

Abstract
Persistent infections with human immunodeficiency viruses and hepatitis B and C viruses are a major cause of morbidity and mortality world-wide. Dendritic cells (DCs), as sentinels of the immune system are crucial in the generation of protective antiviral immunity. Recent advances in the study of the role of DCs during infection with these viruses provide insights into the mechanisms used by these viruses to exploit DC function to evade innate and adaptive immunity. In this review we highlight current knowledge on the interaction between DCs and these viruses and the underlying mechanisms that might influence the outcome of viral infections.
Introduction
The immune response to viral infections is a complex interplay between the virus and the innate and adaptive immune response and is aimed at eradicating the pathogen with minimal damage to the host. Dendritic cells (DCs) are a specialized family of antigen-presenting cells (APCs) that effectively link innate recognition of viruses to the generation of the appropriate type of adaptive immune response 1. DCs are continuously produced from hematopoietic stem cells within the bone marrow and positioned at the different portals of the human body, such as the skin, mucosal surfaces and the blood, giving them the opportunity to instantaneously encounter invading pathogens early in the course of an infection 2.
DCs comprise a heterogeneous family. This heterogeneity arises at several levels, including their anatomical location, phenotype, and function (Table 1) 2. Langerhans cell (LCs) form a long-lived population of stellate DCs in the epidermis. Interstitial DCs comprise the DCs found in all peripheral tissues, excluding the LCs of the epidermis. The hematopoietic stem cell also gives rise to two other main DC subsets in the blood: myeloid (mDCs) and plasmacytoid DCs (pDCs). DCs are equipped with a set of varied pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), through which they sense and process viral information and become activated (Table 1). Following activation, DCs migrate to the regional lymph nodes where they appear as mature interdigitating DCs within the T cell-dependent areas. As a result of viral antigen uptake and presentation on the surface in complex with major histocompatibility complex (MHC) class I and II molecules, DCs trigger an immune response in any T cell that possesses a cognate receptor specific for the viral-peptide-MHC complexes being presented on the DC surface 1.
Table 1.
Subsets of human DCs
| Plasmacytoid DCs | Myeloid subtypes (Stellate DCs) |
||||
|---|---|---|---|---|---|
| Myeloid DCs | Langerhans | Interstitial DCs | Monocyte Monocyte-derived DCs | ||
| Morphology | 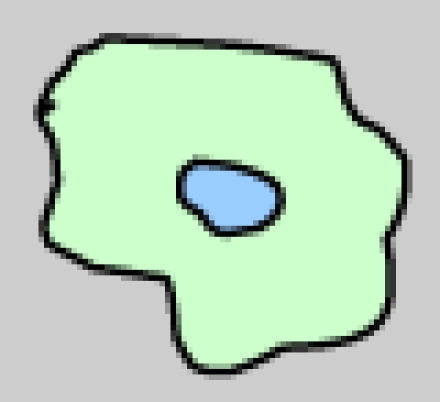 |
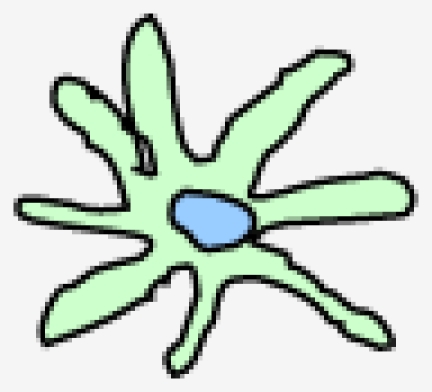 |
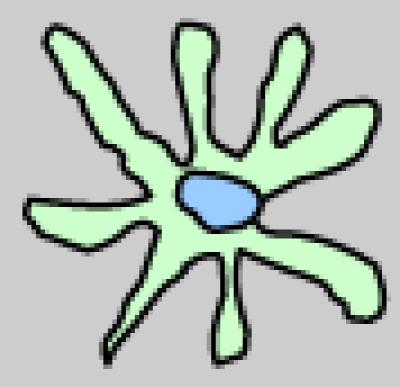 |
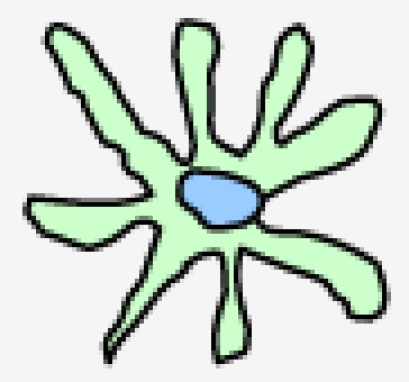 |
 |
| Localisation | Blood | Blood | Epidermal | Dermis and other tissues | in vitro |
| Phenotype | CD1a+ | CD1a+ | CD1a+ | CD1a − | CD1a+ |
| CD1c− | CD1c+ | CD11c+ | CD11c+ | CD1c+ | |
| CD11c− | CD11c+ | Langerin+ | CD68+ | CD11c+ | |
| CD123 high | CD123 low | 1, 2, 3, 6, 7, 8 | Factor XIIIa | CD123 low | |
| CD304 + | CD304 − | Langerin | 1,2, 3, 4, 5, 6, 7, 8, | 1,2, 3, 4, 5, 6, 8, 10 | |
| TLR expression | 1, 6, 7, 9, 10 | 1,2, 3, 4, 5, 6, 8, 10 | + | MR | MR DC-SIGN DCIR |
| C-type lectin expression | DCIR BDCA-2 | DCIR (MR) (DC-SIGN) | + | DC DC-SIGN | + |
| Immunological function | |||||
| CD4+ T cel cell priming | + | + | +/− | + | + |
| CD8+ T cell priming | + | + | + | + | + |
| B cell activation | + | + | + | + | |
| IFN-α production | +++ | + | + | + | |
Abbreviations: BDCA, blood dendritic cell antigen; DCs, dendritic cells; DC-SIGN, dendritic-cell specific ICAM3 grabbing non-integrin; IFN, interferon; DCIR, dendritic cell immunoreceptor; MR, mannose receptor; TLR, Toll-like receptor
The different DC subsets appear to have evolved over time to acquire both distinct and overlapping functions in order to better defend the host. Myeloid DCs and pDCs function in both innate and adaptive immunity and provide a critical link between the two arms of immunity upon viral infection 3. Following activation, mDCs produce IL-12 and IL-15 that in turn stimulate IFN-γ secretion by natural killer (NK) cells, promote the differentiation of CD4+ T helper (Th) cells into Th type 1 cells and CD8+ T cells into cytotoxic T cells that contribute to viral clearance either by killing infected cells directly through the release of cytolytic mediators, e.g. granzyme, or indirectly by secreting Th1-type cytokines that inhibit viral replication (Figure 1). In contrast to mDCs, which may have mainly evolved to prime and activate anti-viral T-cells, pDCs represent the key effector cells in the early anti-viral innate immune responses by producing large amounts of type I interferon upon viral infection. Type I interferons (IFN-α/β) released by pDCs have not only potent antiviral activity but also support subsequent steps of antiviral immunity including activation of natural killer (NK) cell-mediated cytotoxicity and CD4+ and CD8+ T cell differentiation and survival 4. In addition, pDCs also have an overlapping role as antigen-presenting cells 5.
Figure 1. Function of dendritic cells (DCs) in viral immune response.
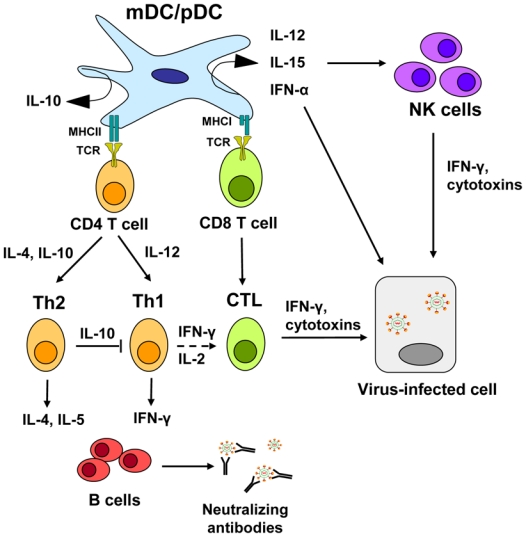
Following uptake of viral antigen, myeloid and plasmacytoid DC subsets (mDC, pDC) migrate to lymphoid tissue to prime naïve CD4+ and CD8+ T cells. In addition, activated DCs produce a range of cytokines, such as IFN-α, IL-12 and IL-15 that in turn activate natural killer (NK) cells and influence T-cell survival and differentiation. Depending on the cytokine signal, CD4+ T cells differentiate into Th1 or Th2 type CD4+ T cells. Th1 mediated IFN-γ secretion stimulates the activation of cytotoxic CD8+ T cells (CTLs) and IgG production in B cells. Th2 mediated cytokine production acts on B cells to simulate IgG production but also has the capacity to inhibit activation of Th1 type T cells. Virus-specific antibodies can be neutralizing, preventing viral re-infection. NK cells and CTLs inhibit viral replication through secretion of IFN-γ or lysis of viral infected cells through the release of cytotoxins (perforin, granzymes). In addition, pDCs are characterized by their ability to produce large amounts of type I IFNs in response to many viruses and, thereby, produces a first strong wave of IFN-α, which not only inhibits viral replication but also is a potent enhancer for NK cell-mediated cytoxicity.
