

Abstract
Alzheimer’s disease (AD), the most common form of dementia, is characterized by the presence of neurofibrillary tangles composed of tau and senile plaques of amyloid-beta peptides (Aβ) derived from amyloid precursor protein (APP). Pin1 is a unique prolyl isomerase that has been shown to protect against age-dependent neurodegeneration by acting on phosphorylated tau and APP to suppress tangle formation and amyloidogenic APP processing. Here we report a functional polymorphism, rs2287839, in the Pin1 promoter that is significantly associated with a 3-year delay in the average age-at-onset (AAO) of late-onset AD in a Chinese population. More significantly, the Pin1 polymorphism rs2287839 is located within the consensus binding motif for the brain-selective transcription factor, AP4 (CAGCTG) and almost completely abolishes the ability of AP4 to bind and suppress the Pin1 promoter, as shown by chromatin immunoprecipitation, electrophoretic mobility shift assay and promoter luciferase assay. Moreover, overexpression or knockdown of AP4 resulted in an 80% reduction or two-fold increase in endogenous Pin1 levels, respectively. Thus, AP4 is a novel transcriptional repressor of Pin1 expression and the Pin1 promoter SNP identified in this study that prevents such suppression is associated with delayed onset of AD. These results indicate that regulation of Pin1 by AP4 plays a critical role in determining AAO of AD and might be a novel therapeutic target to delay the onset of AD.
1. Introduction
Alzheimer’s disease (AD) is by far the most common cause of dementia and it is currently affecting more than 30 million people worldwide (Ferri et al. 2009). Mutations in amyloid precursor protein (APP) gene, presenilin-1 and -2 (PSEN1, PSEN2) genes were identified in genetic studies on familial AD patients, but it only accounted for <5% of all AD cases (Bettens et al.). On the other hand, many genetic association studies including hypothesis-driven association studies (Cathcart et al. 2005; Corder et al. 1993; Rogaeva et al. 2007) and genome-wide association studies (GWAS) (Bertram et al. 2008; Harold et al. 2009; Lambert et al. 2009) were performed to identify the risk predisposition genes for the vast majority of sporadic AD. Age-at-onset (AAO) of AD has also been shown to be modulated by genetic factors (Daw et al. 2000) and the GWAS by Harold et al found that only SNPs at the ApoE locus are associated with AAO (Harold et al. 2009). The prevalence of AD increases with age and doubles every 5 years after the age of 65 (Evans et al. 1989). Identifying the genetic modulators affecting the AAO of AD will be important for the development of treatments that may potentially delay the onset of the disease beyond lifespan. Therefore, even a small difference in the AAO of AD would have a major impact on the prevalence of AD. For example, an intervention that could delay the average AAO of AD by only 2 years has been predicted to reduce the expected prevalence by 23% by 2050 (Brookmeyer et al. 1998). However, not much is known about the genetic factors that affect AAO of AD.
AD is characterized by the presence of amyloid plaques derived from amyloid-β peptides (Aβ) of amyloid precursor protein (APP) and neurofibrillary tangles composed of hyperphosphorylated tau. Pin1 is a unique prolyl isomerase that catalyzes the conversion of cis to trans conformation specifically at certain phosphorylated Ser/Thr-Pro motifs (Lu et al. 1996; Lu et al. 1999; Ranganathan et al. 1997; Yaffe et al. 1997; Zhou et al. 1999). Importantly, such Pin1-catalyzed conformational regulation can have profound effects on many key proteins in many cellular processes and Pin1 aberrations contributes to a growing number of diseases, notably AD, cancer and aging (Lee et al. 2009; Lu et al. 2007; Lu and Zhou 2007). Interestingly, Pin1 is mapped to 19p13.2, which has been identified as a novel locus for late-onset AD that is independent of the effect of ApoE4 (Butler et al. 2009; Wijsman et al. 2004). We and others have shown that Pin1 is highly expressed in most neurons, but is downregulated and/or inhibited by oxidative modifications in AD (Liou et al. 2003; Lu et al. 1999; Sultana et al. 2006; Zhou et al. 2000). Pin1 acts on the pThr231-Pro in tau to restore its microtubule function and to promote its dephosphorylation and degradation (Lim et al. 2008; Lu et al. 1999; Zhou et al. 2000). Furthermore, Pin1 also acts on the phosphorylated Thr668-Pro motif in APP to promote non-amyloidogenic APP processing (Pastorino et al. 2006). Indeed, Pin1 is the first gene, when deleted in mice, that causes tau- and Aβ-related pathologies and neurodegeneration in an age-dependent manner (Liou et al. 2003; Pastorino et al. 2006), resembling many aspects of AD. By contrast, Pin1 overexpression in postnatal neurons suppresses tangle formation and neurodegeneration induced by transgenic overexpression of human tau in mice (Lim et al. 2008). These results suggested Pin1 is both a functional and positional candidate for AD and it plays a pivotal role in protecting against age-dependent neurodegeneration.
