

Abstract
GluK2 is a kainate receptor subunit that is alternatively spliced at the C-terminus. Previous studies implicated GluK2 in autism. In particular, the methionine-to-isoleucine replacement at amino acid residue 867 (M867I) that can only occur in the longest isoform of the human GluK2 (hGluK2), as the disease (autism) mutation, is thought to cause gain-of-function. However, the kinetic properties of the wild-type hGluK2 and the functional consequence of this gain-of-function mutation at the molecular level are not well understood. To investigate whether the M867I mutation affects the channel properties of the human GluK2 kainate receptor, we have systematically characterized the rate and the equilibrium constants pertinent to channel opening and channel desensitization for this mutant and the wild-type hGluK2 receptor, along with the wild-type rat GluK2 kainate receptor (rGluK2) as the control. Our results show that the M867I mutation does not affect either the rate or the equilibrium constants of the channel opening but does slow down the channel desensitization rate by ~1.6-fold at saturating glutamate concentrations. It is possible that a consequence of this mutation on the desensitization rate is linked to facilitating the receptor trafficking and membrane expression, given the close proximity of M867 to the forward trafficking motif in the C-terminal sequence. By comparing the data of the wild-type human and rat GluK2 receptors, we also find that the human GluK2 has a ~3-fold smaller channel-opening rate constant but an identical channel-closing rate constant, and thus a channel-opening probability of 0.85 vs. 0.96 for rGluK2. Furthermore, the intrinsic equilibrium dissociation constant K1 for hGluK2, like the EC50 value, is ~2-fold lower than rGluK2. Our results therefore suggest that the human GluK2 is relatively a slowly activating channel but more sensitive to glutamate, as compared to the rat ortholog, despite the fact that the human and rat forms share 99% sequence homology.
GluK2 (previously known as GluR6) is a subunit of the glutamate ion channel receptor family of kainate subtype (1–3). Within this family, glutamate ion channel receptors are subdivided by their prototypical agonists into N-methyl-D-aspartate (NMDA) receptors, α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate (AMPA) receptors, and kainate receptors (3). Within the kainate receptor subtype, there are five subunits, i.e., GluK1–5. GluK2, like GluK1 and 3, can form functional, homomeric channels, whereas GluK4–5 can only form functional channels by co-assembling with any one of the GluK1–3 subunits (3). Studies have shown that kainate receptors are involved in regulating both excitatory and inhibitory neurotransmission, and the trafficking of kainate receptors is also involved in altering neurotransmission during synaptic plasticity and neuronal development (see a recent review by Jane et al. (1)). On the other hand, abnormal function of kainate receptors is implicated in epilepsy, pain and ischemia, and therefore, kainate receptors are a potential target of therapeutic development (1, 4–6).
Among some specific roles that GluK2 plays (7–9), Jamain et al. (10) hypothesized that human GluK2 (hGluK2) (11) is in linkage disequilibrium with autism, a pervasive developmental disorder (12). This hypothesis is based on a whole-genome linkage scanning study of susceptibility genes of autism, which led to the identification of hGluK2 in chromosome 6q21 (13–15). Screening of 33 autistic patients further revealed a mutation, i.e., methionine-to-isoleucine replacement at amino acid 867 or M867I, in the cytoplasmic C-terminal region of GluK2 (10). A recent study further suggested that the M867I substitution causes gain-of-function or potentiation in that the mutant activity is significantly enhanced as compared with the wild-type GluK2 (16). Specifically, the potentiation is thought to manifest itself in a significant increase in current amplitude and membrane expression of the mutant GluK2, as compared with the wild-type receptor (16). It should be pointed out that Strutz-Seebohm et al. (16) observed the current increase in Xenopus oocytes expressing the wild-type and the mutant GluK2 from the rat origin (rGluK2). The choice of using the rat receptor appeared to be reasonable, given the fact that the rat and the human GluK2 share 99% sequence homology (11), and the M867 is a conserved amino acid residue in both forms. Although the oocyte experiment by Strutz-Seebohm et al. (16) has provided convincing evidence from immunolabeling and fluorescence imagining measurements that the M867I mutation enhances the surface expression of the receptor, the same study did not reveal any quantitative information about the molecular mechanism of macroscopic current augmentation. In fact, there have been very few studies to date for characterizing the kinetic properties of human GluK2.
Based on these early findings, we set out to address two specific questions. First, did the M867I mutation affect the kinetic and equilibrium constants of the hGluK2 channel activities that led to potentiation of macroscopic current amplitude? Second, were there any differences in the kinetic and equilibrium constants between the human and the rat wild-type GluK2? Different from the study by Strutz-Seebohm et al. (16), we constructed the human M867I mutant and compared the results with the human wild-type GluK2. We also collected the data from the wild-type rGluK2 not only to address the second question but also as the control for our study described here (despite the fact that we published the rat data previously) (17). To specifically investigate the potential effect of the M867I mutation on the channel-opening kinetic process of GluK2, we used a laser-pulse photolysis technique, together with a photolabile precursor of glutamate or caged glutamate (18), which provided ~60 microsecond (μs) time resolution (17, 19), suitable for the characterization of the channel-opening kinetic mechanism (17).
EXPERIMENTAL PROCEDURES
cDNA Plasmids
The plasmid DNA (pcDNA3.1-Hyg/HiGluK2) encoding the wild-type hGluK2, the unedited isoform or the Q form, was kindly provided by David Bleakman (Eli Lilly). The cDNA for the M867I mutant hGluK2 was constructed from the wild-type hGluK2 by using QuikChange® Site-Directed Mutagenesis Kit (Stratagene). Two complimentary oligonucleotide primers carrying the M867I mutation were synthesized (Operon) and used to introduce a silent mutation for the construction of Sac I restriction enzyme site without amino acid changes at the 864th and the 865th amino acid residues. The silent mutation facilitated the screening of the mutated plasmid carrying Met-to-Ile at the 867th amino acid position by Sac I digestion. The Met-to-Ile substitution was confirmed by DNA sequencing. It should be noted that the naming of this mutation or M867I follows its original numbering (10), which included the 31 amino-acid signal peptide, rather than just the number of amino acid residues in the mature protein.
Expression of cDNAs and Cell Culture
The cDNA plasmids were propagated through the E. coli host (DH5α) and were purified using a DNA purification kit (QIAGEN, Valencia, CA). All of the receptors were individually expressed in human embryonic kidney (HEK-293S) cells by a standard calcium phosphate transfection method (19). HEK-293S cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin in a 37 °C, 5% CO2, humidified incubator. The weight ratio of the plasmid for any receptor to green fluorescent protein, which was cotransfected as a marker, was 2:1, with a receptor gene being 2–3 μg/35 mm Petri dish. Transfected cells were allowed to grow for >24 h before recording.
Whole-Cell Current Recording
Electrodes for whole-cell recording were made from glass capillaries from World Precision Instruments (Sarasota, FL) and fire polished. The electrode had a resistance about 3 MΩ when filled with the pipet solution. The pipet solution contained (in mM) 110 CsF, 30 CsCl, 4 NaCl, 0.5 CaCl2, 5 EGTA, and 10 HEPES (pH 7.4 adjusted by NaOH). The external cellular solution contained (in mM) 150 NaCl, 3 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4 adjusted by NaOH). All chemicals used for making buffers were from commercial sources. Green fluorescence in the transfected cells was visualized using a Carl Zeiss Axiovert S100 microscope equipped with a fluorescent detection system (Thornwood, NY). The glutamate-induced whole-cell current was recorded using an Axopatch 200B amplifier at a cutoff frequency of 2–20 kHz by a builtin, 4-pole low-pass Bessel filter and digitized at 5–50 kHz sampling frequency using a Digidata 1322A from Molecular Devices (Sunnyvale, CA). pClamp 8 (also from Molecular Devices) was used for data acquisition. All recordings were preformed with transfected HEK-293S cells voltage-clamped at −60 mV and 22 °C.