The importance of DCs in the clearance of viral infection has been shown in several viral infections, such as the common respiratory viral pathogens respiratory syncytial virus (RSV) and influenza virus 6, 7. DCs also play an important part in the control of blood-borne viruses, of which the most common and deadly are the hepatitis B virus (HBV), hepatitis C virus (HCV), and human immunodeficiency viruses (HIV-1,-2). Patients who spontaneously clear HBV and HCV infection exhibit a strong multi-epitope specific CD4+ and CD8+ T cell response that probably reflects efficient priming and activation of anti-viral T cells by DCs 8–11. However, viral clearance after HBV, HCV or HIV infection is not always possible and together these viruses have created a global health problem of substantial proportions. Not only do they establish asymptomatic persistent infections with potential oncogenic sequelae, but they also cause significant morbidity and mortality (Table 2). HIV infection causes AIDS (acquired immune deficiency syndrome) that is characterized by profound immunosuppression and a diverse variety of associated opportunistic infections 12. Worldwide, HBV and HCV have infected more than 370 and 130 million people respectively 13 and are the two major causes of chronic liver disease with its associated complications including liver cirrhosis, liver failure and hepatocellular carcinoma 14. A common denominator in all these persistent infections is the weak and narrowly focused anti-viral T-cell response 8–11.
Table 2.
Clinical and virological features of HCV, HBV, and HIV
| HCV | HBV | HIV | |
|---|---|---|---|
| Structure | 50 nm; enveloped nucleocapsid; positive singlestranded RNA genome | 42 nm; enveloped nucleocapsid; partially double-stranded DNA genome | 120 nm, enveloped nucleocapsid; positive singlestrand RNA virus |
| Family | Flaviviridae | Hepadnaviridae | Retroviridae |
| Entry factors | Glycosaminoglycans, CD81, SR-BI, claudin-1, occludin-1 | unknown | CD4, CCR4, CXCR5, DCSIGN |
| Replication strategy | Synthesis of a complementary negative-strand RNA, using the viral genome as a template, from which genomic positive-strand RNA is produced | Reverse transcription of HBV RNA into a covalently closed circular DNA which serves as a template for HBV transcripts | conversion of the singles-tranded HIV RNA to double-stranded HIV DNA by viral reverse transcriptase, followed by integration of HIV DNA within the host genome |
| Mutation rate | High (1 in 1 000 bases per year) | Low (1 in 100 000 bases per year) | High (1 in 10 000 bases par replication cycle) |
| Genotypes | 6 main genotypes (1 to 6), with several subtypes within each genotype (more than 50 in total) | 8 genotypes (A to H), with 22 subtypes | HIV-1 which include one major group (M) divided into nine subtypes (A to D, F to H, J, K) and two minor groups (O, N) HIV-2 which include 2 groups (A and B) |
| Transmission | Intravenous drug use, blood transfusions, perinatal | intravenous drug use, blood transfusions, perinatal, sexual contact | intravenous drug use, blood transfusions, perinatal, sexual contact |
| Infection site | Liver | Liver | CD4+ T cells |
| Public health impact | |||
| Chronically infected individuals | 130 million | 350 million | 35 million |
| New infections/year | 3 to 4 million | 4 million | 3 million |
| Related deaths/year. | 350 000 | 500 000 to 1.2 million | 2 million |
| Rate of co-infection with HCV | 10 to 30% | 10% | |
| Infection outcome | |||
| Spontaneous recovery | 20% | 90% of adults, 10% of children | 0% |
| Disease outcome | liver cirrhosis (20–30% of chronically infected patients) hepatocellular carcinoma (5% of patients with liver cirrhosis) | liver cirrhosis (2–5% of chronically infected patients) hepatocellular carcinoma (5% of patients with liver cirrhosis) | acquired immunodeficiency syndrome (AIDS); susceptibility to lifethreatening opportunistic infections |
| Available Therapy & recovery rates with therapy | pegylated interferon alpha I combination with ribavirin/HCV clearance in 50%–80% of individuals, depending on HCV genotype liver transplantation/systematic reinfection of the graft |
interferon alpha, nucleoside and nucleotide analogues resulting in efficient control of viral infection liver transplantation/prevention of graft reinfection using antiviral treatment and anti-HBs antibodies |
highly active antiretroviral therapy (HAART: nucleoside analogue reverse transcriptase inhibitors, protease inhibitor and/or nonnucleoside reverse transcriptase inhibitor) No HIV clearance |
| Vaccine | Absent | Present Based on recombinant HBsAg which induce neutralizing HBsAg-specific antibodies and CD4+ and CD8+ T cell responses |
Absent |
Due to their central role in the initiation of the anti-viral immune response, DCs are ideal targets for viruses to exercise their immune evasion strategies and in fact viruses that cause persistent infection appear to have perfected the art of evading the pathogen recognition and elimination properties of the DC (Box 1). Gaining a better understanding of these mechanisms in virus-infected DCs may enable us to better understand virus-host interaction and in turn provide newer perspectives for the therapy of persistent infections as well as the design of vaccines.
Box 1. Viral immune evasion strategies.
As a consequence of co-evolution with their hosts, viruses have developed various immune evasion strategies to ensure their own replication and survival (reviewed in 124).
Antigenic variation
This is an important strategy evolved in particular by RNA viruses to remain evade immune response of the host. Because of the infidelity of their polymerases, many mutations are introduced in the viral genome during the course of replication, resulting in the existence of many different genetic quasispecies in a single host. This makes it difficult for the host to generate the staggeringly complex immune response that is required for virus elimination
Sequestration
Viruses infect non-permissive or semi-permissive host cells to store their genetic information and, thereby, become invisible to the immune system of the host. These viruses stay “latent” with absent or decreased transcription until the virus or cell becomes activated.
Blockade of antigen-presentation by APC
Collectively, viruses encode proteins have the capacity to interfere with almost any step in antigen processing and presentation by APCs, such as prevention of proteasomal antigen fragmentation and transport to the endoplasmic reticulum, interference with MHC I and II molecules expression and localization, downregulation of the expression of co-stimulatory molecules.
Cytokine evasion
Cytokines released by the host in response to viral infections coordinate and control the processes of immune activation and proliferation. It is therefore not surprising that viruses counteract these responses through encoding mimics or homologs of normal cytokines and their receptors, which bind to or replace the normal cellular counterparts as well as interfere with cytokine signaling within the host cell.
Inhibition of apoptosis
Apoptosis, or programmed cell death, is a relatively silent and non-inflammatory process to eliminate virus-infected cells. Viruses encode a variety of proteins to block apoptosis at essentially every step to delay cell death until the viral progeny have been formed and are infectious.
This review highlights the latest advances in our understanding of the interplay between DCs and viruses that cause persistent viral infections. We will focus on the interaction of HBV, HCV and HIV with different subtypes of DCs, outlining diverse outcomes of the virus-DC interaction and its relevance to viral pathogenesis and the mechanisms that the viruses have developed to interfere with the normal response of the host.
Do persistent viruses infect DCs?
The presence of DCs within the skin, the blood and particularly within the mucosal surfaces and their ability to take up antigen at these sites predisposes DCs to function as primary target cells for viruses. It is therefore possible that viruses establish persistence by directly infecting DCs. It is not unreasonable to assume that replication of the viral genome along with the expression of viral antigens would interfere with signaling pathways in DC or directly impair DC function, rendering infected DCs less able to stimulate T cell responses. For example, herpes simplex virus 1 ICP47 protein and human cytomegalovirus US6 protein are known to inhibit loading of antigenic peptides onto MHC class I molecules, thereby interfering with the ability of infected DCs to prime naive T cells efficiently 15.