There have been some major progresses in the genetic studies of rare familiar early onset AD, while the genetics of late-onset AD is more complicated, with many genetic association studies including GWAS being focused on different susceptibility genes for AD (Bertram and Tanzi 2009; Waring and Rosenberg 2008). For example, two GWASs identified an significant association between CLU and the risk of AD (Harold et al. 2009; Lambert et al. 2009). However, another association study does not find a significant association between the common variants in the coding region of CLU and the risk of AD (Guerreiro et al. 2010). Therefore, the results are inconclusive and the functional mechanisms of many reported associations are unclear with a few exceptions such as ApoE (Brouwers et al. 2008). Several common polymorphisms have been identified in the coding and promoter regions of Pin1. Previous studies showed that Pin1 promoter SNP (−842G>C) is associated with reduced level of Pin1 in blood cells and increased risk for AD in an Italian cohort (Segat et al. 2007), although not in other cohorts (Lambert et al. 2006; Nowotny et al. 2007). Further studies have confirmed that this SNP abolishes Pin1 promoter activity and is associated with reduced risk for multiple cancers (Han et al. 2009; Lu et al. 2009), which further supports the opposite effects of Pin1 on AD and cancer (Lu and Zhou 2007). However, it is not known whether Pin1 SNPs affect AAO of AD.
In this study, we identified a functional polymorphism in the Pin1 promoter that affects the binding of transcription factor AP4 and this polymorphism is significantly associated with 3-year delay in the onset of AD. AP4 is a transcription factor selectively expressed in brain (Yap et al. 2003) and belongs to the basic helix-loop-helix leucine-zipper (bHLH-LZ) subgroup of bHLH proteins and recognizes the symmetrical DNA core sequence CAGCTG (Hu et al. 1990). Although AP4 was initially shown to activate transcription of the SV40 promoter (Mermod et al. 1988), more recent studies indicate that AP4 mediates transcriptional repression of cellular and viral genes (Cui et al. 1998; Imai and Okamoto 2006; Jung et al. 2008; Kim et al. 2006). Our current study identified a functional Pin1 promoter SNP that completely abolishes the ability of AP4 to bind to the Pin1 promoter and suppress its expression, leading to increased Pin1 expression. This functional variation may account for the possible delay of the onset of AD.
2. Materials and Methods
2.1. Subjects
A total of 556 Chinese subjects over the age of 65 were recruited from the social centers and residential hostels for the elderly in Hong Kong. The subjects were all volunteers who responded to the announcements at the centers and hostels. The elderly participants and their relatives were informed that a study on cognitive and functional assessment for the early detection of dementia would be conducted, and the interested parties participated. One psychiatrist explained the details of the study and obtained written informed consent from each participant. All subjects were assessed by trained geriatric psychiatrists. The Clinical Dementia Rating (CDR) scale was used to assess the severity of dementia. Subjects with a global CDR of 1 were further assessed by NINCDS-ADRDA criteria and included for the study as AD subjects if they were diagnosed as probable or possible AD. Their cognitive status was assessed using the Cantonese version of the Mini-Mental State Evaluation (MMSE) (Chiu et al. 1994), as well as an extensive neuropshycological evaluation. Functional status was assessed by activities of daily living (ADL) through a caregiver’s observation of the subject’s actual performance and instrumental activities of daily living (IADL). The exclusion criteria included a CDR of 2 or above and a known history of other neurodegenerative disorders or major psychiatric disorders or cancers. Participants with profound communication difficulties were also excluded. All subjects originated from Guangdong province in China and were currently living in Hong Kong. A total of 256 Southern Chinese subjects (85.1% women; age range, 66–95) were diagnosed as possible or probable AD and were included as AD patients in this study. The age-at-onset (AAO) of AD patients was defined as the age at which the family caregivers and/or the individual first noted cognitive and/or behavioral problems sufficient to interfere with independent daily activities. The project was approved by the institutional ethics committee.
2.2. Polymorphisms in Pin1 and Genotyping
Pin1 contains more than 50 SNPs over its genomic span of 14.4 kb (dbSNP). Genotyping data of the Chinese population (CHB) was obtained from HapMap (www.hapmap.org, The International HapMap Consortium, 2003) and was used to analyze the linkage disequilibrium (LD) structure and the selection of the SNP for the current study.
Genomic DNA was extracted from peripheral blood samples using DNA extraction kit according to the manufacturer’s instruction (Roche, U.S.). PCR-RFLP was performed as previously described (Ma et al. 2005). Briefly, primers were designed to incorporate the polymorphism into the restriction site. PCR was performed to amplify the genomic DNA and the PCR product was subjected to restriction enzyme digestion. To validate the genotyping results, genotyping experiments were repeated and direct sequencing was performed in 10% of the samples. The ApoE genotyping was performed as reported earlier (Ma et al. 2005).
2.3. Luciferase Reporter Gene Constructs
Human genomic DNA samples were amplified to generate a 2807 bp fragment (−5430 to −2624) of the Pin1 promoter. The fragment was cloned into the previously generated Pin1 promoter construct (−2300 to +1) (Ryo et al. 2002) with pGL3-Basic firefly luciferase expression vector (Promega, Madison, WI). Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to manufacturer’s instructions to introduce the mutation −5185 (rs2287839) G > C and the sequence of the constructs was verified by DNA sequencing.
2.4. Transient Transfection and Promoter Reporter Assay in Mammalian Cells
Human H4 neuronal cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1mM sodium pyruvate, 100 U/ml of penicillin G and 100 mg/ml streptomycin (Life Technologies, Inc.) at 37°C in 5% CO2. Subconfluent cells cultured in 24-well dishes were transiently co-transfected with pGL3-G or pGL3-C reporter vector, the indicated amounts of AP4 expression plasmid and Renilla luciferase control vector using Lipofectamine and Plus reagent (Invitrogen) according to manufacturer’s instruction. Cells were harvested 48 hour after transfection and were lysed. Firefly and Renilla luciferase activities were measured simultaneously in each sample using the Dual-Luciferase Reporter Assay System according to the manufacturer’s instructions (Promega). Firefly luciferase activities were normalized to Renilla luciferase activity as ‘relative luciferase activity’. All experiments were performed in triplicate.
