

Introduction.a
Over 30 nitrile-containing pharmaceuticals are prescribed for a diverse variety of medicinal indications with more than 20 additional nitrile-containing leads in clinical development. Trends identifying the roles of the nitrile in medical agents have emerged as the number of nitrile-containing pharmaceuticals has increased. Coupled with the increasing number of nitrile-containing agents have been structural advances that provide insight into the binding of small molecule inhibitors. X-ray crystallography in particular, is providing key insight into small molecule-protein interactions through an increasing number of structures with inhibitors bound in the active site. Augmenting the available interactions with current nitrile-containing pharmaceuticals are details from clinical candidates no longer under development. The present review surveys this range of medicinally active nitriles by focusing on the roles of the CN unit.
The prevalence of nitrile-containing pharmaceuticals, and the continued stream of potential agents in the clinic, attests to the biocompatibility of the nitrile functionality.1 The nitrile group is not particularly electrophilic toward free nucleophiles, even glutathione,2 unless activated by adjacent structural elements such as electron withdrawing groups.3 A caveat is the highly orchestrated activation-electrophilic additions such as those being exploited in several amino nitriles for diabetes and osteoporosis treatments that feature a reversible electrophilic attack (vide infra).
The nitrile group is quite robust and, in most cases, is not readily metabolized.4 Metabolically, the nitrile group in most nitrile-containing drugs is passed through the body unchanged.5 In cases of drug metabolism prior to elimination, the formation of glucuronides,6 conjugation with glutathione,7 N-dealkylation,8 N-acetylation,9 hydrolysis,6a-d and oxidation10 typically occurs at sites remote from the nitrile and without modification of the nitrile group.
Release of cyanide from aromatic or fully substituted carbons is not observed11 whereas alkylnitriles bearing an adjacent proton can be oxidized in the liver to cyanohydrins with subsequent cyanide release.12 Mandelonitrile, a cyanohydrin produced by ingesting almonds or some fruit pits, releases cyanide as the main degradation pathway and is responsible for the toxicity of cyanogenic glycosides.13 The potential oxidation and cyanide ejection likely explains why only four of the bioactive nitriles in the review contain an adjacent C-H bond. Epoxidation of alkenenitriles and ring opening can potentially liberate cyanide but the epoxidation is synthetically difficult14 and metabolism at other sites appears more likely given the success of several alkenenitrile-containing pharmaceuticals.
Vildagliptin (1) is a recently released aminonitrile-containing antidiabetic drug in which the nitrile bearing carbon is not fully substituted (Figure 1).15 Perhaps because of a concern for cyanide release, the metabolism has been closely examined in humans. The main metabolite comes from hydrolysis of the nitrile which likely stems from the covalent intermediate formed from this carboxyl transition structure analog. Nitrile hydrolysis is rather rare and, when observed, is a very minor metabolic pathway.16
Figure 1.
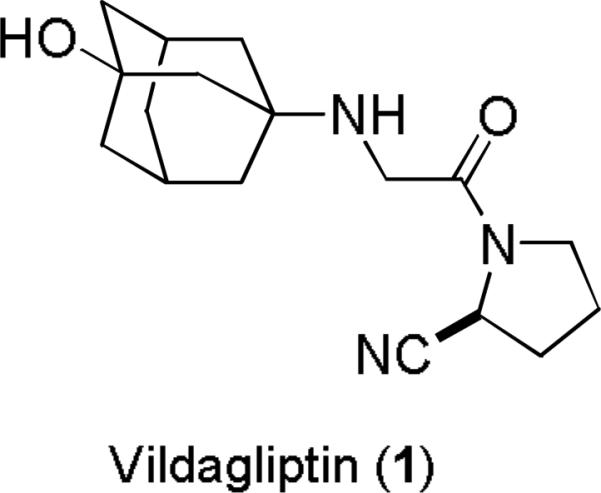
Aminonitrile Vildagliptin
Nitriles are unusual functionalities by virtue of the short, polarized triple bond.17 The linear, rod-like geometry has a cylindrical diameter of 3.6 Å for the π-system18 resulting in a minuscule steric demand along the axis. For comparison, the C≡N unit is essentially eight times smaller than a methyl group!19 Several crystal structures show the nitrile projecting into narrow clefts to make polar interactions or hydrogen bonds in sterically congested environments.20
Nitriles often play a key role as hydrogen bond acceptors.17,21 Several crystal structures show hydrogen bonding between the nitrile nitrogen and amino acids or to water which in turn is bound to the protein backbone. Many hydrogen bonds are between the nitrile and serine or arginine as expected for these hydrogen bond donors. In other clinical candidates, the strong dipole facilitates polar interactions in which the nitrile acts as a hydroxyl or carboxyl isostere.
The review is structured according to the nature of the nitrile-bearing substituent. As the number of nitrile-containing drug leads is vast, the review has focused on launched nitrile-containing pharmaceuticals and currently active clinical candidates. Most nitrile-containing pharmaceuticals are aromatics with aliphatic-, alkene-, and nitrogen-bearing nitriles being progressively less frequent. Within each class, the bioactive nitriles are collated according to common structural elements and mode of action. The hope is that this survey will allow greater deployment of this versatile functionality within drug leads.
Arylnitrile-Containing Pharmaceuticals
By far the largest class of nitrile-containing drugs are comprised of an aromatic core with a nitrile substituent. In many cases the nitrile functions as a ketone bioisostere with the nitrile engaging in non-specific, polar interactions. In other instances the nitrile is relatively remote from the recognition site and may polarize the aromatic π-system to optimize π–π interactions. para-Substituted aryl nitriles are common, possibly because the excellent inductive properties of the nitrile group more strongly polarize the aromatic ring making aromatics less susceptible to oxidative metabolism.
Several substituted benzonitriles have been developed as selective inhibitors of the aromatase enzyme for the treatment of estrogen-dependent diseases. Placement of the nitrile in the para position is essential for inhibition. There is general agreement that the nitrile mimics the carbonyl group of androst-4-ene-3,17-diones by functioning as a hydrogen bond acceptor (cf. 2 and 3, Figure 2).22 4 (fadrozole monohydrochloride), marketed by Novartis as Afema, was one of the first non-steroidal aromatase inhibitors23 for treatment of breast cancer.24 Structure activity relationships identified the efficacy of electron withdrawing groups at C-4 with bromine and nitrile groups being best.25 Subsequent development by Novartis identified 5 (letrozole) as a more potent oral aromatase inhibitor for the adjuvant treatment of hormonally-responsive breast cancer.26 Recently the crystallographic structure of enzyme-bound androstenedione was determined which may aid in designing future members of this class of inhibitors.27
Figure 2.