HIV
Langerhans cells, the professional APCs of the epidermis, were the first DCs reported to be susceptible to HIV-1 infection. Since then mDCs and pDCs isolated from the blood of HIV-infected patients have been shown to be infected by HIV-1 (reviewed in 16). However, HIV replication in DCs is generally less productive, and the frequency of HIV-infected DCs in vivo is often 10–100 times lower 17 when compared to HIV infection rates observed in CD4+ T cells. In vitro studies indicated that on average only 1–3% of mDCs and pDCs from healthy blood donors can be productively infected by both primary and laboratory-adapted HIV, as detected by intracellular staining of HIV p24 protein 18. Immature DCs have been reported to be more susceptible to productive infection than mature DCs 19 which can be partly explained by the enhanced capacity of immature DCs to acquire viral antigen. During maturation of DCs the ability to capture antigens through macropinocytosis and receptor-mediated endocytosis, rapidly declines and the DCs instead assemble complexes of antigen with either MHC class I and MHC II1. Furthermore, HIV replication in pDCs was observed to increase substantially following CD40 ligation 20, a signal physiologically delivered by CD4+ T cells. Thus, HIV replication in pDCs may be triggered through the interaction with activated CD4+ T cells within the extrafollicular T-cell zones of the lymphoid tissue, suggesting that pDCs serve as viral reservoirs for CD4+ T cells.
HCV
HCV genomic RNA has been detected in pDCs and mDCs directly isolated from the blood of HCV-infected patients 21, 22 and it was initially believed that DCs were susceptible to HCV infection. However, using a strand-specific semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR), Goutagny and colleagues observed the replicative intermediate in only a small percentage of DCs isolated from HCV-infected patients (3 of 24 HCV-infected patients) indicating that HCV replication in DCs occurs at a lower frequency when compared to hepatocytes, the main reservoir of HCV 21. Studying HCV in vitro is difficult due to the lack of a robust in vitro propagation system (Box 2). To study HCV infection of DCs in vitro, monocyte-derived DCs (MoDCs) of healthy individuals were incubated with HCV RNA positive serum. The replicative intermediate was subsequently detected in MoDCs, indicating that DCs may support at least the first steps of the viral life-cycle 23. However, following incubation of MoDCs and subsets of blood DC with infectious recombinant HCV neither viral replication nor HCV protein synthesis could be detected 24–28 suggesting that HCV may infect DCs but does not result in a productive infection.
BOX 2. The challenge to develop in vitro models for the study of HCV-DC interaction.
In contrast to HIV and HBV, studies addressing the interaction of HCV with DC have been hampered for a long time because of the lack of a robust cell-culture system that allows the production of recombinant infectious HCV. Various surrogate models have been used to study the virus-host interaction, such as recombinant viral proteins, virus-like particles (VLPs) that are generated by self-assembly of HCV structural proteins and closely mimic the properties of native virions. Furthermore, infectious pseudoviruses consist of unmodified HCV envelope proteins assembled onto retroviral or lentiviral core particles and are replication incompetent 125. They have been used to study HCV entry into target cells. Finally, in 2005 a cell-culture system based on the transfection of JFH1 mRNA (HCV genotype 2a strain) into a highly permissive human hepatoma cell line became available 126–128. Until now, studies addressing the interaction of HCV with DCs are limited to the use of recombinant HCV derived from an HCV genotype 2a strain that is highly adapted to a hepatic cell line. Of note, HCV strains of genotype 1 are more prevalent and are associated with liver disease in most countries of the world 129. Although low levels of infectious cell-culture derived HCV have been obtained from a genotype 1a isolate with five adaptive mutations 130, and progress has been made in the construction of chimeric HCV genomes comprising JFH-1derived replicase proteins and structural proteins from heterologous HCV strains 131, 132, the challenge to develop an in vitro system for higher virus production of other HCV genotypes and novel cell-culture systems allowing the selection of HCV variants particularly adapted to DCs remains.
HBV
Although detection of HBV-DNA in subsets of isolated blood DCs from HBV-infected patients has been proposed to indicate HBV infection 29, additional studies have not revealed the presence of the HBV RNA replicative intermediates in either the blood DC subsets of HBV-infected patients or from DCs infected in vitro with wildtype or recombinant HBV 30, 31. Thus, it is likely that DCs do not support replication and production of HBV viral particles and that the detection of HBV-DNA merely reflects the attachment of the virus to the cell surface or the natural antigen-uptake function of DCs.
In summary, DCs can support the production of HIV particles, although at much lower levels than the CD4+ T cells (the primary targets for HIV), but not HCV and HBV particles, even though HCV may be able to initiate replication. There are three possible explanations for this. First, viral receptors or co-receptors may be absent or present only at a low frequency on DCs. DCs express relatively low levels of the HIV receptor CD4 and the co-receptors CCR5 and CXCR432 and very low levels of the HCV co-entry factor claudin-128. Unlike HIV and HCV, functional receptors mediating entry of HBV have not yet been identified.
Second, the virus may be degraded in intracellular compartments in DCs before it completes its replicative cycle. Antigens can be targeted to different processing pathways after internalization through receptor-mediated endocytosis where the endocytosed antigen undergoes extensive degradation prior to its presentation on the cell surface in association with MHC class I and II molecules 33. DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), a C-type lectin receptor 34, has been shown to promote HIV antigen presentation by MHC class I and II molecules 35, 36. Scavenger receptor class B type I (SR-BI) is known to mediate uptake and presentation of HCV particles by DCs 37.
Third, host factors may block viral replication, or host factors required for replication may be missing in DCs. A family of cellular restriction factors, the APOBEC cytidine deaminase family, functions as a restriction factor that blocks replication of HIV after viral entry 38. Expression levels of APOBEC3G in myeloid DCs correlate with HIV resistance 39, suggesting that cytidine deaminases represent a potent innate barrier to HIV infection. Tang and MacLachlan demonstrated the dependency of HBV replication on the presence of liver specific transcription factors belonging to a family of nuclear hormone receptors 40. The presence of host restrictions factors may be a crucial factor in determining the susceptibility of DC populations to productive infection with persistent viruses.
The role of DCs in viral dissemination
After uptake of viral antigen, activated DCs can traffic extensively from peripheral tissues to secondary lymphoid organs in an effort to present viral antigens to naïve T cells. It is therefore not surprising that persistent viruses exploit this migratory property of DCs to disseminate to more favorable sites of replication.
HIV
It has been known for more than a decade that DCs efficiently transmit HIV to CD4+ T cells. One potential mechanism of HIV transfer from DCs to T cells involves DC-SIGN (reviewed in 16). Binding of HIV by DC-SIGN requires the interaction of the HIV-1 envelope glycoprotein gp120 with the carbohydrate-recognition domain of DC-SIGN. HIV is subsequently internalized into non-lysosomal compartments and transported within DCs before it is transferred to CD4+ T cells in a process termed trans-infection. The sequential endocytosis and exocytosis of intact HIV virions, without viral replication, is called the “trojan horse” model. In this model, virion transmission is thought to occur via the infectious synapse 41, a structure that is formed between the DC and T cells, along with viral receptors, co-receptors and DC-SIGN or other C-type lectins. Because DCs can sequester infectious virus for several days in their endosomal compartments, DCs can carry HIV to interacting T cells in the lymph node, which is the most important site for viral replication and spread 42. Though direct HIV infection of DCs is less efficient than infection of CD4+ T cells, several reports indicate that HIV dissemination may be aided by the transfer of progeny virus from infected DCs to T cells 43, 44, a process known as cis-infection. It is possible that DCs form a long-lived, motile HIV reservoir that helps to disseminate infectious virus through peripheral blood and in lymphoid and non-lymphoid tissues.
The differences between the DC subsets (Table 1) raise the possibility that they have distinct roles in HIV transmission. pDCs have been found to be less efficient in HIV transmission when compared to mDCs 45. In addition, although myeloid DC subsets are known to transfer HIV to activated CD4+ T cells efficiently 16, Langerhans cells appear to prevent HIV transmission by degrading captured HIV particles 46, suggesting that distinct DC subsets can either mediate or prevent HIV-1 transmission.