2.5. Chromatin Immunoprecipitation (ChIP)
ChIP assay was performed according to the manufacturer’s instruction (Upstate Biotechnology). Briefly, H4 cells were cross-linked with 1% formaldehyde for 10 min at 37C, washed twice with ice-cold phosphate-buffered saline, and lysed for 10 min at 1×106 cells in 200 μl of SDS lysis buffer. The extracts were sonicated until the DNA fragments were 500–800 bp in size. Cell extracts were subsequently incubated with 1 mg IgG or antibody against AP4 (Santa Cruz Biotechnology) overnight at 4°C. The extracts were incubated with salmon sperm DNA/protein A-Sepharose beads (Upstate Biotechnology) for 1 hr. After extensive washing of the beads, proteins were eluted and reversed by heating for 4 hrs at 65°C. After DNA purification, 30 cycles of PCR was performed and a PCR product of 220bp was resulted. Primers were as follows: Forward 5′-AAATCCACGATGGGATGAAC-3′ and reverse 5′– ATGGTGCAGTGATGACCAAA-3′.
2.6. Electrophoretic Mobility Shift Assay (EMSA)
Nuclear protein extracts from H4 cells was prepared using the NE-PER kit from Pierce (Milwaukee, MI). EMSA was performed by using a lightshift chemiluminescent electrophoretic mobility shift assay kit (Pierce). Biotin end-labeled oligonucleotides spanning the SNP (G/C) were designed and produced by Sigma. Binding reaction was performed by incubating the oligonucleotides with the nuclear extracts at room temperature for 20 minutes according to manufacturer’s instructions. After the incubation, the DNA-protein complexes were subjected to a 6% native polyacrylamide gel electrophoresis and transferred to a nylon membrane. After the transfer, the membrane was immediately cross-linked for 90 seconds using a commercial UV-light cross-linker instrument equipped with 254 nm bulbs. The bands were detected by streptavidin-HRP/chemiluminescence for biotin-labeled probes. Specific binding of AP4 to Pin1 promoter containing the SNP was evaluated by competition with excess unlabeled double-stranded AP4 consensus oligonucleotides (5′-TAGACAGCTGTCATACAGAGACAA-3′) and mutant consensus probes (5′-TAGACAGCTCTCATACAGAGACAA-3′)
2.7. AP4 overexpression and shRNA-mediated knockdown of AP4
For AP4 overexpression, subconfluent H4 neuronal cells cultured in 6-well dishes were transiently transfected with 360ng of AP4 expression plasmid using Lipofectamine and Plus reagent (Invitrogen) according to manufacturer’s instruction. Cells were harvested 48 hour after transfection and were lysed. For the generation of AP4 stable knockdown H4 cell line, replication-deficient lentivirus was produced in 293T packaging cells with pLKO.1 vector containing the specific shRNA sequence for human AP4 (RNAi Consortium), following the Consortium’s protocol. Viral supernatants were collected after 48 hours of transfection, filtered through a 0.45μm filter and used for infection immediately. H4 cells were transfected with viral supernatant in 10cm plate in the presence of 40μg of polybrene. To eliminate uninfected cells, puromycin (2.5μg/ml) was added to the cells for additional 5 days. Cell lysates were subjected to SDS-PAGE, followed by immunoblot with anti-Pin1, anti-AP4 or anti-tubulin antibodies. Density of each band was quantitated using ImageJ software (National Institutes of Health, Bethesda, MD). Result of the western blot was used to confirm the effect of AP4 overexpression or inhibition in the H4 cells.
2.8. Statistical Analysis
Deviations from Hardy-Weinberg equilibrium for genotypes were assessed by using Pearson chi square tests. Statistical analysis of genotype distribution was performed by chi-square test. Multiple regression analysis was performed to investigate the presence of significant association between AAO of AD and Pin1 SNP and adjusted for the effects of gender and ApoE ε4. Multiple regression analysis was also performed to assess the interaction effect of genotypes of the polymorphism and ApoE on AAO of AD, with status of ApoE and sex included as covariates in the analysis. Disease-free curves (Kaplan Meier and log-rank statistics) were used to compare the age at disease onset of AD patients carrying the GG or CG genotypes of rs2287839 (SPSS for Windows; 13.0). Significance level was set at p<0.05.