Laser-Pulse Photolysis Measurement
The laser-pulse photolysis technique, together with γ-O-(α-carboxy-2-nitrobenzyl)glutamate or caged glutamate (18) (Invitrogen), was used for measuring the rate of GluK2 receptor channel opening (17, 19). We also used the MNI-caged glutamate (or 4-methoxy-7-nitroindolinyl-caged-L-glutamate) (20) for some of our measurements reported in this study. We tested the MNI-caged glutamate extensively with GluK2 receptors and other glutamate receptor channels, and found no difference in the rate constant measurement with the same receptor. Caged glutamate (or free glutamate) was applied to an HEK-293S cell suspended in the extracellular solution using a U-shaped or U-tube flow device (17, 19). The cell was equilibrated with caged glutamate for 250 ms, before irradiated by a laser pulse to liberate free glutamate. A Minilite II pulsed Q-switched Nd:YAG laser (Continuum, Santa Clara, CA), tuned by a third harmonic generator, delivered single pulses at 355 nm with a pulse length of 8 ns. The laser light was coupled into a fiber optic from FiberGuide Industries (Stirling, NJ), and the power was adjusted to be 200–800 μJ. For a laser-pulse photolysis measurement of a channel-opening kinetic rate constant, we also used at least two known concentrations of free glutamate with the same cell before and after a laser flash to calibrate the concentration of photolytically released glutamate (17). The current amplitudes obtained for the flow measurements were compared with the amplitude of the laser measurement with reference to the dose-response relationship. The flow measurement also allowed us to monitor any damage to the receptors and/or the cell for successive laser experiments with the same cell.
Data Analysis
In the laser-pulse photolysis measurement of the channel-opening reaction, we observed that the whole-cell current rise followed a single-exponential rate process (>95%) in the entire range of ligand (glutamate) concentrations we were able to measure (20 μM – 410 μM glutamate or 4% – 80% of the fraction of the open channel population – see the dose-response curve in the Results). As such, the observed rate constant of channel opening, kobs, was empirically determined by eq 1
| (eq 1) |
where It and IA represent the whole-cell current amplitude at time t and the maximum current amplitude, respectively. Furthermore, eq 2 was formulated to count for the relationship of kobs with ligand concentration according to a general mechanism (Figure 1) (all of the terms were defined either in the text or in the legend of Figure 1):
| (eq 2) |
Figure 1.
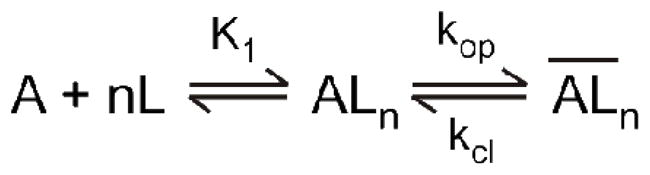
A general mechanism of channel opening for kainate receptors. Here, A stands for the active, unliganded form of the receptor, L the ligand or glutamate, ALn the closed-channel state with n ligand molecules bound, and the open-channel state. The number of glutamate molecules to bind to the receptor and to open its channel, n, can be from 1 to 4, assuming that a receptor is a tetrameric complex and each subunit has one glutamate binding site. It is further assumed that a ligand does not dissociate from the open-channel state. The kop and kcl are the channel-opening and channel-closing rate constants, respectively. For simplicity and without contrary evidence, it is assumed that glutamate binds with equal affinity or K1, the intrinsic equilibrium dissociation constant, at all binding steps.
In deriving eq 2, the rate of ligand binding was assumed to be fast relative to the rate of channel opening. This assumption was again consistent with our observation that the whole-cell current rise (described in detail in the Results) followed a first-order rate law (eq 1) in the entire range of glutamate concentrations not only in this study but also in all of our previous studies of the channel-opening reactions for both kainate receptors (17) and AMPA receptors (19, 21–24). According to eq 2, an analysis of a set of kcl and kop as well as K1 was correlated to a particular number of ligand(s) or n bound to and subsequently opened the receptor channel. Specifically, n ranges from 1 to 4 (Figure 1), based on the notion that a functional receptor channel is a tetramer and each subunit contains a ligand binding site (25–29).
In our experiment, K1 was estimated independently from the dose-response relationship as in eq 3, which was also derived based on the general mechanism of channel opening (Figure 1). In eq 3, IM is the current per mole of receptor, RM the number of moles of receptors on the cell surface, and Φ−1 the channel opening equilibrium constant.
| (eq 3) |
Each data point shown in the plots of this study was an average of at least three measurements collected from at least three cells unless otherwise noted. Linear regression and nonlinear fitting were performed using Origin 7 (Origin Lab, Northampton, MA). Unless otherwise noted, standard error of mean was reported.
RESULTS
Dependence of Desensitization Rate Constants on Glutamate Concentration for the Wild-Type hGluK2 and the M867I Mutant hGluK2
We hypothesized that a macroscopic “potentiation” of the current amplitude, as observed in two-electrode recording of Xenopus oocytes by Strutz-Seebohm et al. (16), could be contributed by the change of gating property of the M867I mutant in that a left-shift of K1 or qualitatively EC50 value (i.e., the ligand concentration that corresponds to 50% of the maximum response) would result in a higher current amplitude at a given glutamate concentration (but not at a saturating concentration for both the wild-type and the mutant). Alternatively, a slower desensitization rate observed at a given glutamate concentration for the mutant would also seemingly generate a higher current amplitude from a solution exchange measurement, because fewer channels would inactivate during the current rise time.
Based on these hypotheses, we first measured the whole-cell current response of the wild-type hGluK2 and the M867I mutant individually expressed in HEK-293 cells. A series of representative whole-cell current response to glutamate at various concentrations rose initially due to channel activation, and then fell to the baseline due to channel desensitization or transient channel inactivation in the continued presence of glutamate (Figure 2A). Yet, either nontransfected HEK-293 cells or GFP-transfected cells but without glutamate receptor genes were not responsive to glutamate application (17). The desensitization or the falling phase of the whole-cell current (as shown in Figure 2A) proceeded with a single first-order rate process for >98 % of the reaction and in the entire range of glutamate concentration for all of the receptors. The desensitization rate constant or kdes for both the wild-type and mutant hGluK2 became larger as higher glutamate concentrations were used (Figure 2B). In both cases, kdes reached a plateau around 2 mM glutamate (Figure 2B). However, the wild-type hGluK2 exhibited a maximum kdes of 196 ± 45 s−1 (average of 12 cells between 2 and 5 mM glutamate, black symbol in Figure 2B) whereas the maximum kdes for the mutant hGluK2 only reached 126 ± 16 s−1 (average of 20 cells between 2 and 5 mM glutamate, red symbol in Figure 2B). Some representative whole-cell traces are in Figure 2D. These results showed that the M836I mutation slowed the rate of desensitization.
Figure 2.

