivity from xylose in TMB 3420 was increased ∼40 times compared to that of the parent strain (0.32 g/g [dry weight {DW}] × h versus 0.007 g/g [DW] × h), and the anaerobic growth rate was increased from ∼0 h−1 to 0.08 h−1. The improved traits of TMB 3420 were readily transferred to the parent strain by reverse engineering of the mutated XR gene. Since integrative vectors were employed in the construction of the library, transfer of the improved phenotype does not require multicopy expression from episomal plasmids.
There is currently great worldwide interest in producing bioethanol through fermentation of lignocellulose biomass derived from forest and agricultural by-products. Utilization of lignocellulose biomass does not compete with food and feed production. However, the overall conversion of lignocellulose to ethanol is more complicated than sucrose- and starch-based ethanol production. A significant fraction of lignocellulose biomass may consist of nonhexose sugars, with the pentose sugar xylose comprising as much as 40% of the total carbohydrate content (8). Pentose sugars such as xylose are not utilized by native Saccharomyces cerevisiae, which is the organism of choice in the sucrose- and starch-based ethanol industries. Fermentation of xylose to ethanol has consequently been achieved in S. cerevisiae by expression of heterologous xylose-catabolizing pathways employing either xylose reductase (XR) and xylitol dehydrogenase (XDH) or xylose isomerase (XI) (12, 16, 39).
A major limitation of xylose fermentation by recombinant S. cerevisiae is the low ethanol productivity compared to glucose fermentation. The metabolic flux from xylose to ethanol is controlled at several levels in the pathway. The affinity of S. cerevisiae transporters for xylose is lower than for glucose (9, 35), and compared to glycolysis, the flux through the pentose phosphate pathway is kinetically constrained (20, 43). Most importantly, however, the activity of the heterologous xylose-catabolizing enzymes, XR/XDH or XI, is low, and expression from strong promoters or multicopy plasmids is required for efficient xylose utilization (19, 21, 27). In addition, the cofactor usage in the XR/XDH pathway is unbalanced, with XR and XDH preferring NADPH and NAD+, respectively. This imbalance leads to by-product formation in terms of xylitol and reduced ethanol yield and ethanol productivity (18, 24). To improve the cofactor balance and the flux through the xylose pathway, the NADH/NADPH specificity of XR and/or XDH has previously been engineered by site-directed mutagenesis (Table 1 and references therein).
TABLE 1.
Kinetics of XR mutantsa
| Enzymeb | Cofactor | kcat (min−1) | KmAc (μM) | KmBd (mM) | KiAe (μM) | Rself | Reference |
|---|---|---|---|---|---|---|---|
| S. stipitis WT | NADPH | 63 × 101 | 2.5 | 47 | −g | − | 44 |
| NADH | 42 × 101 | 31 | 32 | − | |||
| Candida tenius WT | NADPH | 78 × 101 | 3.0 | 96 | 1.0 | 0.03 | 32 |
| NADH | 66 × 101 | 38 | 142 | 19 | |||
| S. stipitis WT | NADPH | 0.30h | 1.0 | 62 | 1.4 | 0.04 | 2 |
| NADH | 0.21h | 29 | 59 | 26 | |||
| C. tenius R276H | NADPH | 90 × 101 | 4.0 | 107 | 3.0 | 0.24 | 32 |
| NADH | 13 × 102 | 15 | 94 | 20 | |||
| S. stipitis R276H | NADPH | 16 | 1.7 | 53 | − | − | 44 |
| NADH | 41 × 101 | 17 | 46 | − | |||
| C. tenius N272D | NADPH | 22 × 102 | 17 | 170 | 21 | 0.23 | 32 |
| NADH | 84 × 101 | 26 | 99 | 60 | |||
| S. stipitis K270R | NADPH | 2.1h | 26 | 468 | 23 | 0.59 | 2 |
| NADH | 0.96h | 63 | 145 | 57 | |||
| C. tenius K270R | NADPH | 16 × 102 | 5.0 | 350 | 3.0 | 1.5 | 32 |
| NADH | 48 × 101 | 21 | 54 | 4.0 | |||
| S. stipitis K270R/N272D | NADPH | 19 × 102 | 2810 | 350 | − | − | 44 |
| NADH | 71 × 101 | 138 | 68 | − | |||
| C. tenius K270R/N272D | NADPH | 38 × 102 | 128 | 722 | 64 | 2.7 | 32 |
| NADH | 72 × 101 | 41 | 106 | 30 |
Uncertainties of values are not reported, since the manner of calculation differed among investigations.
Enzymes are listed in the general order of increasing NADH specificity, Rsel. For the sake of comparison, amino acid residues are numbered in relation to the S. stipitis XR. WT, wild type.
A is the cofactor NADH or NADPH; KmA is the cofactor affinity constant.
B is the substrate xylose; KmB is the substrate affinity constant.
A is the cofactor NADH or NADPH; KiA is the cofactor dissociation constant.
The selectivity ratio, Rsel, is given by [Vmax/KiAKmB]NADH/[Vmax/KiAKmB]NADPH. Vmax is the maximum reaction rate.
−, missing data.
Vmax (U/mg protein) was determined instead of kcat.
The cofactor binding site of XR has been mapped by homology to aldose reductases from different organisms (3, 23, 28, 30) and through analysis of crystal structures (4, 22). Amino acid residues responsible for discriminating between NADH and NADPH binding in XR have been engineered by site-directed mutagenesis to increase the specificity for NADH (23, 32, 44). S. cerevisiae strains harboring mutant XRs with enhanced NADH specificity have thus been demonstrated to exhibit increased ethanol yield and productivity (2, 17, 33, 34, 44). The current study used a random rather than a rational (site-directed) method to engineer XR for ethanol production in S. cerevisiae. The cofactor binding region of XR was mutated by error-prone PCR, and the mutant library was expressed in S. cerevisiae. The resulting S. cerevisiae library was selected in sequential anaerobic batch cultivation in xylose medium. The selected strain harbored two mutated residues within the cofactor binding region of XR and displayed unprecedentedly high anaerobic growth rate and ethanol productivity.