HCV
Compared to HIV research, studies analyzing the in vivo dissemination of hepatotropic viruses by DCs are in their infancy. The HCV envelope glycoprotein E2, HCV virions derived from HCV infected patient serum samples and retroviruses pseudotyped with HCV envelope glycoproteins (HCV pseudovirus) have been shown to bind specifically to DC-SIGN 47–49. Thus, it may be possible that in vivo blood DCs or hepatic DCs within the liver sinusoids bind circulating HCV particles through a DC-SIGN mediated mechanism. Of note, HCV pseudovirus, bound to DC-SIGN expressed on MoDCs, was transmitted efficiently when cocultured with the human hepatocellular carcinoma cell line Huh7, a cell line that supports HCV pseudovirus entry and productive viral replication of recombinant infectious HCV 49, 50. Furthermore, Ludwig and colleagues observed that virus-like particles bound by DC-SIGN, representative of HCV envelope glycoproteins, are targeted to early endosomal vesicles or non-lysosomal compartments in MoDCs. The HCV particles resided in these compartments for over 24h 51, suggesting that HCV can bypass viral antigen processing and presentation pathways in DCs, thereby escaping degradation. Possibly, HCV retained in the non-lysosomal compartments of DCs plays a role in HCV transmission from DCs to hepatocytes. HCV captured by blood DCs or hepatic DCs within the liver sinusoids may allow transfer of the virus to the underlying hepatocytes when DCs traverse the sinusoidal lumen to the hepatic lymph. Besides DCs, liver sinusoidal endothelial cells (LSECs), which form the lining endothelium of the hepatic sinusoid (Fig. 2), have been shown to bind recombinant HCV E2 protein through the interaction of DC-SIGN and DC-SIGNR that are expressed on the cell surface of LSECs 52. However, LSECs have been shown to be unable to support HCV pseudovirus entry and infection with cell-culture derived HCV, suggesting that LSECs are not permissive for HCV infection 52. However, binding of HCV to LSECs may support a model where DC-SIGN mediated binding of HCV on LSECs provides a mechanisms for high affinity binding of circulating HCV within the liver sinusoid that may allow subsequent transfer to the underlying hepatocyte and thereby may increase the rate and efficiency of virus infection of the underlying hepatocytes.
Figure 2. Antigen presentation in the liver results in T cell tolerance.
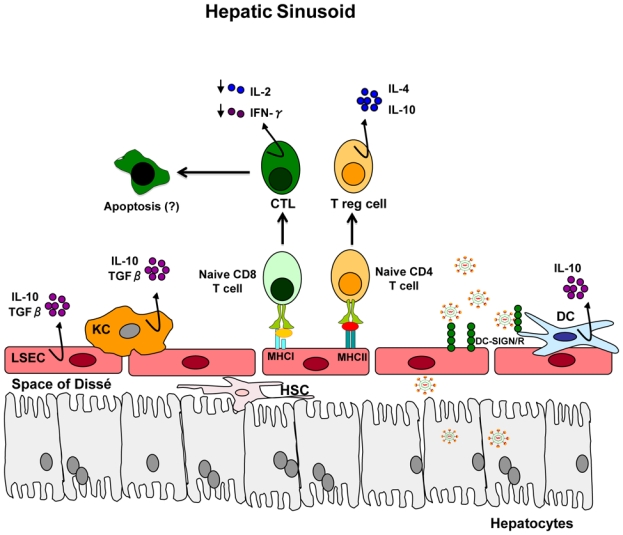
The liver sinusoid is lined by a fenestrated endothelium (liver sinusoidal endothelial cells, LSEC). Kupffer cells (KCs) and immature dendritic cells (DCs) are found in the sinusoids. Hepatic stellate cells (HSC) are located in the sub-endothelial space, known as the Space of Dissé. T cells that recognize antigen in the liver are exposed to immunosuppressive cytokines (IL-10 and TGF-β) that are synthesized by KCs, LSECs and DCs. Interaction of naïve T cells with LSEC results in differentiation of T cells into CD4+ regulatory T cells and impaired cytotoxic CD8+ T cells, followed by cell death. Hepatotropic viruses appear to be captured by DCs and/or LSECs in process that probably involves DC-SIGN or DC-SIGNR (for HCV) or other not yet defined cell-surface molecules (for HBV) for subsequent transfer to the underlying hepatocytes or viral particles may be internalized by hepatic DCs and LSECs for processing and presentation to naïve T cells (Adapted from 116 and 133).
HBV
Although DC-SIGN recognizes a broad range of pathogens ranging from bacteria to viruses, binding of recombinant hepatitis B surface antigen or cell-culture derived HBV particles to DC-SIGN has not been observed so far 53. Interestingly, studies have shown that the enzymatic modification of the N-linked oligosaccharide structures of the HBV antigen appears to prevent recognition by the carbohydrate-recognition domain of DC-SIGN 53, although other mechanisms may also play a role.
Impact of persistent viruses on DC function
Effects of persistent viruses on DCs in vivo
Virus-mediated impairment of DC function is a strategy to attenuate the multiple downstream immune effector mechanisms that depend on optimal DC function and may facilitate establishing viral persistence.
At the primary level, viruses can modulate the frequency of DC subsets by interfering with DC development, causing aberrant trafficking or inducing apoptosis. Significantly lower numbers of blood mDCs and pDCs have been observed in patients infected with HIV 54–59, HCV 60–65, and HBV 66 when compared to healthy individuals. In HIV infection, DC depletion appears to be due to migration of pDCs to the inflamed lymph nodes, where pDCs have been found to be activated and apoptotic, and frequently infected with virus 67, 68, suggesting that HIV-mediated cell death may account for the decreased number of circulating pDCs. In patients infected with HCV or HBV, blood DC subsets have been found to be enriched in the liver 64, 69–71, suggesting that DC migration to the liver results in the observed paucity of circulating DCs. However, lower numbers of circulating DCs have also been observed in patients with non-viral related liver diseases, e.g. granulomatous hepatitis or primary biliary cirrhosis 27, 61, 72 suggesting that low DC counts in virus related liver diseases is a common non-specific feature of inflammatory hepatitis. Interestingly, in vitro studies revealed that sera from HCV-infected patients and HCV envelope glycoprotein E2 itself inhibited in vitro migration of DCs towards CCL21, a CCR7-binding chemokine important for homing to lymph nodes 64. This leads to the hypothesis that after HCV uptake DCs may experience impairment in their ability to migrate to the draining lymph node, causing them to be trapped in the liver and thereby become less able to prime T cell responses. However, the in vivo relevance of this hypothesis remains to be investigated.
The differentiation and activation of virus-specific Th1 type CD4+ T cells and cytotoxic CD8+ T cells are regulated by DC-mediated production of IL-12. DC-mediated production of IL-10, on the other hand, is capable of inhibiting these responses 3. Upregulation of IL-10 production and suppression of Th1-response-promoting IL-12 as well as IFN-α have been documented in MoDCs, mDCs and pDCs isolated from HIV-73–75, HCV-63, 65, 76, 77 and HBV-infected patients 29 in response to various maturation stimuli. Dysregulated cytokine production by DCs may furthermore affect the early anti-viral host defense mediated by NK cells (Fig. 1). Several lines of evidence indicate that NK cell activity is impaired during HIV infection, in part due to a defective pDC function 78, 79. In particular, defective IFN-γ production by NK cell was attributable to impaired pDC function 79. Whether the impaired IFN-γ producing activity by NK cells derived from chronically infected HCV-patients 80 relies also on impaired cell cross talk between DCs and NK remains to be investigated.
DCs isolated from patients infected with HIV 73–75, HCV 63, 81–83 and HBV 29 were less able to stimulate T cell activation and proliferation as seen in a mixed lymphocyte reaction. Less efficient allogeneic T cell stimulation by DCs from HIV- 84 or HCV-infected patients 82 could be reversed by the neutralization of IL-10, suggesting that virus-induced production of IL-10 by DCs may limit T cell proliferation and activation, skewing the immune response towards tolerance. However, several investigators failed to detect an impaired capacity to stimulate allogeneic T cells by DCs ex vivo isolated from HCV-,60–62, 72, 85 and HBV- 86 infected patients. Possible reasons for these contradictory results could be the different experimental and technical settings used (e.g. different isolation protocols, maturation cocktails), differences in response by the DCs depending on their maturation status and uptake of viral antigen and the viral load in the infected patients. In addition, DC dysfunction in hepatotropic viruses may be restricted to the virus-specific response since the strong global immune dysfunction seen in HIV/AIDS is not observed with HBV and HCV infection.