(A) Representative traces of whole-cell current measured from a single HEK-293 cell expressing the wild-type hGluK2. Glutamate was applied at time zero, and the concentrations of glutamate were, from bottom up, 2, 0.5, 0.2, 0.1 and 0.05 mM, respectively. (B) Dependence of desensitization rate constant on glutamate concentration for the wild-type hGluK2 (black circle) and the M867I mutant (red circle). Each data is an average of at least three data points from three cells. A total of 39 and 29 cells were measured for the wild-type and mutant GluK2s, respectively. (C) Dependence of desensitization rate constant on glutamate concentration for the wild-type rGluK2. A total of 52 cells were measured, and each data as shown is an average of at least three data points from three cells. (D) Representative whole-cell current traces for the wild-type rGluK2 (left panel), wild-type hGluK2 (middle panel) and M867I mutant hGluK2 (right panel). All traces were collected at 2 mM glutamate. The desensitization rate constant is, from left to right, 234, 212 and 118 s−1, respectively.
We further characterized the desensitization rate profile for the wild-type, rGluK2, as a control. We found that the rGluK2 desensitized with a glutamate-dependent profile (Figure 2C) similar to the wild-type hGluK2. Furthermore, rGluK2 had the maximum kdes of 230 ± 32 s−1, observed at or above 2 mM glutamate (based on an average of 22 cells between 2 and 5 mM glutamate, in Figure 2C). It should be noted that our kdes of 230 s−1 for rGluK2 is in good agreement with the range of the values published by others for the same receptor (see a list of these values in Table S1, Supporting Information). However, the maximum kdes of 230 s−1 for rGluK2 we observe in this study was slightly larger than the one we reported earlier (17); this is due to an improved solution flow system. The rise time (10–90%) of the whole-cell current using our flow system with saturating glutamate concentrations was 1.0 ± 0.2 ms for rGluK2 (based on the average of the measurement of 15 cells). This value was in good agreement with the one observed in whole-cell recording configuration for rGluK2 or ~1 ms reported by Vivithanaporn et al. (30), for example, but was larger than ~0.5 ms observed in the outside-out configuration for the same receptor (31–32). Similar to the rGluK2, an average rise time of 1.1 ± 0.3 ms for wild-type hGluK2 (20 cells) and 1.0 ± 0.2 ms for the mutant hGluK2 (20 cells) was also observed.
Dependence of the Whole-Cell Current Response on Glutamate Concentration for the Wild-Type hGluK2 and the M867I Mutant
Next we characterized the dose-response relationship for both the wild-type and the mutant hGluK2 in order to estimate K1 (i.e., the intrinsic equilibrium dissociation constant of the ligand as in Figure 1). As seen in Figure 3A, the dose-response relationship between the wild-type hGluK2 and the mutant receptor was statistically indistinguishable. As such, the best fit of the wild-type and the mutant hGluK2 data by eq 3 yielded K1 of 170 ± 23 μM (left panel in Figure 3, and Table 1) with n = 2 (here n represents the number of ligand molecules bound to the receptor to open the channel; see also Figure 1). If the dose-response curve were analyzed separately, K1 would be 180 ± 28 μM for the wild-type and 140 ± 30 μM for the mutant receptor. For the same dose-response data, we also estimated the EC50 value by the Hill equation (33). We found that EC50 = 150 ± 10 μM and the Hill coefficient = 1.1 (Figure S1, Supporting Information). When the dose-response relationships of the wild-type and the mutant hGluK2 receptors were separately analyzed, EC50 of 180 ± 30 μM and the Hill coefficient of 1.4 for the wild-type hGluK2 and EC50 of 130 ± 40 μM and the Hill coefficient of 1.3 for the M867I mutant hGluK2 would be respectively obtained. On the basis of these results, we concluded that the mutation M867I did not affect the dose-response relationship, nor K1 or EC50 value.
Figure 3.

Dose-response relationship of the wild-type hGluK2 (black circle, left panel) and the M867I mutant hGluK2 (red circle, left panel), and the wild-type rGluK2 (right panel). Each data point is an average of at least three measurements from three different cells. A total of 33, 37 and 52 cells were measured for the wild-type human, the mutant and the rat GluK2, respectively. The whole-cell current amplitudes from different cells were normalized to that of the control or 0.5 mM glutamate, and the average of the current amplitude collected from ≥2 mM glutamate concentrations was set to be 100%. The solid line is the nonlinear fit of the combined data based on eq 3 (see the detailed results of the fitting in Table 1 for the human and Table 2 for the rat GluK2). From the combined data, when n = 2, K1 of 170 ± 23 μM, Φ of 0.29 ± 0.03 and IMRM of 125 ± 8 were obtained for the hGluK2 and the M867I mutant (left panel). Likewise, K1 of 300 ± 21 μM, Φ of 0.48 ± 0.73 and IMRM of 161 ± 84 at n = 2 were obtained for the rGluK2 (right panel).
Table 1.
Nonlinear Fitting of the Dose-Response Curve for the Wild-Type and M867I hGluK2
| na | K1 (mM) | Φ | IMRM (nA) | R2 |
|---|---|---|---|---|
| 1 | 0.41 ± 0.06 | 0.73 ± 0.04 | 180 ± 18 | 0.989 |
| 2 | 0.17 ± 0.02 | 0.29 ± 0.03 | 125 ± 8 | 0.986 |
| 3 | 0.07 ± 0.03 | 0.45 ± 0.13 | 140 ± 15 | 0.983 |
| 4 | 0.06 ± 0.03 | 0.49 ± 0.15 | 143 ± 19 | 0.974 |
n was chosen as integer.
The dose-response relationship for the wild-type rGluK2 was further measured as the control. The best fit of the rGluK2 data by eq 3 yielded K1 of 300 ± 21 μM at n = 2 (right panel of Figure 3 and Table 2). Using the Hill equation (33) to fit the same data, we found that EC50 = 260 ± 10 μM and the Hill coefficient = 1.7 for rGluK2 (Figure S2, Supporting Information). These data are close to those we previously published (17). Taken together, our results showed that the EC50 value we determined for rGluK2 was well within the range of values reported for the same receptor in literature (see a list of EC50 values in literature in Table S1, Supporting Information). Furthermore, the comparison of the wild-type data between the human GluK2 and the rat counterpart showed that the K1 and the EC50 values of hGluK2 are lower by ~1.8-fold and ~1.4-fold, respectively.
Table 2.
Nonlinear Fitting of the Dose-Response Curve for the Wild-Type rGluK2
| na | K1 (mM) | Φ | IMRM (nA) | R2 |
|---|---|---|---|---|
| 1 | 1.14 ± 0.38 | 0.50 ± 0.10 | 175± 29 | 0.975 |
| 2 | 0.30 ± 0.21 | 0.48 ± 0.73 | 161 ± 84 | 0.994 |
| 3 | 0.17 ± 0.07 | 0.43 ± 0.40 | 152 ± 47 | 0.996 |
| 4 | 0.11 ± 0.04 | 0.45 ± 0.40 | 153 ± 46 | 0.996 |
n was chosen as integer.
Channel-Opening Rate Constants for the Wild-type hGluK2 and the M867I Mutant
We also measured the channel-closing (kcl) and channel-opening (kop) rate constants for the wild-type and the mutant hGluK2 receptors. The magnitude of kop reflects how fast a channel opens following the binding of agonist, whereas the magnitude of kcl is a measure of how long a channel stays open or the lifetime of the open channel (22). Therefore, a putative potentiation of the current amplitude could be attributed to simply an increase in kop alone, leading to an increase in the total amount of ions passing through within the same lifetime of the open channel. On the other hand, a reduction of kcl alone would lead to lengthening the life time of the open channel, thus increasing the number of the ionic species passing through the channel per unit time. To be able to measure the channel-opening rate process, we used a laser-pulse photolysis technique with the caged glutamate (17) because this technique provided a suitable time resolution for this measurement (22).