MATERIALS AND METHODS
Strains and cultivation conditions.
The strains and plasmids used in this study are summarized in Table 2. Escherichia coli was used for cloning of plasmids and was grown at 37°C in liquid or solid LB medium (10 g/liter tryptone, 5 g/liter yeast extract, 5 g/liter NaCl) supplemented with 100 mg/liter ampicillin. On solid medium, S. cerevisiae strains were grown on YNB plates (6.7 g/liter yeast nitrogen base without amino acids) supplemented with 20 g/liter glucose (the d isomers of all sugars were used). Defined mineral medium was used for liquid cultivation of S. cerevisiae and was composed of 60 g/liter xylose (unless otherwise noted), mineral salts [(NH4)2SO4, 5 g/liter; KH2PO4, 3 g/liter; MgSO4·7H2O, 0.5 g/liter], 0.4 g/liter Tween 80, 0.01 g/liter ergosterol (1), vitamins, and trace elements (41). Identical medium was used for shake flask cultivation and cultivation in an instrumented bioreactor, with the exception that 50 mM potassium hydrogen phthalate, pH 5.5 (13), was added as a buffering agent in shake flasks. Cultivation of S. cerevisiae was performed at 30°C.
TABLE 2.
S. cerevisiae plasmids and strains used in the current study
| Plasmids and strains | Relevant features | Reference |
|---|---|---|
| Plasmids | ||
| YIpOB8 | URA3 TDH3p-XYL1-ADH1t PGK1p-XYL2-PGK1t | O. Bengtsson, M. Bettiga, R. Garcia Sanchez, and M. F. Gorwa-Grauslund, unpublished data |
| YIpDR6 | pOB8 XR N272D P275Q | This work |
| YIpDR7 | pOB8 XR N272D | This work |
| YIpDR8 | pOB8 XR P275Q | This work |
| S. cerevisiae strains | ||
| TMB 3044 | CEN.PK 2-1C ΔGRE3 his3::PGK1p-XKS1-PGK1t TAL1::PGK1p-TAL1-PGK1t TKL1::PGK1p-TKL1-PGK1t RKI1::PGK1p-RKI1-PGK1t RPE1::PGK1p-RPE1-PGK1t leu2::YIplac128 ura3 | 21 |
| TMB 3420 | Population selected from repeated batch cultivations | This work |
| TMB 3421 | TMB 3044 ura3::YIpDR6 | This work |
| TMB 3422 | TMB 3044 ura3::YIpDR7 | This work |
| TMB 3423 | TMB 3044 ura3::YIpDR8 | This work |
| TMB 3424 | TMB 3044 ura3:: YIpOB8 | This work |
Batch cultivation was performed in an instrumented bioreactor (Applikon Biotechnology, AC Schiedam, Netherlands) with a 1-liter working volume. The medium was prepared as described above, with antifoam (Dow Corning) added to the reactor at a final concentration of 0.25 ml/liter. The temperature was maintained at 30°C, and the pH was controlled at 5.5 through addition of 3 M KOH. During aerobic growth, the agitation rate was set to 900 rpm and the culture was sparged with air at 1 liter/min. During anaerobic cultivation, the agitation rate was reduced to 200 rpm, and oxygen-free conditions were maintained by nitrogen sparging at 0.2 liter/min. The off gas was analyzed on line for CO2 production and ethanol evaporation with an Innova 1313 fermentation monitor (LumaSense Technologies, Ballerup, Denmark). Cultures were sampled for high-performance liquid chromatography (HPLC), optical density at 620 nm (OD620), and cell dry-weight (DW) measurements.
Library construction.
A library of randomly mutated Scheffersomyces stipitis (previously Pichia stipitis) XYL1 sequences was generated by error-prone PCR and the MEGAWHOP protocol for whole-plasmid synthesis (29). Primers were constructed to amplify a region between bp +631 and +870, corresponding to the cofactor binding region. Error-prone PCR was conducted using Mutazyme II polymerase (Stratagene, Cedar Creek, TX) according to the manufacturer's instructions. The mutation frequency of the PCR was set to 1.5 nucleotides (nt)/amplicon by adjusting the amount of template DNA in the reaction mixture and verifying the mutation distribution by sequencing in an iterative process. Following error-prone PCR, the amplified section was purified using the E.Z.N.A Cycle-Pure kit (Omega Bio-tek, Doraville, GA) and used as “megaprimer” for reconstruction of the template plasmid YIpOB8 by PCR according to the MEGAWHOP protocol (29). Following PCR, the template DNA was removed by digestion with FastDigest DpnI (Fermentas, Vilnius, Lithuania) for 1 h at 37°C.
The mutated plasmid library was used to transform Electro-10Blue E. coli cells (Stratagene, Cedar Creek, TX) by electroporation (17 kV/cm; 200 Ω; 25 μF) in a 0.1-cm cuvette (6). The size of the library was determined by plating an appropriately diluted volume of the transformation mixture on LB-ampicillin (100-mg/liter) plates. The remainder of the transformed cells were selected overnight in 500 ml liquid LB medium supplemented with ampicillin (100 mg/liter). The resulting E. coli library was stored in 15% glycerol stocks at −80°C, and plasmid DNA was harvested using the QIAfilter Plasmid Mega Kit (Qiagen, Hilden, Germany). The mutated plasmid library was linearized using EcoRV and used for large-scale integrative transformation (10) of S. cerevisiae strain TMB 3044 (Table 2). The size of the library in S. cerevisiae was determined by plating a volume of appropriately diluted cells on YNB plates, whereas the rest of the transformation mixture was directly inoculated in liquid medium for selection (see below). The completion level of the library was calculated from the PCR mutation distribution, the size of the mutagenized gene segment, and the number of transformants, using the PEDEL online tool (31; http://guinevere.otago.ac.nz/aef).