3. Results
3.1. Pin1 SNP rs2287839 is associated with 3-year delay in the average AAO of AD that is independent of ApoE4
Five tagSNPs (rs2287839, rs1077220, rs889162, rs2010457 and rs2287838) were selected by a R2 algorithm among SNPs with minor allele frequencies of at least 5% in Asians (Tang et al. 2006) to cover the Pin1 gene. Genotyping was performed to investigate the association of Pin1 SNPs and the risk or AAO of AD. Although the five tagSNPs selected for the study were not associated with the risk of AD (Table 1A), SNP rs2287839, but not the other SNPs, was significantly associated with AAO of AD (Table 1B). Demographic data of the patients showed the distribution of sex ratio and ApoE genotypes were similar among patients with either CG or GG genotypes of rs2287839 (Table 2A). Data were analyzed using multiple regression taking into account sex and ApoE genotypes and showed these two covariates were not associated with AAO in AD (Table 2B)(p=0.567 & p=0.584) while age was significantly associated with the genotypes (odds ratio = 0.913, 95% CI: 0.839–0.982; p=0.013). AD patients with the CG genotype had a 3-year delay in the age of AD onset compared to those with the GG genotype (Mantel-Cox p-value = 0.017) (Table 2A) (Figure 1). No statistically significant deviation from Hardy–Weinberg equilibrium was found. In addition, the ApoE ε4 allele was over-represented in AD group and ε4+ genotype was significantly associated with AD (p<0.05), consistent with other studies identifying ApoE ε4 as a susceptibility locus for late-onset AD (Farrer et al. 1997). Genotyping result of 300 normal elderly controls from the recruitment procedure of this study showed there was no significant difference in their age of recruitment regarding to their genotypes (p=0.15), suggesting this polymorphism was not associated with overall survival or life expectancy.
Table 1.
Association of Pin1 with disease risk and AAO of AD.
| (A) Genotypic data of 256 AD patients and 300 normal control elderly in relation to Pin1 SNPs. | |
|---|---|
| Chi-square p-value | |
| rs2287839 (C/G) | 0.62 |
| rs1077220 (A/G) | 0.06 |
| rs889162 (C/T) | 0.55 |
| rs2010457 (A/G) | 0.59 |
| rs2287838 (A/G) | 0.16 |
| (B) Association of SNP rs2287839, but not others, with AAO in AD patients | |
|---|---|
| p-value | |
| rs2287839 | 0.01 |
| rs1077220 | 0.50 |
| rs889162 | 0.80 |
| rs2010457 | 0.80 |
| rs2287838 | 0.67 |
Table 2.
| (A) Demographic and genotypic data of 256 AD patients in relation to Pin1 SNP rs2287839. | ||
|---|---|---|
| Genotype of rs2287839 | ||
| CG | GG | |
| Number | 24 (9.4%) | 232 (90.6%) |
| Mean AAO (mean±SD)* | 83.8±6.9 | 80.6±6.4 |
| % Female | 86.5% | 85.1% |
| ApoE ε4 carrier | 20.8% | 21.3% |
| (B) Multiple regression analysis showed that Pin1 SNP rs2287839 was significantly associated with AAO of AD independent of ApoE or sex. | |||||
|---|---|---|---|---|---|
| Variables | B | S.E. | β* | t | p-value |
| Constant | 3.469 | 0.236 | 14.702 | ||
| Age | −0.007 | 0.003 | −0.170 | −2.502 | 0.013 |
| ApoE ε4 | −0.008 | 0.014 | −0.036 | −0.548 | 0.584 |
| Sex | 0.031 | 0.054 | 0.039 | 0.574 | 0.567 |
Mantel-Cox p-value = 0.017
β: coefficient of regression;
Figure 1. Kaplan–Meier survival plots for rs2287839.

256 AD patients diagnosed as probable or possible AD according to NINCDS-ADRAD criteria were recruited and genotyped in this study. The plot showing that AD patients with Pin1 SNP rs2287839 (CG genotype) had a 3-year delay in the age of AD onset compared to those with the wild-type GG genotypes.
3.2. Pin1 SNP rs2287839 severely impairs binding of AP4 to the Pin1 promoter
Bioinformatic analysis using the transcription factor prediction program, TRANSFAC (Heinemeyer et al. 1998) predicted that the Pin1 SNP rs2287839 falls within the consensus binding motif for AP4 transcription factor, CAGCTG. Furthermore, the analysis predicts that the C variant binds weakly with AP4 when compared to the wild-type G. Therefore, the study focused on the functional consequence of this SNP.
Since the SNP was significantly associated with AAO in AD and it is located at the consensus sequence of AP4, ChIP assay was performed to confirm the binding of endogenous AP4 to the AP4-bindnig site (comprise the SNP) on the Pin1 promoter sequence (Figure 2A). We next asked whether the polymorphism abolished the binding site of AP4 in the Pin1 promoter. Using EMSA, double-stranded oligomers containing the SNP with either wild-type G or C variant and the AP4 binding site were used to compare the binding ability of AP4 to the AP4 binding site on the Pin1 promoter. Wild type G allele formed strong complexes with AP4 while the C variant showed 75% (p=0.017) reduction of binding to AP4. In addition, competition EMSA reactions were performed using an excess of unlabelled double stranded oligonucleotide containing wild-type or variant base at the SNP position. The result showed the strong binding between AP4 and Pin1 promoter containing G at the binding site could only be competed by wild-type consensus probe, but not the probe with C variant, in a dose-dependent manner (Figure 2B, C).
Figure 2. Pin1 SNP rs2287839 severely impairs binding of AP4 to the Pin1 promoter.