Laser-pulse photolysis of caged glutamate triggered a rapid rise of whole-cell current (Figure 4A; some representative, raw traces are provided in Figure S3). The time course of the rising phase of the whole-cell current (as in Figure 4A), representing an observed rate constant of channel opening defined by kobs, was adequately described by a single exponential rate process (see the solid line as the fit to this rate process in Figure 4A by eq 1). Accordingly, various kobs values were obtained at corresponding concentrations of photolytically released glutamate. Inspection of kobs values as a function of ligand concentration showed there was not a statistically significant difference between the wild-type and the mutant hGluK2 channels (black and red symbols in Figure 4B respectively). As such, the analysis of kobs was based on the data combined from the mutant and the wild-type receptors. Specifically, by regression analysis of kobs as a function of the glutamate concentration according to eq 2, we found that the best fit of kobs was at n = 2, and the kop and kcl values were (1.9 ± 0.1) × 103 s−1 and (3.3 ± 0.2) × 102 s−1, respectively. If the rate data were separately analyzed by the same way, the best fit yielded kop and kcl of (1.7 ± 0.1) × 103 s−1 and (3.1 ± 0.1) × 102 s−1 for the wild-type receptor, and (2.0 ± 0.1) × 103 s−1 and (3.4 ± 0.3) × 102 s−1 for the mutant, respectively, at n =2 (see Table 3). Based on these results, we concluded that the M867I mutation did not affect the observed rate of channel opening at any given glutamate concentration (Figure 4B) or kobs, nor kop (i.e., the slope of the plot in Figure 4B) or kcl (the intercept of the plot in Figure 4B) regardless of n values (Table 3).
Figure 4.
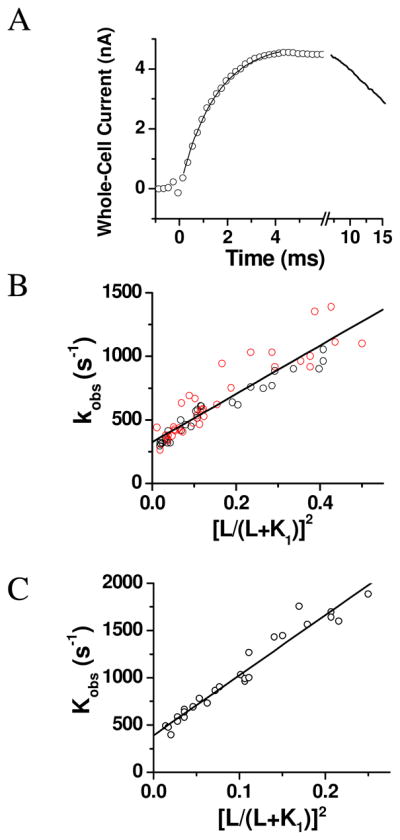
(A) A representative whole-cell current trace generated by the laser-pulse photolysis measurement with an HEK-293 cell expressing the wild-type hGluK2. A pulse of laser was fired at time zero. The concentration of glutamate released was estimated to be 200 μM. The kobs was calculated from the rising phase of the current to be 900 ± 12 s−1 by using eq 1 (the solid line). Note that the current is plotted opposite to the direction that is recorded. (B) Linear plot of the kobs data combined from the wild-type and the M867I mutant hGluK2 as a function of glutamate concentration according to eq 2. Each data point represents a single kobs value obtained at a particular concentration of photolytically released glutamate. Overall, a total of 29 and 36 cells were measured for the wild-type (black circle) and the mutant hGluK2 (red circle). The best fit of kobs was at n = 2, and the kop and kcl values were (1.9 ± 0.1) × 103 s−1 and (3.3 ± 0.2) × 102 s−1, respectively. See Table 3 and Tables S2 and S4 for a detailed description of nonlinear fits. (C) Linear plot of kobs as a function of glutamate concentration by eq 2, based on a total of 26 cell measurements, for the rGluK2. The best fitted kop and kcl values were (6.4 ± 0.1) × 103 s−1 and (3.9 ± 0.3) × 102 s−1, respectively, at n = 2. See Table 4 and Tables S3 and S5 for a detailed description of nonlinear fits.
Table 3.
Fitting of kop and kcl with Different n Values for Wild-Type and M867I hGluK2
| na | Wild-type hGluK2 |
M867I Mutant hGluK2 |
||||
|---|---|---|---|---|---|---|
| kop (× 103 s−1) | kcl (× 102 s−1) | R2 | kop (× 103 s−1) | kcl (× 102 s−1) | R2 | |
| 1 | 1.33 ± 0.50 | 1.08 ± 0.19 | 0.98 | 1.68 ± 0.12 | 0.49 ± 0.46 | 0.93 |
| 2 | 1.72 ± 0.08 | 3.12 ± 0.14 | 0.97 | 1.99 ± 0.14 | 3.43 ± 0.30 | 0.92 |
| 3 | 3.58 ± 0.17 | 3.78 ± 0.18 | 0.95 | 2.74 ± 0.24 | 4.40 ± 0.30 | 0.89 |
| 4 | 3.96 ± 0.33 | 4.11 ± 0.21 | 0.92 | 3.90 ± 0.40 | 4.87 ± 0.32 | 0.86 |
n was chosen as integer, and K1 was chosen to be 170 μM.
We further measured the channel-opening kinetics for the rGluK2 as the control. The best fit of kobs by linear regression using eq. 2 yielded a set of kop and kcl of (6.4 ± 0.3) × 103 s−1 and (3.9 ± 0.3) × 102 s−1, respectively, with n = 2 (Table 4); Figure 4C shows these best fitted values in the linear plot. Based on the kop and kcl values from the human and rat GluK2, as described above, it was apparent that the hGluK2 opened the channel >3-fold slower than the rGluK2, but closed the channel with a similar rate. It should be noted however, that the kop value from our earlier experiment (17) is higher than the one we determined in this study. This difference could be best explained by the fact that in the current study, we were able to extend the measurement of kobs to a much higher glutamate concentration than we were previously (17). Consequently we had a wider range of kobs values vs. glutamate concentrations for analyzing kop from the slope (as in Figures 4B and C). On the other hand, because the analysis of kcl was not as sensitive to the slope or to the range of glutamate concentration, it was not surprising that the kcl value from this study is identical to the value we obtained earlier (17).
Table 4.
Fitting of kop and kcl with Different n Values for Wild-Type rGluK2
| na | kop (× 103 s−1) | kcl (× 102 s−1) | R2 |
|---|---|---|---|
| 1 | 3.89 ± 0.18 | −1.20 ± 0.58 | 0.97 |
| 2 | 6.37 ± 0.28 | 3.91 ± 0.34 | 0.98 |
| 3 | 12.3 ± 0.75 | 5.69 ± 0.38 | 0.96 |
| 4 | 24.4 ± 2.0 | 6.64 ± 0.45 | 0.93 |
n was chosen as integer, and K1 was chosen to be 300 μM.