Selection process.
Following large-scale transformation of TMB 3044, cells were inoculated in an instrumented bioreactor for selection in liquid mineral medium (glucose, 5 g/liter, and xylose, 55 g/liter) under aerobic conditions. After approximately 48 h, the cell density had reached an OD620 of ∼40, and the conditions were changed to anaerobiosis. Repeated batch cultures were then performed by pumping out the spent culture and replacing it with fresh medium (xylose, 60 g/liter) when CO2 production by the culture ceased. Anaerobic selection was continued in successive batches until no further improvement of the growth rate was seen, at which point the resulting population was named TMB 3420 (Table 2).
Reverse construction of selected mutations.
The XR N272D and XR P275Q mutations identified in TMB 3420 were reverse engineered into YIpOB8. The XYL1 gene from TMB 3420 was amplified and used as a “megaprimer” for reconstruction of the YIpOB8 plasmid as described above. The reconstructed plasmid was named YIpDR6 (Table 2) and carried the N272D and P275Q mutations identified in TMB 3420. In addition, the plasmids YIpDR7 and YIpDR8 carrying the individual mutations N272D and P275Q, respectively, were constructed using the YIpOB8 plasmid as a backbone. Plasmids YIpDR6, YIpDR7, and YIpDR8 were transformed into S. cerevisiae strain TMB 3044, yielding strains TMB 3421, TMB 3422, and TMB 3423, respectively (Table 2). A control strain, TMB 3424, carrying the native XYL1 gene, was also constructed by transforming TMB 3044 with YIpOB8.
Enzyme kinetics.
Cells were cultivated in shake flasks in defined mineral medium (xylose, 60 g/liter) and harvested in late exponential phase. Whole-cell protein extract was generated by treating the cells with Y-PER extraction solution (Pierce, Rockford, IL) according to the manufacturer's instructions. The total protein concentration in the extract was determined using Coomassie dye (Bio-Rad, Hercules, CA) and a bovine serum albumin standard (Pierce). XR kinetics were determined using xylose as the substrate, NADH or NADPH as a cofactor, and potassium phosphate (50 mM, pH 7.0) as the buffering agent. Enzyme activities were recorded using an Ultraspec 2100 Pro spectrophotometer (Amersham Biosciences, Uppsala, Sweden) at 340 nm [ɛNAD(P)H = 6.22 mM−1 cm−1] at room temperature (22 ± 1°C). Initial enzymatic rates (v) were fitted to a compulsory-order ternary complex kinetic model (equation 1) (5) using nonlinear regression (Matlab 2007b; Mathworks Inc., Natred, MA). The uncertainties of the fitted parameters were reported in terms of ±1 standard deviation (σ).
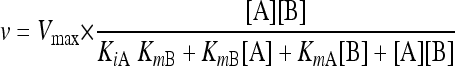 |
(1) |
where Vmax is the maximum reaction rate, A is NADPH or NADH (cofactor), B is xylose (substrate), KiA is the cofactor dissociation constant, KmA is the cofactor affinity constant, and KmB is the substrate affinity constant.
Analysis of substrate and products.
Metabolite concentrations were determined by HPLC using a Waters HPLC system (Milford, MA). An Aminex HPX-87H ion-exchange column (Bio-Rad, Hercules, CA) and a refractive index detector (RID-6a; Shimadzu, Kyoto, Japan) were used for separation and detection, respectively. The column temperature was 45°C, and 5 mM H2SO4 was used as a mobile phase at a flow rate of 0.6 ml/min. Cell dry weight was determined by filtering samples through a preweighed 0.45-μm membrane (Pall Corporation, New York, NY), washing them with distilled water, and drying them in a microwave oven for 8 min at 350 W.
Calculation of metabolic fluxes.
Specific rates of substrate consumption and product formation were determined from the exponential anaerobic growth phase in batch culture. The rates were calculated in Matlab (Matlab R2007b; MathWorks Inc., MA) by fitting the measured metabolite and cell concentrations to equations 2 and 3 using a differential equation solver and nonlinear regression. A pseudo-steady-state assumption was validated by observing constant specific consumption and production rates during 2 or 3 cell duplications, as well as the agreement of measured values between biological replicates. The ethanol production was corrected for evaporation (3 to 5%) based on measured evaporated ethanol in the off gas. Carbon balances in batch cultures closed to 90 to 110%.
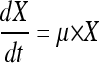 |
(2) |
where X is the biomass (g/liter) and μ is the specific growth rate (h−1).
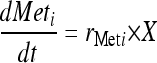 |
(3) |
where  is the concentration of a given metabolite (g/liter) and r is the specific consumption/production rate (g/g [DW] × h).
is the concentration of a given metabolite (g/liter) and r is the specific consumption/production rate (g/g [DW] × h).
RESULTS
Library construction.