(A) Chromatin immunoprecipitation (ChIP) assay on H4 neuronal cells showing AP4 binds to the AP4-binding site on the Pin1 promoter. (B) Allele-specific binding of AP4. EMSA analysis of allele-specific effect of G (wild-type) and C (mutant) on the interaction of nuclear protein complexes extracted from H4 cells. Wild type G allele (Lane 2 and 3) formed strong complexes with AP4, while C variant (Lane 4 and 5) showed 75% reduction of binding to AP4. Competition experiments were performed with increasing concentrations (100–1000 fold) of unlabeled oligonucleotides (Lane 6–9, 11–14). (C) The images were quantified from three independent experiments using Image J analysis, with the wild-type binding being defined as 100% (Lane 1). Values are mean ± SEM from three independent experiments.
3.3. Pin1 SNP rs2287839 completely abolishes the ability of AP4 to repress the Pin1 promoter
To determine how the binding of AP4 to the Pin1 promoter affects the expression of Pin1, wild-type G or C variant promoter constructs were co-transfected with different doses of AP4 in H4 cells. The result revealed a dose-dependent repression of transactivation activity in wild-type but not the one with C variant. In addition, a significant increase in baseline Pin1 transcriptional activity was observed for the promoter with C variant (p=0.03) (Figure 3A). To show that the presence of the AP4 binding site at the SNP is important in regulating the activity of Pin1 promoter, either constructs containing the wild-type G allele of rs2287839 or the promoter spanning 2.3 kb upstream from the transcription site were cotransfected with different dose of AP4. A dose-dependent effect of AP4 repression was only observed in the 5.8 kb promoter containing the wild-type G allele but not the 2.3 kb promoter or the promoter with the C variant.
Figure 3. Pin1 SNP rs2287839 completely abolishes the ability of AP4 to repress the Pin1 promoter.

(A) Graph showing transcriptional activity of wild-type G or mutant C Pin1 promoter (5.8 kb) luciferase reporter cotransfected with increasing doses of AP4 DNA. AP4 suppresses wild-type Pin1 promoter activity in a dose-dependent manner, whereas the Pin1 mutant promoter showed significantly higher activity and its activity is not affected by AP4, even at high doses. (B) Graph showing transcriptional activity of long (5.8 kb) promoter containing the AP4 binding site or short (2.3 kb) promoter without the AP4 binding site. Note, the dose of AP4 differs between panels A and B. Results showed the AP4 binding site in the long promoter is important for the response to AP4. Values are mean ±SEM from three independent experiments. *p<0.05; ** p<0.001.
3.4. AP4 overexpression or knockdown suppresses or enhances Pin1 protein levels, respectively
To assess whether AP4 regulates Pin1 expression in H4 cells, we studied both the effect of overexpression and knockdown of AP4 in H4 cells. When AP4 expression was inhibited by shRNA stable transfection, there was a 2-fold increase in the expression of Pin1 protein. On the other hand, cells overexpressing AP4 showed only 80% reduction in Pin1 protein expression when compared to the cells transfected with empty vector (Figure 4). These results further reinforced the findings on the repressor property of AP4 on the expression of Pin1. Since the SNP, rs2287839, is the only one SNP overlaps with the binding motif of AP4 in the proximal promoter of Pin1, the data suggested that this polymorphism might have a key role in the binding of AP4 to Pin1 promoter and might be a key genetic determinant in the regulation of downstream pathway.
Figure 4. AP4 overexpression or knockdown suppresses or enhances Pin1 protein levels, respectively.
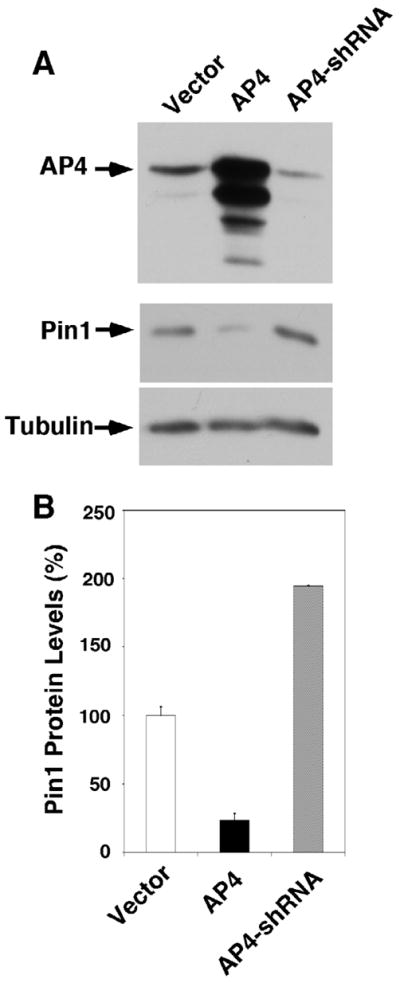
(A) Overexpression of AP4 in H4 neuronal cells significantly suppressed the expression on Pin1 whereas AP4 knockdown by shRNA increased the expression of Pin1. (B) Graph obtained from three independent experiments showing overexpression of AP4 resulted in an 80% reduction of Pin1 expression but AP4 knockdown increased the expression of Pin1 by two fold.
4. Discussion
Pin1 is tightly regulated on multiple levels and its deregulation has an important role in pathological conditions including AD (Butterfield et al. 2006; Lu 2004; Lu et al. 2007; Lu and Zhou 2007; Maudsley and Mattson 2006; Wulf et al. 2005). Aβ and neurofibrillary tangles are the hallmarks of AD and Pin1-null mice develop progressive age-dependent neuropathy including hyperpohosphorylation of tau, tau filaments formation and neurodegneration (Liou et al. 2003). In addition, it was shown that Pin1 knockout alone or in combination with APP mutant overexpression in mice increases amyloidogenic APP processing and causes an age-dependent increase in the amount of insoluble Aβ42 in the brain (Pastorino et al. 2006).