To gain a different perspective on the impact of persistent viruses on DCs, DC numbers and function have been studied before and during antiviral therapy. Highly Active Anti-Retroviral Therapy (HAART) for HIV infection resulted in an increase in pDC number and restoration of IFN-α production to normal levels 78, 87 indicating that anti-retroviral therapy that reduces viral load has the capacity to reconstitute the function of DCs. Likewise, following pegylated-interferon-α and ribavirin therapy for HCV infection, the frequency of pDCs in individuals with viral clearance increased significantly, and reached levels observed in healthy controls 88. Therapy for HBV infection with the nucleotide analogue adefovir dipivoxil increased the frequency of mDCs, the T-cell stimulatory capacity and the capacity to produce IL-12 by mDC, whereas the production of IL-10 decreased 30. This functional recovery of mDCs coincided with a significant reduction in viral load underscoring the importance of viral load reduction in antiviral regimens which serves as the first step in a multistep process that culminates in the restoration of impaired immune responses during recovery from persistent viral infections.
In vitro studies to investigate molecular mechanisms of virus-DC interaction
To clarify the impact of persistent viruses on DC function and to identify the molecular mechanisms involved in this interaction, DC subsets isolated from healthy individuals have been exposed directly to recombinant infectious virus or viral proteins. In contrast to HIV 89 and other viruses, such as influenza and human herpesvirus type 125, 90–92, recombinant and serum-derived HCV and HBV have been shown to be poor inducers of IFN-α production in pDCs 24, 25, 65, 90 suggesting that these viruses may use this mechanism to downregulate downstream effector functions dependent on pDC-mediated IFN-α production. In pDCs, TLR7 and TLR9 detect viral RNA and DNA, respectively, in endosomal compartments, leading to the activation of nuclear factor kappaB (NF-kappaB) and IFN regulatory factors (IRFs) 4. IFN-α production induced by the TLR9 agonist CpG, but not by the TLR7 agonist resiquimod, was inhibited by HCV 24, 25, 90 and HBV 93. Similarly, HIV inhibited TLR9-mediated IFN-α production 59, 94 indicating that impairment of IFN-α production in pDCs is a universally used strategy by persistent viruses. What are the underlying mechanisms responsible for the virus-mediated interference of the IFN-α pathway in pDCs? A possible mechanism could be related to virus cross-linkage of cell surface receptors that down-regulate IFN-α production, such as the C-type lectins BDCA2 (blood dendritic cell antigen 2) and DCIR (dendritic cell immuno receptor). It has been shown that BDCA-2 and DCIR ligation and cross-linking results in the inhibition of CpG-mediated induction of IFN-α by pDCs 95, 96. The viral envelope proteins HBsAg and HIV gp120 can directly impair TLR9-mediated IFN-α production by pDCs through binding of BDCA293, 94. Although HCV core 24 and recombinant non-infectious HCV particles 24, 25, 90, composed of HCV core and the envelope glycoproteins E1 and E2, also blocked TLR9-mediated IFN-α production it is not known whether the interaction occurs also via BDCA2.
Recent studies indicate that persistent viruses may target immunosuppressive enzymes in DCs to actively suppress anti-viral T cell immune responses. The tryptophan catabolizing enzyme indoleamine 2,3-dioxygenase (IDO), seems to be a central feature of the suppressive function of DCs. DC-mediated IDO activity has been associated with inhibition of T cell proliferation and function 97. In vitro activated human T cells underwent cell-cycle arrest when deprived of tryptophan 98 and T cells became susceptible to apoptosis in vitro and in vivo in response to the toxic metabolites generated during tryptophan degradation 99. Direct exposure of DCs to HIV induces IDO leading to the inhibition of CD4+ T cell proliferation in vitro 100. Moreover, HIV-stimulated IDO activity in pDCs induced the differentiation of naïve T cells into Tregs with suppressive function 101, suggesting that HIV induced IDO activity may contribute to viral persistence by suppressing virus-specific T-cell responses. In SIV-infected macaques peak IDO activity coincided with an increase in plasma viremia and the transient expansion of the regulatory FoxP3+CD25+CD8+ T-cell subset that may participate in dampening the CD4+ Tcell SIV-specific response 102. Since enhanced IDO activity has been observed in patients infected with HIV, HCV and HBV 103–105, the role of DC-mediated IDO production in viral persistence merits further investigation.
In summary, several lines of evidence indicate that viruses efficiently target DC function to attenuate the anti-viral host immune response and establish persistence. However, is there also a role for DCs in disease progression? Chronic HBV and HCV infections are major risk factors for hepatocellular carcinoma (HCC) development 14. There is increasing evidence that a long-standing, inflammatory injury is an important procarcinogenic factor in many different cancer types, including HCC 106. The host immune responses to hepatitis viruses by DCs are fairly weak and often fail to control or completely clear infection, resulting in chronic stimulation of the antigen-specific immune response in persistently infected patients. Chronic antigen-stimulation at the infection sites and continuous infiltration of DCs into liver tissue may perpetuate a long-lasting chronic inflammatory process by the continued expression of pro-inflammatory cytokines, the attendant activation of liver NK cells and recruitment of T cells. These events may affect many cellular pathways that ultimately result in fibrosis, cirrhosis, and/or HCC. In HIV infection, it is widely accepted that chronic immune activation has a central role in driving progression to AIDS 107. Recent reports indicate that chronic pDC stimulation and IFN-α production are associated with a higher risk for HIV disease progression 108, 109, underscoring the role of pDCs in disease progression. A detailed comparison of the complex processes that govern homeostasis and immune activation mechanisms in health and persistent viral infection may help define the contribution of DCs to disease pathogenesis.
The role of hepatic APCs in HBV and HCV infection
The liver has several cell populations that can act as APCs. Besides liver-resident DCs, liver sinusoidal endothelial cells (LSECs), stellate cells and Kupffer cells (KCs) 110 (Figure 2), can also present antigens and influence the generation and maintenance of the anti-viral immune responses. However, the liver –specific immune system is maintained at a baseline state of tolerance as evidenced by the spontaneous acceptance of liver allografts 111. Several liver APCs exist in a state of active tolerance and contribute to the tolerogenic liver environment by the continuous secretion of immunosuppressive cytokines, e.g. IL-10 and TGF-β 112. This raises the question of whether the tolerogenic properties of the liver APCs contribute to the persistence of hepatotropic viruses and whether the liver presents unique environments for immune evasion.
Due to the difficulty in gaining access to liver biopsies and the challenge of isolating adequate numbers of APCs from tissue with high purity, limited information is available regarding the role of the hepatic APCs in viral infection. Royer and colleagues incubated isolated KCs with sera from patients containing HCV RNA 113. Genomic HCV RNA disappeared within a few days of infection and the replicative intermediate could not be detected, suggesting that KCs do not support HCV replication. Similarly, isolated LSECs were unable to support infection by cell-culture derived HCV and HBV, suggesting that LSEC are not permissive for hepatotropic viruses 52,114.
Analysis of liver biopsy samples obtained from patients with chronic HCV infection demonstrated that most KCs express high levels of co-stimulatory molecules and MHC class I and II molecules and formed clusters with CD4+ T cells, thereby acquiring the phenotype of an effective APC 115. Since KCs are able to move across the sinusoidal wall into the liver parenchyma, activation of KCs might likely reflect phagocytosis of HCV-infected apoptotic hepatocytes. Even though there is little doubt that liver resident DCs can take up viral particles, available evidence indicates that this may not necessarily translate to efficient T cell priming and activation. In vivo studies have revealed that while hepatic DCs and LSECs present exogenous antigen to naive T cells, the resulting activated T cells either fail to differentiate into effector T cells or acquire an immunosuppressive phenotype (For reviews, see ref 112, 116). It is possible that uptake of viral particles by liver APCs primes regulatory CD4+ T cells and impairs CD8+ T cells that finally fails to eradicate the virus from the liver. Since antigen-specific CD8+ T cells in the liver of patients with chronic HCV infection frequently become dysfunctional and unable to secrete IFN-γ or IL-2117, the role of hepatic DCs in HCV-specific T cell priming merits further investigations.