Error-prone PCR was used to generate random mutations in the S. stipitis XYL1 gene. To maximize the number of relevant mutants, the mutation window was restricted to ∼250 bp surrounding the cofactor binding site of the protein (4, 22). Site-directed mutagenesis in this region has been found to change the cofactor specificity of the enzyme (Table 1 and references therein) and to increase the ethanol yield and productivity of strains expressing the mutated enzymes (2, 17, 33, 34, 44). The degree of completion of the resulting library was calculated based on the number of transformants, the length of the mutation window, and the mutation frequency (Table 3) (7, 31). Accordingly, the intermediate E. coli library contained 100% and 69% of all possible nucleotide sequences containing 1 and 2 mutations, respectively. The S. cerevisiae library contained 100% and 39% of all nucleotide sequences with 1 and 2 mutations, respectively, which corresponds to 55% of all possible single-amino-acid substitutions within the window of mutagenesis (Table 3). To approach completion of the library, it would have been necessary to increase the transformation frequency in E. coli and S. cerevisiae. By decreasing the size of the transforming plasmid, the transformation frequency in E. coli would have been increased (15, 36), whereas using an episomal instead of an integrative plasmid would have significantly increased the transformation frequency in S. cerevisiae (11). However, chromosomal integration of the library in S. cerevisiae was deliberately chosen to enable selection for enzymes with sufficiently high activity when expressed from a single gene copy.
TABLE 3.
Characteristics of constructed XYL1 librariesa
| Mutagenesis range |
E. coli sublibrary |
S. cerevisiae library |
||
|---|---|---|---|---|
| Size (CFU) | % Completion | Size (CFU) | % Completion | |
| +631-+870 | 1.3 × 106 | DNA (1), 100; DNA (2), 69 | 6.3 × 105 | DNA (1), 100; DNA (2), 39; protein (1), 55 |
The completion levels of the E. coli sublibrary and the S. cerevisiae library refer to the predicted contents of discrete mutations of a certain kind divided by the number of possible variants. DNA (1) refers to the fraction of all possible single-nucleotide mutations, DNA (2) refers to the fraction of all possible two-nucleotide mutations, and protein (1) refers to the fraction of all possible single-amino-acid mutations. The numbers were calculated using the PEDEL online tool (31).
Selection for mutations enabling anaerobic growth on xylose.
The S. cerevisiae library expressing mutated XYL1 genes was selected during sequential anaerobic batch cultivation on xylose as the sole carbon source. Anaerobic batch cultivation was used to select for a high growth rate, which is coupled with high ethanol productivity. Altogether, 10 batches were performed over a total time of 1,700 h (∼10 weeks). The largest increase in the growth rate was seen in the first half of the selection process, during which the apparent growth rate was increased four times (Fig. 1). During the remaining selection process, the growth rate increased only an additional 20%. In contrast, the substrate affinity of the cells continued to increase, as evidenced by longer exponential growth phases and higher cell concentrations in stationary phase (Fig. 1). Once the increase in growth performance was only incremental, the selection process was terminated to avoid selecting for adaptive mutations unrelated to XR. Sequencing of more than 20 individual clones from the selected S. cerevisiae population, TMB 3420, identified two conserved mutations in the XYL1 gene, A814G and C824A, corresponding to XR N272D and P275Q at the amino acid level.
FIG. 1.

Growth performance of an S. cerevisiae population expressing the XYL1 library during selection in successive anaerobic batches on 60 g/liter xylose.
Metabolic rates of strains carrying XR mutations.
The XR mutations identified in the selected population, TMB 3420, were reverse engineered in the parental strain, TMB 3044, yielding strains TMB 3421 (N272D/P275Q), TMB 3422 (N272D), and TMB 3423 (P275Q) (Table 2). The metabolic rates of the selected population, TMB 3420, were compared to those of the reverse-engineered strains and a control strain, TMB 3424, carrying the native S. stipitis XYL1 gene (Table 4). The strains were first cultivated under aerobic conditions in 60 g/liter xylose for 2 or 3 generations, after which the conditions were changed to anaerobiosis.
TABLE 4.
Metabolite consumption and production rates and yields during anaerobic growth on 60 g/liter xylosea
| Strain | μ | rs | rxol | rg | ra | re | Yse | Ysxol |
|---|---|---|---|---|---|---|---|---|
| TMB 3420 (selected population) | 0.079 ± 0.001 | 0.89 ± 0.03 | 0.12 ± 0.02 | 0.045 ± 0.005 | 0 | 0.32 ± 0.02 | 0.36 | 0.13 |
| TMB 3421 (N272D/P275Q) | 0.053 ± 0.08 | 0.57 ± 0.02 | 0.15 ± 0.02 | 0.018 ± 0.003 | 0 | 0.20 ± 0.02 | 0.35 | 0.26 |
| TMB 3422 (N272D) | 0.068 ± 0.04 | 0.62 ± 0.01 | 0.15 ± 0.03 | 0.017 ± 0.004 | 0 | 0.230 ± 0.003 | 0.37 | 0.24 |
| TMB 3423 (P275Q) | 0 | 0.097 ± 0.007 | 0.055 ± 0.009 | 0.0021 ± 0.0001 | 0.003 ± 0.003 | 0.017 ± 0.001 | 0.18 | 0.57 |
| TMB 3424 (control) | 0 | 0.06 ± 0.01 | 0.032 ± 0.008 | 0.002 ± 0.001 | 0 | 0.007 ± 0.002 | 0.12 | 0.53 |
μ, specific growth rate (h−1); r, specific consumption/production rates (g/g[DW]×h): rs, specific substrate consumption rate; rxol, specific xylitol production rate; rg, specific glycerol production rate; ra, specific acetate production rate; re, specific ethanol production rate; Y, yields (g/g): Yse, ethanol yield on consumed substrate; Ysxol, xylitol yield on consumed substrate. The uncertainty of the determined rates equals the deviation of the mean of two biological replicates.