Estimation of the Minimal Number of Glutamate Molecules Bound to One Receptor and to Open its Channel and K1 Values Using the Rate Data for rGluK2 and hGluK2
In the study described above (Figure 4), we were able to measure kobs for both the wild-type and the mutant hGluK2 receptors up to 80% of the fraction of the open-channel population or up to 410 μM glutamate photolytically released from laser photolysis. The wide range of kobs values/glutamate concentrations permitted us to simultaneously evaluate, by eq 2, kop and K1 as well as n, the number of glutamate molecules bound to the receptor to open the channel. To achieve a better estimate of n, K1 and kop by nonlinear regression using eq 2, we decided to first fix the value of kcl so that we reduced one variable. This was possible because when L ≪ K1, eq 2 was reduced to kobs ≈ kcl, suggesting that (a) kobs at a low glutamate concentration would reflect kcl, and (b) the value of kcl was independent of whatever the n value might be as in eq 2 (22, 34). Based on this rationale, we identified kobs of 310 s−1 as kcl for the wild-type and mutant hGluK2 receptors. By nonlinear regression, we found that the fitted n values were always close to 2 (Table S2). Furthermore, choosing different kcl values around 310 s−1 did not significantly affect the output of n, nor K1 or kop values (Table 5). These results suggested that n = 2 was the minimal number best fitted to our data in the entire range of glutamate concentrations for the human GluK2 receptors (Table 5). A similar result or n ≈ 2 was also observed when we fitted kobs vs. glutamate concentration for the rat GluK2 either by fixing the kcl at 400 s−1 (Table S3 in Supporting Information) or by varying kcl value around 400 s−1 (Table 6).
Table 5.
Nonlinear Fitting for hGluK2 with fixed kcl
| kcla (s−1) | n | kop (× 103 s−1) | K1 (mM) | R2 |
|---|---|---|---|---|
| 100 | 1.6 ± 0.3 | 1.6 ± 0.1 | 0.16 ± 0.04 | 0.87 |
| 200 | 1.6 ± 0.4 | 1.6 ± 0.3 | 0.14 ± 0.04 | 0.88 |
| 250 | 1.8 ± 0.2 | 1.9 ± 0.1 | 0.13 ± 0.02 | 0.88 |
| 310 | 1.8 ± 0.3 | 1.8 ± 0.2 | 0.17 ± 0.03 | 0.88 |
| 350 | 2.1 ± 0.4 | 1.8 ± 0.2 | 0.16 ± 0.02 | 0.86 |
| 400 | 2.4 ± 0.2 | 1.8 ± 0.3 | 0.15 ± 0.03 | 0.86 |
kcl is fixed as values shown.
Table 6.
Nonlinear Fitting for rGluK2 with fixed kcl
| kcla (s−1) | n | kop (× 103 s−1) | K1 (mM) | R2 |
|---|---|---|---|---|
| 100 | 1.7 ± 0.5 | 6.4 ± 0.3 | 0.31 ± 0.15 | 0.94 |
| 250 | 1.8 ± 0.4 | 6.4 ± 0.4 | 0.31 ± 0.15 | 0.95 |
| 300 | 1.8 ± 0.4 | 6.3 ± 0.4 | 0.31 ± 0.14 | 0.95 |
| 350 | 1.9 ± 0.3 | 6.4 ± 0.4 | 0.31 ± 0.12 | 0.96 |
| 400 | 2.0 ± 0.3 | 6.3 ± 0.3 | 0.30 ± 0.12 | 0.96 |
| 450 | 2.0 ± 0.3 | 6.1 ± 0.4 | 0.30 ± 0.12 | 0.95 |
kcl is fixed as values shown.
It should be noted that the evaluation of n as described above was based on the rate data, independent from the analysis of the dose-response relationship (as in Figure 3). Using the same approach, we also independently evaluated K1, the intrinsic equilibrium dissociation constant (in Figure 1). Specifically, nonlinear regression of kobs vs. glutamate concentration by the use of eq 2 yielded a K1 of 170 μM on average for the wild-type hGluK2 and the mutant receptor (Table S2, where kcl was fixed at 310 s−1). This value was in good agreement with the K1 value of 170 ± 20 μM obtained from the dose-response measurement (left panel of Figure 3 and Table 1). Based on these results, we further constrained K1 to be 170 μM, in addition to a fixed kcl, in fitting kobs as a function of glutamate concentration. We found the values of n were close to 2, and that kop was relatively invariant, the average of which was 2,000 s−1 (Table S4 in Supporting Information). Likewise, nonlinear regression analysis of the rGluK2 data yielded an average of n = 2 and K1 = 300 μM when kcl was fixed at 400 s−1 (Table S3 in Supporting Information). When K1 was also constrained to be 300 μM, kop for rGluK2 was found to be 7,000 s−1 (Table S5 in Supporting Information).
Taken together, our analysis of kobs as a function of glutamate concentration provided an independent way, as compared to the dose-response relationship, to estimate both n and K1, yet the results from both the rate and the amplitude measurements were consistent with each other. Specifically, K1 was shown consistently different in that K1 was 170 μM for hGluK2 but was 300 μM for rGluK2 (Table 7). On the other hand, n = 2 was consistently shown as the best fitted value for both rGluK2 and hGluK2 from both the rate and the amplitude measurements. We favor, therefore, an interpretation of n = 2 as the minimal number of glutamate molecules bound to a receptor to open the channel. This conclusion was also consistent with the results from a study of an AMPA receptor mutant where kobs was collected up to saturation (34), and from studies of both AMPA and kainate receptor channels (17, 19, 22). Furthermore, as is known in kainate receptors, channel activation by activating only a subset of subunits, instead of all four, in a tetrameric complex can occur (35). Thus, we further favor an interpretation of kop and kcl at n = 2 (summarized in Table 7) being the representative values of the channel-opening and channel-closing rate constants for the human and the rat GluK2 receptors.
Table 7.
Comparison between Rat and Human Wild-Type GluK2
| Receptor | kop (× 103 s−1) | kcl (× 102 s−1) | Pop | K1 (mM) |
|---|---|---|---|---|
| rGluK2 | 6.4 ± 0.3 | 3.9 ± 0.3 | 0.94 | 0.30 ± 0.21 |
| hGluK2 | 1.9 ± 0.1 | 3.3 ± 0.2 | 0.85 | 0.17 ± 0.02 |
Measurement of the Channel-Opening Rate Constant
To characterize the mechanism of channel opening for GluK2 receptors, we took into account of several experimental observations in our kinetic data analysis to obtain kop and kcl. The rising phase of the whole-cell current (as in Figure 4A) obeyed a single exponential rate law and did so in the entire concentration range of glutamate (i.e., 20–410 μM). This observation was consistent with the assumption that the channel-opening rate or the rate of transition from the closed-channel form, ALn, to the open-channel form, , was slow as compared with all preceding steps involving glutamate binding (Figure 1). On the basis of this assumption, eq 2 was derived. If, however, the channel-opening rate was either comparable with or faster than the ligand-binding rate, a single-exponential rate process would be inadequate to describe the bi-molecular association rate process or would be broken down in a certain concentration range. For instance, when the ligand concentration is too low such that the ligand association rate dominates the rate process, the rising phase of the current will reflect ligand binding instead of channel opening (19, 22). To ensure that ligand binding was always fast so that the channel-opening rate was the observed kinetic event and thus was measurable, we did not use any kobs values at any glutamate concentrations lower than ~4% of the fraction of the open-channel form (22). For hGluK2 receptors, 4% of the fraction of the open-channel form correlated to be ~20 μM (see also the legend of Figure 4B), and at this fraction or concentration, we considered kobs reflected mainly kcl. Among several lines of supporting evidence (19, 22), we note in particular that the kcl value we calculate using eq 2 is generally similar with the lifetime determined by single-channel recording of the same AMPA channel with glutamate being the agonist. Furthermore, assuming τ is the lifetime expressed in time constant, then kcl = 1/τ (21–22, 27, 36).
DISCUSSION
The first question of this study is whether the M867I mutation, presumably tied to autism, affects the channel gating properties of the human GluK2 kainate receptor. To address this question, we have systematically characterized the rate and the equilibrium constants that are pertinent to the channel opening and channel desensitization for this mutant and the wild-type human GluK2 receptor. In addition, because we use the rat GluK2 kainate receptor as the control for our study, we are able to answer the second question of this study: is the human wild-type GluK2 functionally same as the rat ortholog? The second question is important because to date there has been a very few studies of the channel gating properties of the human GluK2, and the current understanding of the structure-function relationship of kainate receptors in general and GluK2 in particular comes almost entirely from the rat forms.