Our study identified a significant association between the promoter polymorphism (rs2287839) of Pin1 and the AAO of AD. AD patients with the CG genotype at rs2287839 showed 3-year delay in AAO when compared to patients with the GG genotype. The promoter SNP is located at the binding site of AP4 (CAGCTG). Previous studies showed AP4 is a repressor that negatively regulates the expression of target genes (Jung et al. 2008; Kim et al. 2006), but its connection with Pin1 is not known. Our result on AP4 RNAi and AP4 overexpression in H4 cells showed AP4 was able to repress the expression of Pin1 through the AP4 binding site in the promoter region of Pin1. However, the base change at the binding site from G to C of the SNP, rs2287839, almost completely abolished the AP4 binding to the Pin1 promoter and Pin1 expression was unresponsive to the repressive effect of AP4.
A number of studies have attempted to map the loci contributing to AAO in AD and a modest effect of AAO difference was observed on chromosome 19 (Dickson et al. 2008; Holmans et al. 2005; Li et al. 2002). However, due to the strong effect of ApoE, it might mask the effect of other genes on the same chromosome accounting for AAO in AD. Genetic association studies on AAO of AD identified several possible polymorphisms on different genes such as interleukin-1β (IL-1β) (Sciacca et al. 2003), ubiquilin 1 (UBQLN1) (Kamboh et al. 2006) and glutathione S-transferase omega-1 (GSTO1) (Li et al. 2003). However, the functional mechanisms or pathways were not investigated in those studies and further investigations are required to confirm those associations.
Predisposition to disease is only one mode of genetic expression; severity and AAO of disease may also be determined by genetic factors. Emerging data suggest Pin1 is involved in the pathogenesis of AD. Previous studies have shown that Pin1 binds to Tau in a phosphorylation-dependent manner specifically to its pThr231 residue (Lim and Lu 2005; Lu et al. 1999) and the level of pThr-231 was correlated with the progression of AD (Kohnken et al. 2000). In addition, it was demonstrated that Pin1 catalyzes the cis/trans isomerization of pThr668-Pro which is important in regulating APP processing and Aβ production (Pastorino et al. 2006). Several groups have investigated the risk of AD and the polymorphisms in the promoter region (−842 and −667) of Pin1, however the results were not conclusive (Lambert et al. 2006; Nowotny et al. 2007; Segat et al. 2007). It is possible that the undetected population structure in studies with heterogeneous population of subjects can lead to false positive results or failures in detecting the association with disease (Marchini et al. 2004) and this effect increased with samples size which might explain the conflicting results among the studies on Pin1 promoter polymorphisms.
It has been shown that Chinese and Japanese subpopulations are more homogeneous and have a lower degree of population admixture (Marchini et al. 2004). In our study, all AD patients were Chinese from Guangdong province of China and this might provide a homogeneous population for the better detection of association between genetic determinants and AD (Ma et al. 2005; Ma et al. 2009; Ma et al. 2008). Nowotny et al performed a comprehensive study on Pin1 polymorphisms and the risk of AD by using tagSNP strategy and failed to detect the association with the six SNPs selected (Nowotny et al. 2007). Interestingly, the SNP we identified to be associated with AAO of AD was also included in their study but it was not associated with the risk of AD in either their study or our study, although they did not examine the association with AAO of AD (Nowotny et al. 2007). In addition, this SNP is located downstream of the putative transcribed region of human Beacon/Ubl5 gene and it was reported to be associated with some metabolic phenotypes in combination with another SNP in Beacon gene (Jowett et al. 2004). We genotyped 300 normal control elderly for this SNP and it showed no association with overall survival, suggesting this SNP is not associated with life span, which would be expected if this SNP was associated with metabolic phenotypes. Our genotyping and functional data suggested the base change of the SNP abolished the binding site of AP4 and increased the expression level of Pin1, resulting in 3-year delay of onset of the disease. Since Pin1 is important in modulating APP processing and has an inverse relationship with tau accumulation, increased Pin1 expression resulting from the base substitution of the SNP might delay the pathological process of AD, thus delaying the AAO of AD. Our results suggested a possible regulation mechanism for Pin1 and a promising target for novel treatment to delay the onset of AD.