The selected population, TMB 3420, and two of the reverse-engineered strains, TMB 3421 (N272D/P275Q) and TMB 3422 (N272D), displayed higher aerobic growth rates (μ = 0.24 ± 0.01 h−1) than the control strain and the reverse-engineered strain TMB 3423 (P275Q) (μ = 0.14 ± 0.01 h−1) (Table 4). Under anaerobic conditions, the difference between the strains became even more pronounced, as TMB 3420, TMB 3421 (N272D/P275Q), and TMB 3422 (N272D) grew anaerobically (μ = 0.053 to 0.079 h−1), whereas the control strain and strain TMB 3423 (P275Q) failed to grow (Table 4). Substrate uptake rates and product formation rates were consequently significantly higher in the selected population, TMB 3420, and the reverse-engineered strains TMB 3421 and TMB 3422 than in the control strain and the reverse-engineered strain TMB 3423 carrying only the P275Q mutation. In terms of product yields, a similar trend was observed, as TMB 3420, TMB 3421, and TMB 3422 displayed increased ethanol yields (Yse) and reduced xylitol yields (Ysxol) compared to the control strain and TMB 3423 (Table 4).
The ethanol productivity (re) of strain TMB 3420 (re = 0.32 g/g [DW] × h) and strains TMB 3421 and TMB 3422 (re = 0.20 to 0.22 g/g [DW] × h) was significantly higher than that of any reported strain using XR and XDH for xylose catabolism and was only comparable to strains expressing XI from multicopy plasmids (26, 27). The metabolic rates and the growth rate of the selected population, TMB 3420, were higher than those of any of the reverse-engineered strains, suggesting that the displayed phenotype cannot be completely explained by the mutations in XR. It is likely that the additional improvement results from adaptation during the selection process, as has been previously reported for evolutionary strain engineering (37, 45).
Enzyme kinetics of mutated XR.
Xylose reductase kinetics was assayed to investigate the relationship between the metabolic rates (Table 4) of strains TMB 3420-TMB 3424 and their respective XR activities (Table 5). The kinetic parameters of the native XR (TMB 3424) were comparable to previously reported values (Table 1 and references therein) and differed significantly from the kinetics of the XRs isolated from the selected population, TMB 3420, and the reverse-engineered strains TMB 3421 (N272D/P275Q) and TMB 3422 (N272D). In particular, the mutated XRs of TMB 3420, TMB 3421, and TMB 3422 exhibited approximately 10-times-higher Vmax than the native enzyme (Table 5). The higher Vmax of the mutant XRs coincides with loosened cofactor binding (KmA) in these enzymes (Table 5), which is in agreement with basic enzyme kinetics (5) and previous investigations (Table 1 and references therein).
TABLE 5.
Kinetic constants obtained from crude extracts for XR mutants constructed in the current studya
| Strain | Cofactor | Vmax (U/mg protein) | KmA (μM) | KmB (mM) | KiA (μM) | Rsel |
|---|---|---|---|---|---|---|
| TMB 3420 (selected population) | NADPH | 2.8 ± 0.1 | 28 ± 3 | 90 ± 20 | 40 ± 20 | 1.0 |
| NADH | 1.24 ± 0.05 | 17 ± 2 | 70 ± 10 | 23 ± 9 | ||
| TMB 3421 (N272D/P275Q) | NADPH | 3.51 ± 0.35 | 40 ± 10 | 50 ± 30 | 100 ± 100 | 0.7 |
| NADH | 2.0 ± 0.2 | 26 ± 8 | 80 ± 40 | 50 ± 50 | ||
| TMB 3422 (N272D) | NADPH | 2.4 ± 0.1 | 24 ± 5 | 40 ± 10 | 50 ± 30 | 0.8 |
| NADH | 1.09 ± 0.05 | 4 ± 2 | 36 ± 8 | 30 ± 10 | ||
| TMB 3423 (P275Q) | NADPH | 0.36 ± 0.01 | 5.5 ± 0.9 | 72 ± 9 | −3 ± 1 | NAb |
| NADH | 0.38 ± 0.09 | −2 ± 5 | 160 ± 70 | 6 ± 5 | ||
| TMB 3424 (control) | NADPH | 0.28 ± 0.02 | 1.6 ± 0.6 | 35 ± 8 | 1 ± 1 | 0.1 |
| NADH | 0.21 ± 0.05 | 9 ± 5 | 70 ± 40 | 3 ± 5 |
The uncertainty of the presented values is given in terms of 1 standard deviation (σ). For definitions of kinetic constants, see Table 1.
NA, not applicable.
The relative efficiency of NADH/NADPH utilization, here referred to as the selectivity ratio (Rsel), was calculated using equation 4 (5).
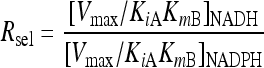 |
(4) |
For definitions of the parameters, see equation 1.
Although this equation technically is valid only at low substrate and cofactor concentrations, it has been useful to approximate the overall cofactor specificity of XR (32). For XRs isolated from TMB 3420, TMB 3421, and TMB 3422, Rsel was 7 to 10 times higher than for the native enzyme (Table 5). The higher Rsel values of the mutated enzymes suggest increased NADH utilization, which in turn leads to reduced cofactor imbalance in the XR/XDH reaction couple. On a metabolic level, the altered cofactor balance is reflected by significantly reduced xylitol production in the strains expressing the mutant XRs (Table 4).
The kinetics of the XR P275Q mutant did not fit the mechanism in equation 1 as well as the other enzymes, which was indicated by negative values of two of the fitted parameters, as well as generally higher uncertainties. Nevertheless, the XR P275Q mutant appeared much more similar to the native enzyme than to the mutant enzymes of TMB 3420, TMB 3421, and TMB 3422 (Table 5).