For the first question, our results show that the M867I mutation does not affect either kop or kcl. The absence of any effect on these two parameters rules out the possibility that an enhancement of macroscopic current amplitude by the M867I mutation is a result of either a reduction of kcl, which would lengthen the life time of the open channel, or an increase of kop, which would accelerate the opening of the channel. In either event, a greater number of ions or a larger amount of charge would pass through the open channel, leading to higher current amplitude. The absence of any effect on K1 (or qualitatively on EC50 value), i.e. a left-shift of K1 on the dose-response curve, further rules out the possibility that at a given glutamate concentration (but not at saturating ones) a larger current amplitude would be observed because a greater percentage of the channels would open in response to the binding of glutamate. What we did find is that the channel desensitization rate of the M867I mutant is slower than the wild-type hGluK2. Our result thus suggests that higher current amplitude is expected in a solution exchange measurement because of a lesser amount of the current reduction within the rising phase of the macroscopic current. Furthermore, because the kop and kcl values for the M867I mutant and the wild-type hGluK2 are the same, the channel-opening probability (Pop) for the mutant and the wild-type receptor will have to be the same (i.e., based on the experimentally determined kop and kcl values, Pop is calculated to be 0.85 – see Table 7) (22). Pop is defined as the probability that a channel will open once it is bound with ligand(s), in this case, glutamate (22, 32). These results therefore suggest that at the receptor level, the only change due to the M867I mutation is the decrease of the rate of channel desensitization. However, our finding that the M867I mutant exhibits a 1.6-fold slower rate of channel desensitization as compared with the wild-type hGluK2 receptor, determined at saturating glutamate concentrations (see Figure 2D), is unlikely to account for the enhancement of the current amplitude from the two-electrode recording of Xenopus oocytes by Strutz-Seebohm et al. (16). The oocyte measurement provided an estimate of the steady-state current amplitude, due to a much larger size of an oocyte and a much slower time course of solution exchange (16).
The change of the desensitization rate constant by the isoleucine substitution at M867 in hGluK2, measured at saturating glutamate concentrations, is close to 2 fold (see Figure 2D). It is unclear, however, whether the change of this magnitude is significant in the context of autism and what the functional consequence(s) of slowing down the channel desensitization is, due to this mutation presumably linked to autism. Interestingly, M867 is located immediately next to a 13-amino acid sequence motif or residues 868-880 (37). Yan et al. (37) identified this motif in the C-terminus of the rat GluK2 as the determinant for receptor trafficking/surface expression, and this sequence motif is conserved in the human GluK2 or precisely hGluK2a, the longest of the three C-terminal isoforms of the hGluK2. In other words, hGluK2a is the only isoform that contains both M867 in its 908 amino acid residues (10) and this trafficking motif. Not surprisingly, hGluK2a is found to be the only isoform capable of efficiently trafficking to the plasma membrane (38). It is therefore conceivable that the location of M867 based on its close proximity to the trafficking motif positions this residue for its role in facilitating receptor trafficking. It should be pointed that Strutz-Seebohm et al. (16) have shown convincingly by immunochemical labeling and fluorescence imaging measurements that the M867I mutant exhibits a higher receptor surface expression. In this context, it is also interesting to note that serine 868 or S868, the one next to M867, is a known protein phosphorylation site; and S868 with another serine residue, i.e. S856, forms a vital component of rGluK2 potentiation of current amplitude upon phosphorylation by protein kinase A (PKA) (39). Furthermore, Yan et al. (37) also reported that this forward trafficking sequence motif in the C-terminus of rGluK2 may affect the channel desensitization in that truncating the amino acid sequences altogether but outside this domain does not affect the channel desensitization but deletion of the amino acid residues within this trafficking sequence segment leads to slowing down the channel desensitization. After all, single amino acid residues in the C-terminus, such as C871 and R873, do play a role in the plasma membrane expression of rGluK2 (37). Interestingly, the M867I mutation positively regulates the receptor trafficking and surface expression (16), but negatively affects the rate of channel desensitization. Furthermore, because of the close proximity, the M867I mutation may affect either the phosphorylation at S868 specifically or the C-terminal domain in such a way that the receptor trafficking and surface expression is enhanced. The latter scenario may involve complex protein-protein interactions in the GluK2 C-terminal domain (37, 40), and the GluK2 C-terminus itself is known to be critically involved in both regulating receptor trafficking/surface expression (37, 41) and agonist-evoked endocytosis (42). Obviously a putative role of this mutation in enhancing receptor trafficking that may ultimately link to autistic condition needs to be examined in the future.
The second question we are interested in addressing is whether the kinetic properties of the human GluK2 are the same as the rat receptor, given that the rat GluK2 has been extensively studied but there has been no systematic kinetic study of the human GluK2. Under the same condition that we studied the human GluK2, we find kop and kcl of (6.4 ± 0.3) × 103 s−1 and (3.9 ± 0.3) × 102 s−1, respectively, for rGluK2 (Table 7). Based on the kop and kcl values, Pop is found to be 0.94 for rGluK2 (Table 7), similar to the value we reported before (17). As is shown in Table 7, the rat GluK2 exhibits a set of parameters that are all larger than the human GluK2 except the kcl value. However, among those parameters that are different, the most significant is the rate of the channel opening: the human receptor is ~3-fold slower, whereas the channel-closing rate constants for the two receptors are about the same (Table 7). Thus, the Pop of the hGluK2 is lower, i.e., Pop = 0.85 vs. Pop = 0.94 for the rGluK2 (Table 7). The smaller Pop for the human GluK2, as compared to the Pop of the rat form, which is near unity, suggests that there is a greater possibility to enhance the activity of the human channel or improve the Pop value by a regulatory mechanism. In the rat GluK2, PKA phosphorylation is such a mechanism and the phosphorylation leads to increase of Pop (32). The second most significant difference is that the K1 value of the human GluK2 is ~2-fold smaller. This difference indicates that at a given glutamate concentration (so long that concentration is not saturating to the human form, such as < 1 mM; see the dose-response curves in Figure 3), binding of glutamate to the human receptor will elicit a greater portion of the channels to open than the rat receptor. It should be noted that 1 mM glutamate is a relevant, synaptic concentration (43). Taken together, the results from this study show that the human GluK2 is relatively a slow-working channel as compared to the rat ortholog, but is more sensitive to glutamate so that the human channel plays a relatively more important role in a low glutamate concentration at which a greater population of the channel is activated for neurotransmission. As observed in a study of rat hippocampal neuron slices, the time course of the kainate receptor-mediated excitatory postsynaptic potential (EPSP) is sufficiently slow to allow substantial tonic depolarization during modest activation of presynaptic afferents (44). In this context, the slower human GluK2 receptor, as compared to the rat form, should be even more sensitive to temporal information in the afferent input, because the human GluK2 has a slower channel-opening rate or kop but a smaller K1 or EC50 value than the rat form. Thus, the kinetically slower human form of GluK2 may be more relevant for synaptic transmission under a condition where low glutamate concentrations are prevalent, such as a spillover or the diffusion of glutamate from distant sites (45–46).