Nitrile-Containing Aromatase and Aldolase Inhibitors.
Structurally related to 4 and 5 is 6 (finrozole) which functions both as an aromatase28 and aldosterone inhibitor.29 Interest in 6 was stimulated by the implication of aldosterone's role in several pathogenic diseases, and in regulating sodium and potassium balance, extracellular fluid volume, and blood pressure. The nitrile group of 6 mimics the steroidal carbonyl by acting as a hydrogen bond acceptor. Separation of the most active finrazole enantiomer was achieved using monoclonal antibodies with a recent crystal structure showing the nitrile interacting with main chain phenylalanine and histidine residues.30
Among the numerous non-steroidal androgen receptor antagonists31 is 7 (bicalutamide, Figure 3). Launched by AstraZeneca for the treatment of advanced prostate cancer,32 7 has good oral bioavailability with minimal activity toward other steroid receptors. The crystal structure of the stronger-binding33 R-enantiomer shows the nitrile participating in a hydrogen bond to arginine and to a water molecule bound in the active site.34 The hydrogen bonding, and positioning of 7, show the nitrile mimicking the 3-keto functionality of dihydrotestosterone.
Figure 3.

Non-steroidal Receptor Antagonists in which Nitriles Function as Carbonyl Bioisosteres.
Several androgen receptor antagonists are in various stages of clinical trials for a variety of indications. Effort to use structurally related antagonists for the topical treatment of acne and hair loss led to the development of 8 (RU-58841)35 which was later superseded by 9 (PF-0998425).36 Like 7, the nitrile of 9 interacts with an arginine residue and has polar interactions with glutamine and leucine at the binding site.36
An excellent example of the equivalency of complex aryl nitriles and steroids is apparent in the comparative cocrystal structures of the human progesterone ligand binding domain with 10 (progesterone, Figure 4a) and 11 (tanaproget, Figure 4b). 11 is one of a potentially new class37 of non-steroidal contraceptives in clinical trials.38 The key interaction with Gln 725 and Arg 766 is a hydrogen bond to the enone carbonyl of 10 which is exceptionally well mimicked by a similar interaction with the nitrile group of 11.39 Hydrogen bonding to the nitrile explains the superior efficacy of this functionality over other electron withdrawing groups within this small binding pocket.
Figure 4.