DISCUSSION
The XR activity tightly controls the flux of xylose in recombinant xylose-utilizing S. cerevisiae (19, 21). The Vmax of XR is ∼3 to 10 times lower than those of most glycolytic enzymes, and the substrate affinity, KmB, is at least 2 orders of magnitude higher (14, 38). In addition, the different cofactor utilization of XR and XDH reduces ethanol yield and ethanol productivity (24). In the current work, a novel strain with superior ethanol productivity (re = 0.32 g/g [DW] × h) and anaerobic growth rate (μ = 0.08 h−1) was generated by a combination of random mutagenesis of XR and selection for anaerobic xylose growth. In contrast to previous studies (32, 44), this setup allowed selection among hundreds of thousands of enzyme variants for mutations that benefit ethanol productivity in vivo. Strains TMB 3420, TMB 3421, and TMB 3422, consequently, have the highest ethanol productivity reported for any xylose-utilizing S. cerevisiae strain employing the XR/XDH pathway (Table 4) (21, 24, 33, 37, 40, 42).
Population TMB 3420 was selected following repeated anaerobic batch cultivation. Two XR mutations, N272D and P275Q, were found in all sequenced clones of TMB 3420, which indicated that the population had reached genetic homogeneity. Compared to the control strain harboring the native XR, metabolic rates and anaerobic growth rates were improved by more than an order of magnitude in TMB 3420 (Table 4). The improved fermentative properties of TMB 3420 were readily reverse engineered in the parental strain by introducing the XR N272D mutation alone (TMB 3422) or together with P275Q (TMB 3421). Introducing the P275Q mutation alone (TMB 3423) only marginally improved the metabolic rates (Table 4), indicating that this mutation was not necessary.
A direct relationship between XR kinetics and overall metabolic rates was demonstrated in this study (Tables 4 and 5). The XR kinetics of TMB 3423 (P275Q) were similar to those of the control strain, which supports the idea that N272D was the primarily selected mutation. The mutant enzymes of TMB 3420, TMB 3421, and TMB 3422 exhibited significantly increased NADH specificities (Rsel) and maximum reaction rates (Vmax) compared to the control strain (Table 5). The increased Rsel of the XRs of TMB 3420, TMB 3421, and TMB 3422 suggests that cofactor recycling between XR and XDH was improved in these strains, which was reflected in the reduced xylitol secretion (Tables 4 and 5). Similarly, the higher Vmax of the XRs of TMB 3420, TMB 3421, and TMB 3422 was reflected in an increased growth rate and ethanol productivity in these strains (Tables 4 and 5). Previously, the low Vmax of native XR has been overcome by introducing multiple gene copies, which in an analogous way resulted in increased metabolic rates (19, 21).
The difference in metabolic rates between the selected population, TMB 3420, and TMB 3421/TMB 3422 was small compared to the control strain and TMB 3423 (P275Q), but not negligible (Table 4). In contrast to the control strain and TMB 3423, the difference in metabolic rates between TMB 3420 and TMB 3421/TMB 3422 (Table 4) could not be correlated with the XR kinetics (Table 5). It is thus likely that the additional improvement of TMB 3420 results from nonspecific adaptive changes elsewhere in the genome. Although this study shows that adaptation is an efficient way to increase metabolic rates, such improvement is only meaningful in a final production strain. In fact, the selection process was deliberately terminated as soon as enrichment for beneficial XR mutations was complete to minimize adaptive changes of unknown molecular origin which could not be transferred between strains.
Interestingly, the N272D mutation responsible for the increased ethanol productivity in the current study was also constructed in a previous study by site-directed mutagenesis (32) (Table 1). Although this coincidence is a testament to the careful enzymatic design conducted by Petschacher and coworkers, the N272D mutation was found in their continued work to have comparatively minor metabolic effects (25, 33). The difference is most likely related to the different S. cerevisiae strains used in these investigations. For instance, the nonoxidative pentose phosphate pathway was not overexpressed in the Petschacher strain, which is a prerequisite for efficient xylose utilization (20, 27).
The currently employed selection method offers an advantage over site-directed mutagenesis, as it selects for mutations that increase in vivo ethanol productivity. The more closely the selection step mimics the final application (i.e., industrial ethanol production), the more relevant the selected mutations will be. In this context, the decision to use integrative rather than episomal vectors in construction of the library was made in consideration of the final application. Although episomal vectors would have increased the size of the library by increasing the transformation efficiency (10), the selection pressure for high Vmax would have been reduced by higher background expression from multiple gene copies. Mutations selected using episomal plasmids would thus not be optimal for industrial yeast strains with chromosomally integrated genetic constructs, and a successful transfer of the improved phenotype would not be guaranteed.
Acknowledgments
The Swedish Energy Agency (Energimyndigheten) is gratefully acknowledged for funding this research.
Cyril Rols is acknowledged for excellent technical assistance.
Footnotes
Published ahead of print on 1 October 2010.
REFERENCES
- 1.Andreasen, A. A., and T. J. Stier. 1953. Anaerobic nutrition of Saccharomyces cerevisiae. I. Ergosterol requirement for growth in a defined medium. J. Cell Physiol. 41:23-36. [DOI] [PubMed] [Google Scholar]
- 2.Bengtsson, O., B. Hahn-Hägerdal, and M. F. Gorwa-Grauslund. 2009. Xylose reductase from Pichia stipitis with altered coenzyme preference improves ethanolic xylose fermentation by recombinant Saccharomyces cerevisiae. Biotechnol. Biofuels 2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohren, K. M., J. L. Page, R. Shankar, S. P. Henry, and K. H. Gabbay. 1991. Expression of human aldose and aldehyde reductases. Site-directed mutagenesis of a critical lysine 262. J. Biol. Chem. 266:24031-24037. [PubMed] [Google Scholar]
- 4.Carugo, O., and P. Argos. 1997. NADP-dependent enzymes. I. Conserved stereochemistry of cofactor binding. Proteins 28:10-28. [DOI] [PubMed] [Google Scholar]
- 5.Cornish-Bowden, A. 2004. Fundamentals of enzyme kinetics, 3rd ed. Portland Press Ltd., London, United Kingdom.