Our finding that the human and rat forms of wild-type GluK2 have different kinetic and equilibrium constants is surprising, given the fact that the amino acid sequences of the human and rat GluK2 are almost 99% identical (11). Among 908 amino acid residues in total (or GluK2a for both the human and the rat forms), the rat and human GluK2 receptors differ by only ten amino acid residues. Specifically, seven amino acids that are different are located in the signal peptide region (i.e., within the first 31 amino acids, which are not present in the mature protein). In the human receptor, position 494 is an alanine whereas it is a valine in the rat. This residue is located in the extracellular ligand binding domain from the crystal structure obtained from the S1S2 soluble portion of the rGluK2 (47). This residue situates near the edge of the D1 domain and outside of the binding cleft. The last two remaining amino acid residues are I567 and Y571 in hGluK2 but are V567 and C571 in rGluK2. These two amino acid residues are located in the putative M1 transmembrane domain (47). At this time, however, how this very limited number of amino acid differences between the human and rat forms gives rise to different functions is unclear.
The rapid kinetic characterization of the human GluK2 kainate receptor here we describe represents another study in our effort to find the similarity and difference in the channel-opening rate constants between the kainate and AMPA receptor subtypes. In a more significant way, the results of the human GluK2, like the rat GluK2 results we published before (17) and we have collected from this study again as the control, demonstrate that both the rat and human GluK2 receptors exhibit a much slower rate of opening their channels in response to binding of glutamate, and a much slower rate of closing the open channels than AMPA receptors do (19, 21–24). Furthermore, both hGluK2 and rGluK2 desensitize relatively less rapidly than AMPA receptors (19, 21–24). Therefore, our finding that kainate receptors are kinetically slower than AMPA receptors may be useful in understanding the role of kainate receptors in mediating synaptic plasticity, which is much less known than the role of AMPA receptors (1–2, 48). For example, our results may account for slow kinetics or the time course of the rise and decay of the kainate excitatory postsynaptic current (EPSC) as compared with AMPA receptor EPSC (46, 49). When AMPA and kainate receptors are colocalized at a synapse (46), it is therefore expected that the AMPA channels are first to be activated by synaptically released glutamate and first to complete channel desensitization. Our data also help clarify that a smaller size of kainate EPSC should not be due to a low channel activity of kainate receptors (46). This is because the kainate receptors, or precisely GluK2 (in both human and rat forms), have a lower K1 value or qualitatively the EC50 value than AMPA receptors, and thus, at a given glutamate concentration of no more than 1 mM at synapses (43), binding of glutamate to either hGluK2 or rGluK2 induces a greater proportion of the channels to open but at a cost of opening the channel with a slower rate (i.e., with a smaller kop value), as compared with AMPA receptors (19).
It should be noted that some of the points we raised above are a matter of speculation, based on the data of a single kainate receptor or hGluK2 we present here. It is known that heteromeric channels assembled from GluK2 with other kainate receptor subunits (50–51) and even with different, alternatively spliced variants of GluK2 generated at the C-terminus (52) show different kinetic properties from the homomeric GluK2 channels. On the other hand, our current knowledge of kainate receptors is, to a large extent, based on GluK2, one of the best characterized subunits of kainate receptors. That knowledge, however, is further based on the rat ortholog of GluK2. For the human GluK2, few functional studies have ever been carried out since the cloning of the human GluK2 gene and its splicing variants (11, 38, 53). Therefore, our finding that the human and rat GluK2 have different channel gating properties suggests much more studies are needed to understand the human kainate receptors. And much more studies are also needed to continue to explore the putative involvement of the human GluK2 in not only autism but also in several other neurological diseases, such as epilepsy and Huntington disease (4).
Supplementary Material
Acknowledgments
We thank David Bleakman at Eli Lilly for the human GluK2 cDNA and Nathalie Strutz-Seebohm at Universität Tübingen, Germany, for the rat M867I mutant cDNA plasmid. We are grateful for insightful comments from anonymous reviewers.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Caged glutamate
γ-O-(α-carboxy-2-nitrobenzyl)-glutamate
- HEK-293 cells
human embryonic kidney cells
- hGluK2
human wild-type GluK2 kainate receptor
- NMDA
N-methyl-D-aspartic acid
- rGluK2
rat GluK2 kainate receptor
Footnotes
This work was supported in part by grants from the National Institutes of Health (R01 NS060812), Department of Defense (W81XWH-04-1-0106), and the Muscular Dystrophy Association.
SUPPORTING INFORMATION AVAILABLE
The supporting information includes a figure showing Hill equation fit to the dose-response relationship, and a table showing the results of the nonlinear regression data for the best fit of n, K1 and kop to the observed rate constant of channel opening as a function of glutamate concentration. The supporting information includes an additional table showing the nonlinear regression fitting results by constraining both kcl and K1 in order to better estimate n and kop values. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jane DE, Lodge D, Collingridge GL. Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology. 2009;56:90–113. doi: 10.1016/j.neuropharm.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 2.Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. Epub 2005 Dec 2019. [DOI] [PubMed] [Google Scholar]
- 3.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 4.Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience. 2009;158:309–323. doi: 10.1016/j.neuroscience.2008.02.066. [DOI] [PubMed] [Google Scholar]
- 5.Swanson GT. Targeting AMPA and kainate receptors in neurological disease: therapies on the horizon? Neuropsychopharmacology. 2009;34:249–250. doi: 10.1038/npp.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bleakman D, Gates MR, Ogden AM, Mackowiak M. Kainate receptor agonists, antagonists and allosteric modulators. Curr Pharm Des. 2002;8:873–885. doi: 10.2174/1381612024607108. [DOI] [PubMed] [Google Scholar]
- 7.Contractor A, Swanson GT, Sailer A, O’Gorman S, Heinemann SF. Identification of the kainate receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus [In Process Citation] J Neurosci. 2000;20:8269–8278. doi: 10.1523/JNEUROSCI.20-22-08269.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- 9.Martin S, Henley JM. Activity-dependent endocytic sorting of kainate receptors to recycling or degradation pathways. EMBO J. 2004;23:4749–4759. doi: 10.1038/sj.emboj.7600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B, Gillberg C, Leboyer M, Bourgeron T. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry. 2002;7:302–310. doi: 10.1038/sj.mp.4000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paschen W, Blackstone CD, Huganir RL, Ross CA. Human GluR6 kainate receptor (GRIK2): molecular cloning, expression, polymorphism, and chromosomal assignment. Genomics. 1994;20:435–440. doi: 10.1006/geno.1994.1198. [DOI] [PubMed] [Google Scholar]