Co-crystallizations in the Human Progesterone Ligand Binding Domain.
Inhibition of farnesyltransferase has become an important target for preventing oncogenesis by disrupting cell signaling. 12 (BMS-214662) is a farnesyltransferase inhibitor40 that entered early clinical trials41 for chronic myeloid leukemia (Figure 5).42 Crystallization of 12 complexed with mammalian farnesyltransferase shows aromatic π-interactions within a deep hydrophobic cleft that are critical for binding.43 No specific interactions of the nitrile were identified but for 12 the nitrile group improves pharmacokinetic properties. Solubility studies revealed that the nitrile substituent in 12 was nearly 10-fold more soluble than the corresponding bromo analog.44
Figure 5.

Nitrile-Containing Farnesyltransferase Inhibitors.
13 (L-778,123) is a dual inhibitor of farnesyltransferase and geranylgeranyltransferase which entered phase I trials for the treatment of pancreatic cancer, non-small cell lung cancer, and head and neck cancer.45 Two crystal structures of bound 13 show polar interactions of the nitrile nitrogen with glutamine and arginine in the two enzymes.46
14 (neratinib) is an irreversible epidermal growth factor receptor (EGFR) inhibitor currently in phase II trials for patients with breast cancer47 and non-small cell lung cancer (Figure 6).48 The related antineoplastic agent 15 (Pelitinib, EKB-569) entered phase I clinical trials for treating solid tumors49 and cancers resistant to treatment with gefitinib or erlotinib.50 Crystalization of 14 in a mutant kinase confirms the irreversible inhibition through Michael addition of cysteine to the enamide.51 The structure reveals a polar interaction between the nitrile and a key methionine residue postulated as being critical for the remarkable selectivity exhibited over vascular epidermal growth factor receptor-2 (VEGFR-2). 15 also acts as an irreversible Michael acceptor.52 16 (bosutinib, SKI-606) is a kinase inhibitor in phase III clinical trials for treating chronic myelogenous leukemia in patients resistant to other tyrosine kinase inhibitors.53 Docking studies identified a key hydrogen bond between threonine and the nitrile nitrogen of 16 which is a common motif in these kinase inhibitors.54
Figure 6.

Nitrile-Containing Kinase Inhibitors.
Early structure activity relationships for the neratinib family of kinase inhibitors was guided by recognition that the quinazoline-based inhibitors functioned through a water-bound hydrogen bond bridged to a proximal threonine residue (17, Figure 7). Modeling indicated that substitution of the azomethine-water aggregate for a sp2-CN unit, 18, would displace water and allow direct hydrogen bonding between the nitrile and the amino acid.55 This strategy was successfully applied to quinazoline analogs56 leading to identification of 1450 and 15.57 Crystallographically guided lead optimization led to a similar substitution in quinazoline and benztriazine inhibitors of scytalone dehydratase.58
Figure 7.

Nitrile-Substituted Cyanoquinoline as an Azomethine-Water Bioisostere.
19 (milrinone), marketed as Primacor, is a phosphodiesterase inhibitor used for treating heart failure,59 particularly when conventional treatment with vasodilators and diuretics is ineffective (Figure 8).60 19 shares some structural homology with thyroxine and stimulates the myocardial membrane in a similar manner to the hormone. 20 (Olprinone) is a structurally related phosphodiesterase inhibitor used for heart failure which both enhances myocardial contraction and acts as a vasodilator.61 X-ray crystallography of human transthyretin with 20 shows the cyanopyridone ring bound in the hormone pocket.62 The nitrile binds deeper in the position occupied by iodine while binding closer and tighter to the amino acids lining the channel. Replacing the nitrile with an amino group maintains activity but with fewer contacts to residues in the binding pocket. A bromine substituent fits better in the same pocket suggesting that the nitrile has a similar polarizing influence but with greater hydrophilicity.63 The interchange of nitrile and iodine or bromine as non-classical isosteres64 in imidazoles,65 has been observed during lead optimization of other inodilators.66
Figure 8.