Current research has not focused on the ability of the hepatocyte to act as an antigen presenting cell in HCV infection. In general, hepatocytes are not easily accessible to naïve T cells because LSECs may form an effective barrier between hepatocytes and the sinusoidal lumen 118. However, electron microscopy analysis has shown that hepatocytes have microvilli that project into the sinusoidal lumen through the fenestrations in the endothelium, allowing contact between hepatocytes and circulating T cells in the lumen 119. Hepatocytes normally do not express MHC class II molecules; however in clinical hepatitis, aberrant expression of MHC class II molecules has been demonstrated 120, 121. It is therefore possible that MHC class II-expressing hepatocytes may stimulate CD4+ T cells and/or shape the antiviral immune response of pre-activated CD4+ T cells. Additional studies in transgenic mice demonstrated that CD8+T cells might be directly activated by hepatocytes. However, activation of CD8+ T cells by hepatocytes appeared to favor an impaired cytotoxic CD8+ T-cell response and reduced survival, possibly caused by a lack of co-stimulatory molecules 122, 123. Thus, presentation of viral antigens by hepatocytes may influence the anti-viral immune response and appears to promote CD8+ T-cell helplessness.
Conclusions and Perspectives
Accumulating evidence indicates that HIV, HCV and HBV target DC function to disturb the generation of a strong anti-viral innate and adaptive immune response, facilitating viral persistence. All three viruses appear to employ similar strategies to attenuate the potent, antiviral IFN-α response in pDCs. In addition, these viruses appear to affect the ability of the mDCs to produce key cytokines, which are essential for the development and activation of an effective T cell response. Not only do these viruses override the natural anti-viral activity of the DCs but they also use the DCs as vehicles for widespread dissemination within the host.
In the future, key issues in improving our understanding of the interplay between persistent viruses and DCs are the characterization of the intracellular compartments and molecular mechanisms required for virus acquisition, processing and presentation by DCs, the identification of mechanisms regulating the balance between intrahepatic tolerance and immunity and the role of DCs in aiding viral transmission during infection with HBV and HCV. Knowledge of these mechanisms will not only help in understanding viral pathogenesis but can also be used to design strategies that manipulate the immune system towards generating a protective immune response that controls viral replication without the attendant immunopathology.
Table 3.
Effects of persistent viruses on DC number and function
| HCV | HBV | HIV | ||
|---|---|---|---|---|
| DC number: | ||||
| In blood | Decreased | Decreased | Decreased | |
| At infection site | Enriched (in liver) | Enriched (in liver) | Enriched (lymphoid tissue) | |
| Affected DC function: | Effect on downstream immune function/disease activity: | |||
| IFN-α production | Reduced | Reduced | Reduced | (1) suboptimal T and NK cell activation (2)reduced pDC and T cell survival |
| IL-12 production | Reduced | Reduced | Reduced | (1) suboptimal differentiation of T helper (Th) type 1 cells (2)decreased IFN-γ production by CD8+T cell and NK cells |
| IL-10 production | Increased | Increased | Increased | Inhibition of (1) DC activation (2) Th type 1 cytokine production (3)CD8+ T cell function |
| IDO induction | not known | not known | Yes | (1) suppression of T cell proliferation and function (2) T cell apoptosis |
| T cell stimulation | Reduced/normal | Reduced/normal | Reduced | (1) decreased control of viral replication |
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH, the CONECTUS program from University of Strasbourg, the Agence Nationale de la Recherche and the Agence Nationale de Recherche sur le SIDA et les Hépatites Virales.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. This article describes elegantly the basics of dendritic cell function. [DOI] [PubMed] [Google Scholar]
- 2.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741–50. doi: 10.1016/j.immuni.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. This article gives an overview about the development and role of the different dendritic cell subests. [DOI] [PubMed] [Google Scholar]
- 4.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 5.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 6.Smit JJ, et al. The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS One. 2008;3:e1720. doi: 10.1371/journal.pone.0001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GeurtsvanKessel CH, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med. 2008;205:1621–34. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–29. doi: 10.1038/nri1573. This article compares clinical, virological and immunological features of hepatitis B and hepatitis C virus infection. Together with Ref. 11 an excellent overview of the anti-viral adaptive immune response in persistent viral infections is provided. [DOI] [PubMed] [Google Scholar]
- 9.Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 10.Gandhi RT, Walker BD. Immunologic control of HIV-1. Annu Rev Med. 2002;53:149–72. doi: 10.1146/annurev.med.53.082901.104011. [DOI] [PubMed] [Google Scholar]
- 11.McMichael AJ, Rowland-Jones SL. Cellular immune responses to HIV. Nature. 2001;410:980–7. doi: 10.1038/35073658. [DOI] [PubMed] [Google Scholar]
- 12.Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368:489–504. doi: 10.1016/S0140-6736(06)69157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alter MJ. Epidemiology of viral hepatitis and HIV co-infection. J Hepatol. 2006;44:S6–9. doi: 10.1016/j.jhep.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–6. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 15.Bauer D, Tampe R. Herpes viral proteins blocking the transporter associated with antigen processing TAP--from genes to function and structure. Curr Top Microbiol Immunol. 2002;269:87–99. [PubMed] [Google Scholar]
- 16.Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6:859–68. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIlroy D, et al. Infection frequency of dendritic cells and CD4+ T lymphocytes in spleens of human immunodeficiency virus-positive patients. J Virol. 1995;69:4737–45. doi: 10.1128/jvi.69.8.4737-4745.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smed-Sorensen A, et al. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol. 2005;79:8861–9. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canque B, et al. The susceptibility to X4 and R5 human immunodeficiency virus-1 strains of dendritic cells derived in vitro from CD34(+) hematopoietic progenitor cells is primarily determined by their maturation stage. Blood. 1999;93:3866–75. [PubMed] [Google Scholar]
- 20.Fong L, Mengozzi M, Abbey NW, Herndier BG, Engleman EG. Productive infection of plasmacytoid dendritic cells with human immunodeficiency virus type 1 is triggered by CD40 ligation. J Virol. 2002;76:11033–41. doi: 10.1128/JVI.76.21.11033-11041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goutagny N, et al. Evidence of viral replication in circulating dendritic cells during hepatitis C virus infection. J Infect Dis. 2003;187:1951–8. doi: 10.1086/375350. [DOI] [PubMed] [Google Scholar]
- 22.Rodrigue-Gervais IG, et al. Poly(I:C) and lipopolysaccharide innate sensing functions of circulating human myeloid dendritic cells are affected in vivo in hepatitis C virus-infected patients. J Virol. 2007;81:5537–46. doi: 10.1128/JVI.01741-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navas MC, et al. Dendritic cell susceptibility to hepatitis C virus genotype 1 infection. J Med Virol. 2002;67:152–61. doi: 10.1002/jmv.2204. [DOI] [PubMed] [Google Scholar]
- 24.Liang H, et al. Differential effects of hepatitis C virus JFH1 on human myeloid and plasmacytoid dendritic cells. J Virol. 2009 doi: 10.1128/JVI.02671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology. 2008;47:385–95. doi: 10.1002/hep.21996. References 24, 25, and 82 provide first evidence that HCV impairs the production of interferon α in plasmacytoid DCs. [DOI] [PubMed] [Google Scholar]
- 26.Ebihara T, Shingai M, Matsumoto M, Wakita T, Seya T. Hepatitis C virus-infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology. 2008;48:48–58. doi: 10.1002/hep.22337. [DOI] [PubMed] [Google Scholar]
- 27.Decalf J, et al. Plasmacytoid dendritic cells initiate a complex chemokine and cytokine network and are a viable drug target in chronic HCV patients. J Exp Med. 2007;204:2423–37. doi: 10.1084/jem.20070814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marukian S, et al. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology. 2008 doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Molen RG, et al. Functional impairment of myeloid and plasmacytoid dendritic cells of patients with chronic hepatitis B. Hepatology. 2004;40:738–46. doi: 10.1002/hep.20366. [DOI] [PubMed] [Google Scholar]
- 30.van der Molen RG, Sprengers D, Biesta PJ, Kusters JG, Janssen HL. Favorable effect of adefovir on the number and functionality of myeloid dendritic cells of patients with chronic HBV. Hepatology. 2006;44:907–14. doi: 10.1002/hep.21340. [DOI] [PubMed] [Google Scholar]
- 31.Untergasser A, et al. Dendritic cells take up viral antigens but do not support the early steps of hepatitis B virus infection. Hepatology. 2006;43:539–47. doi: 10.1002/hep.21048. [DOI] [PubMed] [Google Scholar]