Acknowledgments
J. D. is a VA Career Development Awardee. The work was supported by NIH grant R01AG017870 and R01GM058556 to K.P.L.
Footnotes
Disclosure Statement
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83(5):623–632. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Genome-wide association studies in Alzheimer’s disease. Hum Mol Genet. 2009;18(R2):R137–145. doi: 10.1093/hmg/ddp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettens K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum Mol Genet. 2010 doi: 10.1093/hmg/ddq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88(9):1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med. 2008;40(8):562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- Butler AW, Ng MY, Hamshere ML, Forabosco P, Wroe R, Al-Chalabi A, Lewis CM, Powell JF. Meta-analysis of linkage studies for Alzheimer’s disease--a web resource. Neurobiol Aging. 2009;30(7):1037–1047. doi: 10.1016/j.neurobiolaging.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R. Pin1 in Alzheimer’s disease. J Neurochem. 2006;98(6):1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- Cathcart HM, Huang R, Lanham IS, Corder EH, Poduslo SE. Cystatin C as a risk factor for Alzheimer disease. Neurology. 2005;64(4):755–757. doi: 10.1212/01.WNL.0000151980.42337.44. [DOI] [PubMed] [Google Scholar]
- Chiu HFK, Lee HC, Chung WS, Kwong PK. Reliability and validity of the Cantonese version of the Min-Mental State Examination - A preliminary study. Journal of Hong Kong College of Psychiatrists. 1994;4(suppl 2):25–28. [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cui Y, Narayanan CS, Zhou J, Kumar A. Exon-I is involved in positive as well as negative regulation of human angiotensinogen gene expression. Gene. 1998;224(1–2):97–107. doi: 10.1016/s0378-1119(98)00512-5. [DOI] [PubMed] [Google Scholar]
- Daw EW, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD, Wijsman EM. The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet. 2000;66(1):196–204. doi: 10.1086/302710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MR, Li J, Wiener HW, Perry RT, Blacker D, Bassett SS, Go RC. A genomic scan for age at onset of Alzheimer’s disease in 437 families from the NIMH Genetic Initiative. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(6):784–792. doi: 10.1002/ajmg.b.30689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, Hebert LE, Hennekens CH, Taylor JO. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA. 1989;262(18):2551–2556. [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- Ferri CP, Sousa R, Albanese E, Ribeiro WS, Honyashiki M. Alzheimer’s Disease International World Alzheimer Report 2009. London: Alzheimer’s Disease International; 2009. [Google Scholar]
- Guerreiro RJ, Beck J, Gibbs JR, Santana I, Rossor MN, Schott JM, Nalls MA, Ribeiro H, Santiago B, Fox NC, Oliveira C, Collinge J, Mead S, Singleton A, Hardy J. Genetic variability in CLU and its association with Alzheimer’s disease. PLoS One. 2010;5(3):e9510. doi: 10.1371/journal.pone.0009510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han CH, Lu J, Wei Q, Bondy ML, Brewster AM, Yu TK, Buchholz TA, Arun BK, Wang LE. The functional promoter polymorphism (−842G>C) in the PIN1 gene is associated with decreased risk of breast cancer in non-Hispanic white women 55 years and younger. Breast Cancer Res Treat. 2009 doi: 10.1007/s10549-009-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26(1):362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmans P, Hamshere M, Hollingworth P, Rice F, Tunstall N, Jones S, Moore P, Wavrant DeVrieze F, Myers A, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, O’Donovan M, Jones L, Hardy J, Goate A, Lovestone S, Owen M, Williams J. Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005;135B(1):24–32. doi: 10.1002/ajmg.b.30114. [DOI] [PubMed] [Google Scholar]
- Hu YF, Luscher B, Admon A, Mermod N, Tjian R. Transcription factor AP-4 contains multiple dimerization domains that regulate dimer specificity. Genes Dev. 1990;4(10):1741–1752. doi: 10.1101/gad.4.10.1741. [DOI] [PubMed] [Google Scholar]
- Imai K, Okamoto T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J Biol Chem. 2006;281(18):12495–12505. doi: 10.1074/jbc.M511773200. [DOI] [PubMed] [Google Scholar]
- Jowett JB, Elliott KS, Curran JE, Hunt N, Walder KR, Collier GR, Zimmet PZ, Blangero J. Genetic variation in BEACON influences quantitative variation in metabolic syndrome-related phenotypes. Diabetes. 2004;53(9):2467–2472. doi: 10.2337/diabetes.53.9.2467. [DOI] [PubMed] [Google Scholar]
- Jung P, Menssen A, Mayr D, Hermeking H. AP4 encodes a c-MYC-inducible repressor of p21. Proc Natl Acad Sci U S A. 2008;105(39):15046–15051. doi: 10.1073/pnas.0801773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboh MI, Minster RL, Feingold E, DeKosky ST. Genetic association of ubiquilin with Alzheimer’s disease and related quantitative measures. Mol Psychiatry. 2006;11(3):273–279. doi: 10.1038/sj.mp.4001775. [DOI] [PubMed] [Google Scholar]
- Kim MY, Jeong BC, Lee JH, Kee HJ, Kook H, Kim NS, Kim YH, Kim JK, Ahn KY, Kim KK. A repressor complex, AP4 transcription factor and geminin, negatively regulates expression of target genes in nonneuronal cells. Proc Natl Acad Sci U S A. 2006;103(35):13074–13079. doi: 10.1073/pnas.0601915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohnken R, Buerger K, Zinkowski R, Miller C, Kerkman D, DeBernardis J, Shen J, Moller HJ, Davies P, Hampel H. Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer’s disease patients. Neurosci Lett. 2000;287(3):187–190. doi: 10.1016/s0304-3940(00)01178-2. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Bensemain F, Chapuis J, Cottel D, Amouyel P. Association study of the PIN1 gene with Alzheimer’s disease. Neurosci Lett. 2006;402(3):259–261. doi: 10.1016/j.neulet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ, Lu KP. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat Cell Biol. 2009;11(1):97–105. doi: 10.1038/ncb1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YJ, Oliveira SA, Xu P, Martin ER, Stenger JE, Scherzer CR, Hauser MA, Scott WK, Small GW, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Goetz CG, Mastaglia F, Middleton LT, Roses AD, Saunders AM, Schmechel DE, Gullans SR, Haines JL, Gilbert JR, Vance JM, Pericak-Vance MA, Hulette C, Welsh-Bohmer KA. Glutathione S-transferase omega-1 modifies age-at-onset of Alzheimer disease and Parkinson disease. Hum Mol Genet. 2003;12(24):3259–3267. doi: 10.1093/hmg/ddg357. [DOI] [PubMed] [Google Scholar]
- Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen FA, Jr, Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70(4):985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, Finn G, Pastorino L, Lee VM, Lu KP. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest. 2008;118(5):1877–1889. doi: 10.1172/JCI34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lu KP. Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta. 2005;1739(2–3):311–322. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424(6948):556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- Lu J, Hu Z, Wei S, Wang LE, Liu Z, El-Naggar AK, Sturgis EM, Wei Q. A novel functional variant (−842G>C) in the PIN1 promoter contributes to decreased risk of squamous cell carcinoma of the head and neck by diminishing the promoter activity. Carcinogenesis. 2009;30(10):1717–1721. doi: 10.1093/carcin/bgp171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem Sci. 2004;29(4):200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3(10):619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380(6574):544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8(11):904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399(6738):784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- Ma SL, Tang NL, Lam LC, Chiu HF. The association between promoter polymorphism of the interleukin-10 gene and Alzheimer’s disease. Neurobiol Aging. 2005;26(7):1005–1010. doi: 10.1016/j.neurobiolaging.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Ma SL, Tang NL, Tam CW, Lui VW, Lau ES, Zhang YP, Chiu HF, Lam LC. Polymorphisms of the estrogen receptor alpha (ESR1) gene and the risk of Alzheimer’s disease in a southern Chinese community. Int Psychogeriatr. 2009;21(5):977–986. doi: 10.1017/S1041610209990068. [DOI] [PubMed] [Google Scholar]
- Ma SL, Tang NL, Zhang YP, Ji LD, Tam CW, Lui VW, Chiu HF, Lam LC. Association of prostaglandin-endoperoxide synthase 2 (PTGS2) polymorphisms and Alzheimer’s disease in Chinese. Neurobiol Aging. 2008;29(6):856–860. doi: 10.1016/j.neurobiolaging.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36(5):512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Mattson MP. Protein twists and turns in Alzheimer disease. Nat Med. 2006;12(4):392–393. doi: 10.1038/nm0406-392. [DOI] [PubMed] [Google Scholar]
- Mermod N, Williams TJ, Tjian R. Enhancer binding factors AP-4 and AP-1 act in concert to activate SV40 late transcription in vitro. Nature. 1988;332(6164):557–561. doi: 10.1038/332557a0. [DOI] [PubMed] [Google Scholar]
- Nowotny P, Bertelsen S, Hinrichs AL, Kauwe JS, Mayo K, Jacquart S, Morris JC, Goate A. Association studies between common variants in prolyl isomerase Pin1 and the risk for late-onset Alzheimer’s disease. Neurosci Lett. 2007;419(1):15–17. doi: 10.1016/j.neulet.2007.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440(7083):528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89(6):875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22(15):5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacca FL, Ferri C, Licastro F, Veglia F, Biunno I, Gavazzi A, Calabrese E, Martinelli Boneschi F, Sorbi S, Mariani C, Franceschi M, Grimaldi LM. Interleukin-1B polymorphism is associated with age at onset of Alzheimer’s disease. Neurobiol Aging. 2003;24(7):927–931. doi: 10.1016/s0197-4580(03)00011-3. [DOI] [PubMed] [Google Scholar]
- Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici M, Arosio B, Crovella S. PIN1 promoter polymorphisms are associated with Alzheimer’s disease. Neurobiol Aging. 2007;28(1):69–74. doi: 10.1016/j.neurobiolaging.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006;27(7):918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Tang NL, Pharoah PD, Ma SL, Easton DF. Evaluation of an algorithm of tagging SNPs selection by linkage disequilibrium. Clin Biochem. 2006;39(3):240–243. doi: 10.1016/j.clinbiochem.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Waring SC, Rosenberg RN. Genome-wide association studies in Alzheimer disease. Arch Neurol. 2008;65(3):329–334. doi: 10.1001/archneur.65.3.329. [DOI] [PubMed] [Google Scholar]
- Wijsman EM, Daw EW, Yu CE, Payami H, Steinbart EJ, Nochlin D, Conlon EM, Bird TD, Schellenberg GD. Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am J Hum Genet. 2004;75(3):398–409. doi: 10.1086/423393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulf G, Finn G, Suizu F, Lu KP. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat Cell Biol. 2005;7(5):435–441. doi: 10.1038/ncb0505-435. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278(5345):1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- Yap CC, Murate M, Kishigami S, Muto Y, Kishida H, Hashikawa T, Yano R. Adaptor protein complex-4 (AP-4) is expressed in the central nervous system neurons and interacts with glutamate receptor delta2. Mol Cell Neurosci. 2003;24(2):283–295. doi: 10.1016/s1044-7431(03)00164-7. [DOI] [PubMed] [Google Scholar]
- Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6(4):873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- Zhou XZ, Lu PJ, Wulf G, Lu KP. Phosphorylation-dependent prolyl isomerization: a novel signaling regulatory mechanism. Cell Mol Life Sci. 1999;56(9–10):788–806. doi: 10.1007/s000180050026. [DOI] [PMC free article] [PubMed] [Google Scholar]