- 12.Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathol. 2007;17:434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, Penet C, Feingold J, Brice A, Leboyer M. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8:805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- 14.Shuang M, Liu J, Jia MX, Yang JZ, Wu SP, Gong XH, Ling YS, Ruan Y, Yang XL, Zhang D. Family-based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am J Med Genet B Neuropsychiatr Genet. 2004;131B:48–50. doi: 10.1002/ajmg.b.30025. [DOI] [PubMed] [Google Scholar]
- 15.Kim SA, Kim JH, Park M, Cho IH, Yoo HJ. Family-based association study between GRIK2 polymorphisms and autism spectrum disorders in the Korean trios. Neurosci Res. 2007;58:332–335. doi: 10.1016/j.neures.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Strutz-Seebohm N, Korniychuk G, Schwarz R, Baltaev R, Ureche ON, Mack AF, Ma ZL, Hollmann M, Lang F, Seebohm G. Functional significance of the kainate receptor GluR6(M836I) mutation that is linked to autism. Cell Physiol Biochem. 2006;18:287–294. doi: 10.1159/000097675. [DOI] [PubMed] [Google Scholar]
- 17.Li G, Oswald RE, Niu L. Channel-opening kinetics of GluR6 kainate receptor. Biochemistry. 2003;42:12367–12375. doi: 10.1021/bi034797t. [DOI] [PubMed] [Google Scholar]
- 18.Wieboldt R, Gee KR, Niu L, Ramesh D, Carpenter BK, Hess GP. Photolabile precursors of glutamate: synthesis, photochemical properties, and activation of glutamate receptors on a microsecond time scale. Proc Natl Acad Sci U S A. 1994;91:8752–8756. doi: 10.1073/pnas.91.19.8752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei W, Huang Z, Wang C, Han Y, Park JS, Niu L. Flip and flop: a molecular determinant for AMPA receptor channel opening. Biochemistry. 2009;48:3767–3777. doi: 10.1021/bi8015907. [DOI] [PubMed] [Google Scholar]
- 20.Canepari M, Nelson L, Papageorgiou G, Corrie JE, Ogden D. Photochemical and pharmacological evaluation of 7-nitroindolinyl-and 4-methoxy-7-nitroindolinyl-amino acids as novel, fast caged neurotransmitters. J Neurosci Methods. 2001;112:29–42. doi: 10.1016/s0165-0270(01)00451-4. [DOI] [PubMed] [Google Scholar]
- 21.Li G, Pei W, Niu L. Channel-opening kinetics of GluR2Q(flip) AMPA receptor: a laser-pulse photolysis study. Biochemistry. 2003;42:12358–12366. doi: 10.1021/bi0347961. [DOI] [PubMed] [Google Scholar]
- 22.Li G, Niu L. How fast does the GluR1Qflip channel open? J Biol Chem. 2004;279:3990–3997. doi: 10.1074/jbc.M310410200. Epub 2003 Nov 3910. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Sheng Z, Huang Z, Niu L. Kinetic mechanism of channel opening of the GluRDflip AMPA receptor. Biochemistry. 2005;44:5835–5841. doi: 10.1021/bi047413n. [DOI] [PubMed] [Google Scholar]
- 24.Pei W, Huang Z, Niu L. GluR3 flip and flop: differences in channel opening kinetics. Biochemistry. 2007;46:2027–2036. doi: 10.1021/bi062213s. [DOI] [PubMed] [Google Scholar]
- 25.Mano I, Teichberg VI. A tetrameric subunit stoichiometry for a glutamate receptor-channel complex. Neuroreport. 1998;9:327–331. doi: 10.1097/00001756-199801260-00027. [DOI] [PubMed] [Google Scholar]
- 26.Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- 27.Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- 28.Mansour M, Nagarajan N, Nehring RB, Clements JD, Rosenmund C. Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron. 2001;32:841–853. doi: 10.1016/s0896-6273(01)00520-7. [DOI] [PubMed] [Google Scholar]
- 29.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivithanaporn P, Lash LL, Marszalec W, Swanson GT. Critical roles for the M3-S2 transduction linker domain in kainate receptor assembly and postassembly trafficking. J Neurosci. 2007;27:10423–10433. doi: 10.1523/JNEUROSCI.2674-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bufler J, Heckmann M, Jahn K, Franke C. Distribution of desensitization time constants of mouse embryonic-like nicotinic and homomeric GLUR6 glutamate receptor channels. Neurosci Lett. 1997;221:173–176. doi: 10.1016/s0304-3940(96)13320-6. [DOI] [PubMed] [Google Scholar]
- 32.Traynelis SF, Wahl P. Control of rat GluR6 glutamate receptor open probability by protein kinase A and calcineurin. J Physiol. 1997;503:513–531. doi: 10.1111/j.1469-7793.1997.513bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loftfield RB, Eigner EA. Molecular order of participation of inhibitors (or activators) in biological systems. Science. 1969;164:305–308. doi: 10.1126/science.164.3877.305. [DOI] [PubMed] [Google Scholar]
- 34.Pei W, Ritz M, McCarthy M, Huang Z, Niu L. Receptor occupancy and channel-opening kinetics: a study of GLUR1 L497Y AMPA receptor. J Biol Chem. 2007;282:22731–22736. doi: 10.1074/jbc.M611821200. [DOI] [PubMed] [Google Scholar]
- 35.Swanson GT, Green T, Sakai R, Contractor A, Che W, Kamiya H, Heinemann SF. Differential activation of individual subunits in heteromeric kainate receptors. Neuron. 2002;34:589–598. doi: 10.1016/s0896-6273(02)00676-1. [DOI] [PubMed] [Google Scholar]
- 36.Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan S, Sanders JM, Xu J, Zhu Y, Contractor A, Swanson GT. A C-terminal determinant of GluR6 kainate receptor trafficking. J Neurosci. 2004;24:679–691. doi: 10.1523/JNEUROSCI.4985-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbon A, Gervasoni A, LaVia L, Orlandi C, Jaskolski F, Perrais D, Barlati S. Human GluR6c, a functional splicing variants of GluR6, is mainly expressed in non-nervous cells. Neurosci Lett. 2008;434:77–82. doi: 10.1016/j.neulet.2008.01.049. [DOI] [PubMed] [Google Scholar]
- 39.Kornreich BG, Niu L, Roberson MS, Oswald RE. Identification of C-terminal domain residues involved in protein kinase A-mediated potentiation of kainate receptor subtype 6. Neuroscience. 2007;146:1158–1168. doi: 10.1016/j.neuroscience.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laezza F, Wilding TJ, Sequeira S, Coussen F, Zhang XZ, Hill-Robinson R, Mulle C, Huettner JE, Craig AM. KRIP6: a novel BTB/kelch protein regulating function of kainate receptors. Mol Cell Neurosci. 2007;34:539–550. doi: 10.1016/j.mcn.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nasu-Nishimura Y, Jaffe H, Isaac JT, Roche KW. Differential regulation of kainate receptor trafficking by phosphorylation of distinct sites on GluR6. J Biol Chem. 2010;285:2847–2856. doi: 10.1074/jbc.M109.081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 44.Frerking M, Ohliger-Frerking P. AMPA receptors and kainate receptors encode different features of afferent activity. J Neurosci. 2002;22:7434–7443. doi: 10.1523/JNEUROSCI.22-17-07434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen TA, DiGregorio DA, Silver RA. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron. 2004;42:757–771. doi: 10.1016/j.neuron.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- 47.Mayer ML. Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron. 2005;45:539–552. doi: 10.1016/j.neuron.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 48.Mellor JR. Synaptic plasticity of kainate receptors. Biochem Soc Trans. 2006;34:949–951. doi: 10.1042/BST0340949. [DOI] [PubMed] [Google Scholar]
- 49.Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- 50.Paternain AV, Herrera MT, Nieto MA, Lerma J. GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J Neurosci. 2000;20:196–205. doi: 10.1523/JNEUROSCI.20-01-00196.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanders JM, Ito K, Settimo L, Pentikainen OT, Shoji M, Sasaki M, Johnson MS, Sakai R, Swanson GT. Divergent pharmacological activity of novel marine-derived excitatory amino acids on glutamate receptors. J Pharmacol Exp Ther. 2005;314:1068–1078. doi: 10.1124/jpet.105.086389. [DOI] [PubMed] [Google Scholar]
- 52.Coussen F, Perrais D, Jaskolski F, Sachidhanandam S, Normand E, Bockaert J, Marin P, Mulle C. Co-assembly of two GluR6 kainate receptor splice variants within a functional protein complex. Neuron. 2005;47:555–566. doi: 10.1016/j.neuron.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 53.Barbon A, Vallini I, Barlati S. Genomic organization of the human GRIK2 gene and evidence for multiple splicing variants. Gene. 2001;274:187–197. doi: 10.1016/s0378-1119(01)00611-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.