- 32.Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A. 1999;96:5215–20. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen PE. Recent advances in antigen processing and presentation. Nat Immunol. 2007;8:1041–8. doi: 10.1038/ni1516. [DOI] [PubMed] [Google Scholar]
- 34.Cambi A, Koopman M, Figdor CG. How C-type lectins detect pathogens. Cell Microbiol. 2005;7:481–8. doi: 10.1111/j.1462-5822.2005.00506.x. [DOI] [PubMed] [Google Scholar]
- 35.Moris A, et al. DC-SIGN promotes exogenous MHC-I-restricted HIV-1 antigen presentation. Blood. 2004;103:2648–54. doi: 10.1182/blood-2003-07-2532. [DOI] [PubMed] [Google Scholar]
- 36.Smith AL, et al. Leukocyte-specific protein 1 interacts with DC-SIGN and mediates transport of HIV to the proteasome in dendritic cells. J Exp Med. 2007;204:421–30. doi: 10.1084/jem.20061604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barth H, et al. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J Virol. 2008;82:3466–79. doi: 10.1128/JVI.02478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu YL, et al. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–14. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 39.Peng G, et al. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007;110:393–400. doi: 10.1182/blood-2006-10-051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang H, McLachlan A. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci U S A. 2001;98:1841–6. doi: 10.1073/pnas.041479698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piguet V, Sattentau Q. Dangerous liaisons at the virological synapse. J Clin Invest. 2004;114:605–10. doi: 10.1172/JCI22812. This article describes cell to cell transmission of retroviruses through the virological synapse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geijtenbeek TB, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–97. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 43.Burleigh L, et al. Infection of dendritic cells (DCs), not DC-SIGN-mediated internalization of human immunodeficiency virus, is required for long-term transfer of virus to T cells. J Virol. 2006;80:2949–57. doi: 10.1128/JVI.80.6.2949-2957.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turville SG, et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood. 2004;103:2170–9. doi: 10.1182/blood-2003-09-3129. [DOI] [PubMed] [Google Scholar]
- 45.Groot F, van Capel TM, Kapsenberg ML, Berkhout B, de Jong EC. Opposing roles of blood myeloid and plasmacytoid dendritic cells in HIV-1 infection of T cells: transmission facilitation versus replication inhibition. Blood. 2006;108:1957–64. doi: 10.1182/blood-2006-03-010918. [DOI] [PubMed] [Google Scholar]
- 46.de Witte L, et al. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med. 2007;13:367–71. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 47.Gardner JP, et al. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc Natl Acad Sci U S A. 2003;100:4498–503. doi: 10.1073/pnas.0831128100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pohlmann S, et al. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77:4070–80. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lozach PY, et al. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J Biol Chem. 2003;278:20358–66. doi: 10.1074/jbc.M301284200. [DOI] [PubMed] [Google Scholar]
- 50.Cormier EG, et al. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc Natl Acad Sci U S A. 2004;101:14067–72. doi: 10.1073/pnas.0405695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ludwig IS, et al. Hepatitis C virus targets DC-SIGN and L-SIGN to escape lysosomal degradation. J Virol. 2004;78:8322–32. doi: 10.1128/JVI.78.15.8322-8332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai WK, et al. Expression of DC-SIGN and DC-SIGNR on human sinusoidal endothelium: a role for capturing hepatitis C virus particles. Am J Pathol. 2006;169:200–8. doi: 10.2353/ajpath.2006.051191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Op den Brouw ML, et al. Branched oligosaccharide structures on HBV prevent interaction with both DC-SIGN and L-SIGN. J Viral Hepat. 2008;15:675–83. doi: 10.1111/j.1365-2893.2008.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donaghy H, et al. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood. 2001;98:2574–6. doi: 10.1182/blood.v98.8.2574. [DOI] [PubMed] [Google Scholar]
- 55.Pacanowski J, et al. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood. 2001;98:3016–21. doi: 10.1182/blood.v98.10.3016. [DOI] [PubMed] [Google Scholar]
- 56.Grassi F, et al. Depletion in blood CD11c-positive dendritic cells from HIV-infected patients. Aids. 1999;13:759–66. doi: 10.1097/00002030-199905070-00004. [DOI] [PubMed] [Google Scholar]
- 57.Chehimi J, et al. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168:4796–801. doi: 10.4049/jimmunol.168.9.4796. [DOI] [PubMed] [Google Scholar]
- 58.Soumelis V, et al. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 2001;98:906–12. doi: 10.1182/blood.v98.4.906. [DOI] [PubMed] [Google Scholar]
- 59.Cavaleiro R, et al. Major depletion of plasmacytoid dendritic cells in HIV-2 infection, an attenuated form of HIV disease. PLoS Pathog. 2009;5:e1000667. doi: 10.1371/journal.ppat.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ulsenheimer A, et al. Plasmacytoid dendritic cells in acute and chronic hepatitis C virus infection. Hepatology. 2005;41:643–51. doi: 10.1002/hep.20592. [DOI] [PubMed] [Google Scholar]
- 61.Wertheimer AM, Bakke A, Rosen HR. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology. 2004;40:335–45. doi: 10.1002/hep.20306. [DOI] [PubMed] [Google Scholar]
- 62.Longman RS, Talal AH, Jacobson IM, Rice CM, Albert ML. Normal functional capacity in circulating myeloid and plasmacytoid dendritic cells in patients with chronic hepatitis C. J Infect Dis. 2005;192:497–503. doi: 10.1086/431523. [DOI] [PubMed] [Google Scholar]
- 63.Kanto T, et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis. 2004;190:1919–26. doi: 10.1086/425425. [DOI] [PubMed] [Google Scholar]
- 64.Nattermann J, et al. Hepatitis C virus E2 and CD81 interaction may be associated with altered trafficking of dendritic cells in chronic hepatitis C. Hepatology. 2006;44:945–54. doi: 10.1002/hep.21350. [DOI] [PubMed] [Google Scholar]
- 65.Dolganiuc A, et al. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177:6758–68. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- 66.Xie Q, et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect. 2009;11:515–23. doi: 10.1016/j.micinf.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 67.Brown KN, Wijewardana V, Liu X, Barratt-Boyes SM. Rapid influx and death of plasmacytoid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog. 2009;5:e1000413. doi: 10.1371/journal.ppat.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meyers JH, et al. Impact of HIV on cell survival and antiviral activity of plasmacytoid dendritic cells. PLoS One. 2007;2:e458. doi: 10.1371/journal.pone.0000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kunitani H, Shimizu Y, Murata H, Higuchi K, Watanabe A. Phenotypic analysis of circulating and intrahepatic dendritic cell subsets in patients with chronic liver diseases. J Hepatol. 2002;36:734–41. doi: 10.1016/s0168-8278(02)00062-4. [DOI] [PubMed] [Google Scholar]
- 70.Lai WK, et al. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J Hepatol. 2007;47:338–47. doi: 10.1016/j.jhep.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Z, et al. Severe dendritic cell perturbation is actively involved in the pathogenesis of acute-on-chronic hepatitis B liver failure. J Hepatol. 2008;49:396–406. doi: 10.1016/j.jhep.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 72.Longman RS, Talal AH, Jacobson IM, Albert ML, Rice CM. Presence of functional dendritic cells in patients chronically infected with hepatitis C virus. Blood. 2004;103:1026–9. doi: 10.1182/blood-2003-04-1339. [DOI] [PubMed] [Google Scholar]
- 73.Donaghy H, Gazzard B, Gotch F, Patterson S. Dysfunction and infection of freshly isolated blood myeloid and plasmacytoid dendritic cells in patients infected with HIV-1. Blood. 2003;101:4505–11. doi: 10.1182/blood-2002-10-3189. [DOI] [PubMed] [Google Scholar]
- 74.Chougnet CA, Margolis D, Landay AL, Kessler HA, Shearer GM. Contribution of prostaglandin E2 to the interleukin-12 defect in HIV-infected patients. Aids. 1996;10:1043–5. doi: 10.1097/00002030-199610090-00018. [DOI] [PubMed] [Google Scholar]
- 75.Chehimi J, et al. Impaired interleukin 12 production in human immunodeficiency virus-infected patients. J Exp Med. 1994;179:1361–6. doi: 10.1084/jem.179.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanto T, et al. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162:5584–91. [PubMed] [Google Scholar]
- 77.Dolganiuc A, et al. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170:5615–24. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- 78.Chehimi J, et al. Baseline viral load and immune activation determine the extent of reconstitution of innate immune effectors in HIV-1-infected subjects undergoing antiretroviral treatment. J Immunol. 2007;179:2642–50. doi: 10.4049/jimmunol.179.4.2642. [DOI] [PubMed] [Google Scholar]