- 6.Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16:6127-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firth, A. E., and W. M. Patrick. 2005. Statistics of protein library construction. Bioinformatics 21:3314-3315. [DOI] [PubMed] [Google Scholar]
- 8.Galbe, M., and G. Zacchi. 2007. Pretreatment of lignocellulosic materials for efficient bioethanol production. Biofuels 108:41-65. [DOI] [PubMed] [Google Scholar]
- 9.Gárdonyi, M., M. Jeppsson, G. Liden, M. F. Gorwa-Grausland, and B. Hahn-Hägerdal. 2003. Control of xylose consumption by xylose transport in recombinant Saccharomyces cerevisiae. Biotechnol. Bioeng. 82:818-824. [DOI] [PubMed] [Google Scholar]
- 10.Gietz, R. D., and R. H. Schiestl. 2007. Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2:38-41. [DOI] [PubMed] [Google Scholar]
- 11.Gietz, R. D., R. H. Schiestl, A. R. Willems, and R. A. Woods. 1995. Studies on the transformation of intact yeast cells by the Liac/S-DNA/PEG procedure. Yeast 11:355-360. [DOI] [PubMed] [Google Scholar]
- 12.Hahn-Hägerdal, B., K. Karhumaa, M. Jeppsson, and M. F. Gorwa-Grauslund. 2007. Metabolic engineering for pentose utilization in Saccharomyces cerevisiae. Adv. Biochem. Eng. Biotechnol. 108:147-177. [DOI] [PubMed] [Google Scholar]
- 13.Hahn-Hägerdal, B., K. Karhumaa, C. U. Larsson, M. F. Gorwa-Grauslund, J. Gorgens, and W. H. van Zyl. 2005. Role of cultivation media in the development of yeast strains for large scale industrial use. Microb. Cell Fact. 4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hynne, F., S. Dano, and P. G. Sorensen. 2001. Full-scale model of glycolysis in Saccharomyces cerevisiae. Biophys. Chem. 94:121-163. [DOI] [PubMed] [Google Scholar]
- 15.Inoue, H., H. Nojima, and H. Okayama. 1990. High-efficiency transformation of Escherichia coli with plasmids. Gene 96:23-28. [DOI] [PubMed] [Google Scholar]
- 16.Jeffries, T. W. 2006. Engineering yeasts for xylose metabolism. Curr. Opin. Biotechnol. 17:320-326. [DOI] [PubMed] [Google Scholar]
- 17.Jeppsson, M., O. Bengtsson, K. Franke, H. Lee, B. Hahn-Hägerdal, and M. F. Gorwa-Grauslund. 2006. The expression of a Pichia stipitis xylose reductase mutant with higher KM for NADPH increases ethanol production from xylose in recombinant Saccharomyces cerevisiae. Biotechnol. Bioeng. 93:665-673. [DOI] [PubMed] [Google Scholar]
- 18.Jeppsson, M., B. Johansson, B. Hahn-Hägerdal, and M. F. Gorwa-Grauslund. 2002. Reduced oxidative pentose phosphate pathway flux in recombinant xylose-utilizing Saccharomyces cerevisiae strains improves the ethanol yield from xylose. Appl. Environ. Microbiol. 68:1604-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeppsson, M., K. Träff, B. Johansson, B. Hahn-Hägerdal, and M. F. Gorwa-Grauslund. 2003. Effect of enhanced xylose reductase activity on xylose consumption and product distribution in xylose-fermenting recombinant Saccharomyces cerevisiae. FEMS Yeast Res. 3:167-175. [DOI] [PubMed] [Google Scholar]
- 20.Johansson, B., and B. Hahn-Hägerdal. 2002. The non-oxidative pentose phosphate pathway controls the fermentation rate of xylulose but not of xylose in Saccharomyces cerevisiae TMB3001. FEMS Yeast Res. 2:277-282. [DOI] [PubMed] [Google Scholar]
- 21.Karhumaa, K., B. Hahn-Hägerdal, and M. F. Gorwa-Grauslund. 2005. Investigation of limiting metabolic steps in the utilization of xylose by recombinant Saccharomyces cerevisiae using metabolic engineering. Yeast 22:359-368. [DOI] [PubMed] [Google Scholar]
- 22.Kavanagh, K. L., M. Klimacek, B. Nidetzky, and D. K. Wilson. 2003. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochem. J. 373:319-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostrzynska, M., C. R. Sopher, and H. Lee. 1998. Mutational analysis of the role of the conserved lysine-270 in the Pichia stipitis xylose reductase. FEMS Microbiol. Lett. 159:107-112. [DOI] [PubMed] [Google Scholar]
- 24.Kötter, P., and M. Ciriacy. 1993. Xylose fermentation by Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 38:776-783. [Google Scholar]
- 25.Krahulec, S., B. Petschacher, M. Wallner, K. Longus, M. Klimacek, and B. Nidetzky. 2010. Fermentation of mixed glucose-xylose substrates by engineered strains of Saccharomyces cerevisiae: role of the coenzyme specificity of xylose reductase, and effect of glucose on xylose utilization. Microb. Cell Fact. 9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuyper, M., H. R. Harhangi, A. K. Stave, A. A. Winkler, M. S. M. Jetten, W. de Laat, J. J. J. den Ridder, H. J. M. Op den Camp, J. P. van Dijken, and J. T. Pronk. 2003. High-level functional expression of a fungal xylose isomerase: the key to efficient ethanolic fermentation of xylose by Saccharomyces cerevisiae? FEMS Yeast Res. 4:69-78. [DOI] [PubMed] [Google Scholar]
- 27.Kuyper, M., M. M. Hartog, M. J. Toirkens, M. J. Almering, A. A. Winkler, J. P. van Dijken, and J. T. Pronk. 2005. Metabolic engineering of a xylose-isomerase-expressing Saccharomyces cerevisiae strain for rapid anaerobic xylose fermentation. FEMS Yeast Res. 5:399-409. [DOI] [PubMed] [Google Scholar]
- 28.Lee, J. K., B. S. Koo, and S. Y. Kim. 2003. Cloning and characterization of the XYL1 gene, encoding an NADH-preferring xylose reductase from Candida parapsilosis, and its functional expression in Candida tropicalis. Appl. Environ. Microbiol. 69:6179-6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazaki, K., and M. Takenouchi. 2002. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques 33:1033-1034, 1036-1038. [DOI] [PubMed] [Google Scholar]
- 30.Morjana, N. A., C. Lyons, and T. G. Flynn. 1989. Aldose reductase from human psoas muscle. Affinity labeling of an active site lysine by pyridoxal 5′-phosphate and pyridoxal 5′-diphospho-5′-adenosine. J. Biol. Chem. 264:2912-2919. [PubMed] [Google Scholar]
- 31.Patrick, W. M., and A. E. Firth. 2005. Strategies and computational tools for improving randomized protein libraries. Biomol. Eng. 22:105-112. [DOI] [PubMed] [Google Scholar]
- 32.Petschacher, B., S. Leitgeb, K. L. Kavanagh, D. K. Wilson, and B. Nidetzky. 2005. The coenzyme specificity of Candida tenuis xylose reductase (AKR2B5) explored by site-directed mutagenesis and X-ray crystallography. Biochem. J. 385:75-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petschacher, B., and B. Nidetzky. 2008. Altering the coenzyme preference of xylose reductase to favor utilization of NADH enhances ethanol yield from xylose in a metabolically engineered strain of Saccharomyces cerevisiae. Microb. Cell Fact. 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Runquist, D., B. Hahn-Hagerdal, and M. Bettiga. 2009. Increased expression of the oxidative pentose phosphate pathway and gluconeogenesis in anaerobically growing xylose-utilizing Saccharomyces cerevisiae. Microb. Cell Fact. 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saloheimo, A., J. Rauta, O. V. Stasyk, A. A. Sibirny, M. Penttila, and L. Ruohonen. 2007. Xylose transport studies with xylose-utilizing Saccharomyces cerevisiae strains expressing heterologous and homologous permeases. Appl. Microbiol. Biotechnol. 74:1041-1052. [DOI] [PubMed] [Google Scholar]
- 36.Siguret, V., A. S. Ribba, G. Cherel, D. Meyer, and G. Pietu. 1994. Effect of plasmid size on transformation efficiency by electroporation of Escherichia coli DH5-alpha. Biotechniques 16:422-424. [PubMed] [Google Scholar]
- 37.Sonderegger, M., and U. Sauer. 2003. Evolutionary engineering of Saccharomyces cerevisiae for anaerobic growth on xylose. Appl. Environ. Microbiol. 69:1990-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teusink, B., J. Passarge, C. A. Reijenga, E. Esgalhado, C. C. van der Weijden, M. Schepper, M. C. Walsh, B. M. Bakker, K. van Dam, H. V. Westerhoff, et al. 2000. Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 267:5313-5329. [DOI] [PubMed] [Google Scholar]
- 39.van Maris, A. J., A. A. Winkler, M. Kuyper, W. T. de Laat, J. P. van Dijken, and J. T. Pronk. 2007. Development of efficient xylose fermentation in Saccharomyces cerevisiae: xylose isomerase as a key component. Adv. Biochem. Eng. Biotechnol. 108:179-204. [DOI] [PubMed] [Google Scholar]
- 40.van Zyl, W. H., A. Eliasson, T. Hobley, and B. Hahn-Hagerdal. 1999. Xylose utilisation by recombinant strains of Saccharomyces cerevisiae on different carbon sources. Appl. Microbiol. Biotechnol. 52:829-833. [DOI] [PubMed] [Google Scholar]
- 41.Verduyn, C., E. Postma, W. A. Scheffers, and J. P. van Dijken. 1992. Effect of benzoic acid on metabolic fluxes in yeasts—a continuous culture study on the regulation of respiration and alcoholic fermentation. Yeast 8:501-517. [DOI] [PubMed] [Google Scholar]
- 42.Wahlbom, C. F., W. H. van Zyl, L. J. Jonsson, B. Hahn-Hägerdal, and R. R. Otero. 2003. Generation of the improved recombinant xylose-utilizing Saccharomyces cerevisiae TMB 3400 by random mutagenesis and physiological comparison with Pichia stipitis CBS 6054. FEMS Yeast Res. 3:319-326. [DOI] [PubMed] [Google Scholar]
- 43.Wang, P. Y., and H. Schneider. 1980. Growth of yeasts on D-xylulose 1. Can. J. Microbiol. 26:1165-1168. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe, S., A. Abu Saleh, S. P. Pack, N. Annaluru, T. Kodaki, and K. Makino. 2007. Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein-engineered NADH-preferring xylose reductase from Pichia stipitis. Microbiology 153:3044-3054. [DOI] [PubMed] [Google Scholar]
- 45.Wisselink, H. W., M. J. Toirkens, Q. Wu, J. T. Pronk, and A. J. van Maris. 2009. Novel evolutionary engineering approach for accelerated utilization of glucose, xylose, and arabinose mixtures by engineered Saccharomyces cerevisiae strains. Appl. Environ. Microbiol. 75:907-914. [DOI] [PMC free article] [PubMed] [Google Scholar]