- 79.Conry SJ, et al. Impaired plasmacytoid dendritic cell (PDC)-NK cell activity in viremic human immunodeficiency virus infection attributable to impairments in both PDC and NK cell function. J Virol. 2009;83:11175–87. doi: 10.1128/JVI.00753-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahlenstiel G, et al. Natural Killer Cells are Polarized towards Cytotoxicity in Chronic Hepatitis C in an Interferon-alpha-Dependent Manner. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bain C, et al. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120:512–24. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- 82.Dolganiuc A, Paek E, Kodys K, Thomas J, Szabo G. Myeloid dendritic cells of patients with chronic HCV infection induce proliferation of regulatory T lymphocytes. Gastroenterology. 2008;135:2119–27. doi: 10.1053/j.gastro.2008.07.082. [DOI] [PubMed] [Google Scholar]
- 83.Auffermann-Gretzinger S, Keeffe EB, Levy S. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood. 2001;97:3171–6. doi: 10.1182/blood.v97.10.3171. [DOI] [PubMed] [Google Scholar]
- 84.Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, Steinman RM. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc Natl Acad Sci U S A. 2004;101:7669–74. doi: 10.1073/pnas.0402431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Piccioli D, et al. Comparable functions of plasmacytoid and monocyte-derived dendritic cells in chronic hepatitis C patients and healthy donors. J Hepatol. 2005;42:61–7. doi: 10.1016/j.jhep.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 86.Tavakoli S, et al. Peripheral blood dendritic cells are phenotypically and functionally intact in chronic hepatitis B virus (HBV) infection. Clin Exp Immunol. 2008;151:61–70. doi: 10.1111/j.1365-2249.2007.03547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kamga I, et al. Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J Infect Dis. 2005;192:303–10. doi: 10.1086/430931. [DOI] [PubMed] [Google Scholar]
- 88.Mengshol JA, et al. Impaired plasmacytoid dendritic cell maturation and differential chemotaxis in chronic hepatitis C virus: associations with antiviral treatment outcomes. Gut. 2009;58:964–73. doi: 10.1136/gut.2008.168948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beignon AS, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–75. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gondois-Rey F, et al. Hepatitis C virus is a weak inducer of interferon alpha in plasmacytoid dendritic cells in comparison with influenza and human herpesvirus type-1. PLoS ONE. 2009;4:e4319. doi: 10.1371/journal.pone.0004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krug A, et al. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–7. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 92.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–20. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu Y, et al. HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol Immunol. 2009 doi: 10.1016/j.molimm.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 94.Martinelli E, et al. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2007;104:3396–401. doi: 10.1073/pnas.0611353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dzionek A, et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J Exp Med. 2001;194:1823–34. doi: 10.1084/jem.194.12.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meyer-Wentrup F, et al. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood. 2008;111:4245–53. doi: 10.1182/blood-2007-03-081398. [DOI] [PubMed] [Google Scholar]
- 97.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 98.Munn DH, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–72. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fallarino F, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–77. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 100.Boasso A, et al. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood. 2007;109:3351–9. doi: 10.1182/blood-2006-07-034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Manches O, et al. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J Clin Invest. 2008;118:3431–9. doi: 10.1172/JCI34823. This articles provides evidence that immunodeficiency viruses induce suppressive T cells through the induction of indoleamine-2,3-dioxygenase in plasmacytoid dendritic cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malleret B, et al. Primary infection with simian immunodeficiency virus: plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood. 2008;112:4598–608. doi: 10.1182/blood-2008-06-162651. [DOI] [PubMed] [Google Scholar]
- 103.Boasso A, et al. Regulatory T-cell markers, indoleamine 2,3-dioxygenase, and virus levels in spleen and gut during progressive simian immunodeficiency virus infection. J Virol. 2007;81:11593–603. doi: 10.1128/JVI.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Larrea E, et al. Upregulation of indoleamine 2,3-dioxygenase in hepatitis C virus infection. J Virol. 2007;81:3662–6. doi: 10.1128/JVI.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen YB, et al. Immunosuppressive effect of IDO on T cells in patients with chronic hepatitis B*. Hepatol Res. 2009;39:463–8. doi: 10.1111/j.1872-034X.2008.00476.x. [DOI] [PubMed] [Google Scholar]
- 106.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med. 2006;12:289–95. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- 108.Mandl JN, et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008;14:1077–87. doi: 10.1038/nm.1871. [DOI] [PubMed] [Google Scholar]
- 109.Meier A, et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med. 2009;15:955–9. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–63. doi: 10.1146/annurev.immunol.021908.132629. This article gives an overview over the unique environment and composition of antigen-presenting cells in the liver. Antigen presentation in the liver often results in tolerance due to the dominant presence of immunosuppressive cyotkines and tolerogenic antigen-presenting cells. [DOI] [PubMed] [Google Scholar]
- 111.Qian S, et al. Murine liver allograft transplantation: tolerance and donor cell chimerism. Hepatology. 1994;19:916–24. doi: 10.1002/hep.1840190418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sumpter TL, Abe M, Tokita D, Thomson AW. Dendritic cells, the liver, and transplantation. Hepatology. 2007;46:2021–31. doi: 10.1002/hep.21974. [DOI] [PubMed] [Google Scholar]
- 113.Royer C, et al. A study of susceptibility of primary human Kupffer cells to hepatitis C virus. J Hepatol. 2003;38:250–6. doi: 10.1016/s0168-8278(02)00418-x. [DOI] [PubMed] [Google Scholar]
- 114.Breiner KM, Schaller H, Knolle PA. Endothelial cell-mediated uptake of a hepatitis B virus: a new concept of liver targeting of hepatotropic microorganisms. Hepatology. 2001;34:803–8. doi: 10.1053/jhep.2001.27810. [DOI] [PubMed] [Google Scholar]
- 115.Burgio VL, et al. Expression of co-stimulatory molecules by Kupffer cells in chronic hepatitis of hepatitis C virus etiology. Hepatology. 1998;27:1600–6. doi: 10.1002/hep.510270620. [DOI] [PubMed] [Google Scholar]
- 116.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 117.Radziewicz H, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–53. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Knolle PA, Limmer A. Neighborhood politics: the immunoregulatory function of organ-resident liver endothelial cells. Trends Immunol. 2001;22:432–7. doi: 10.1016/s1471-4906(01)01957-3. [DOI] [PubMed] [Google Scholar]
- 119.Warren A, et al. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology. 2006;44:1182–90. doi: 10.1002/hep.21378. [DOI] [PubMed] [Google Scholar]
- 120.Dienes HP, Hutteroth T, Hess G, Meuer SC. Immunoelectron microscopic observations on the inflammatory infiltrates and HLA antigens in hepatitis B and non-A, non-B. Hepatology. 1987;7:1317–25. doi: 10.1002/hep.1840070623. [DOI] [PubMed] [Google Scholar]
- 121.Franco A, et al. Expression of class I and class II major histocompatibility complex antigens on human hepatocytes. Hepatology. 1988;8:449–54. doi: 10.1002/hep.1840080302. [DOI] [PubMed] [Google Scholar]
- 122.Bertolino P, Trescol-Biemont MC, Rabourdin-Combe C. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur J Immunol. 1998;28:221–36. doi: 10.1002/(SICI)1521-4141(199801)28:01<221::AID-IMMU221>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 123.Bowen DG, et al. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J Clin Invest. 2004;114:701–12. doi: 10.1172/JCI21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hilleman MR. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc Natl Acad Sci U S A. 2004;101 (Suppl 2):14560–6. doi: 10.1073/pnas.0404758101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barth H, Liang TJ, Baumert TF. Hepatitis C virus entry: molecular biology and clinical implications. Hepatology. 2006;44:527–35. doi: 10.1002/hep.21321. [DOI] [PubMed] [Google Scholar]
- 126.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 127.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9. doi: 10.1073/pnas.0503596102. References 126, 127 and 128 describe for the first time a cell culture system that allows the generation of recombinant infectious hepatitis C virus. This system provides a powerful tool for studying the viral life cycle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pawlotsky JM. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004;12:96–102. doi: 10.1016/j.tim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 130.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci U S A. 2006;103:2310–5. doi: 10.1073/pnas.0510727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gottwein JM, et al. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49:364–77. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 132.Pietschmann T, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103:7408–13. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]