Nitrile-Containing Cardiovascular Agents.
21 (cromakalim) is a potassium-dependent ATP channel opener used to treat hypertension.67 This “first generation” treatment lacks specificity which led to the search for selective agents with decoupled anti-ischemic and vasorelaxant activity.68 Of the numerous analogs pursued, 22 (BMS-191095) is particularly promising with over 4000-fold improved selectivity for the ischemic myocardium than 21.69 The receptor is assumed to have a π-interaction to the aromatic core and hydrogen bonding with the nitrile.70 Replacement of the aromatic nitrile by iodine affords an equipotent analog again indicating some similarity between nitrile and halogen groups.71 The hydrogen bonding, however, is not as critical as in previous examples because replacing the nitrile with iodine maintains the potency. Structure activity series within cromakalim-type potassium ion channel openers have shown that the cyanophenyl ring can be replaced with an N-6 pyridyl ring indicative of hydrogen bonding analogous to that observed in cyanoquinolines (cf. Figure 7).67a,71
Several phenyl-substituted nitriles have been developed for treatment of mood disorders (Figure 9). Although the receptors are usually known, in most instances the precise binding, in terms of interactions, is not well understood. ±23 (citalopram) is a selective serotonin reuptake inhibitor prescribed for depression which has recently been superseded by the more efficacious single enantiomer 23 (escitalopram).72 Molecular modeling suggests that the two enantiomers bind in the human serotonin transporter with opposite orientations of the aromatic groups and with an interaction between the nitrile and a phenylalanine residue.73 24 (RS-8359) is a selective and reversible monoamine oxygenase inhibitor under examination for treatment of depression.74 Clinical studies75 demonstrate different efficacies for the two enantiomers of 24 and an in vivo epimerization through oxidation and reduction of the benzylic alcohol.76
Figure 9.

Nitrile-Containing CNS Drugs.
25 (vilazodone) is a dual-acting serotonergic antidepressant that has completed Phase III clinical trials.77 During screening of a series of 25 analogs the most efficacious leads contained electron withdrawing groups with the nitrile or fluorine substituents being most potent. Computed molecular electrostatic potentials and dipole moments showed a strong homology suggesting that the nitrile can function as a fluorine bioisostere.78 Electrostatic mapping performed during the development of the muscarinic agonist, sabcomeline, generated similar electrostatic potential maps for the respective imidoyl nitrile, fluoride, and chloride again suggesting the nitrile as a halogen bioisostere.79
26 (pericyazine) is a phenothiazine antipsychotic thought to act on several receptors in the brain but for which no specific binding is available.80 27 (cyamemazine) is an antipsychotic widely used in France to minimize withdrawal symptoms after drug addiction.81 The mechanism of cyamemazine's anxiolytic action, and therefore the role of the functional groups, is unknown.
28 (zaleplon) is a non-benzodiazepine hypnotic drug used for treating insomnia.82 Binding assays show that 28 selectively binds to GABAA receptors containing the α1 subunit, though the exact interactions are currently unknown.83 29 (donitriptan) is a 5-HT1B agonist84 which entered phase II trials for the treatment of migraines.85
A series of nitrile-containing aromatics, some containing two nitriles, are ushering in a potentially revolutionary approach for treating AIDS.86 30 (etravirine) is the first of this new type of non-nucleoside reverse transcriptase inhibitor (NNRTI) of HIV to be launched (Figure 10).87 31 (dapivirine)88 and 32 (rilpivirine) are among the many etravirine analogs under development, with 32 being touted as among “the most potent anti-HIV agent(s) ever discovered.”89 The nitrile groups of 32 project deep into the binding pocket with the flexible pyrimidine allowing conformational mobility, and potency, even with several mutation-induced changes of the binding pocket.90 Crystallographic analysis of HIV-1 inhibitor complexes indicate that one nitrile binds to a water molecule that bridges to amino acids on the main chain.91
Figure 10.

Nitrile-Containing Anti-HIV Agents.
33 (lersivirine, UK-453,061) is a novel NNRTI currently in phase II clinical trials.92 Structural optimization identified the nitrile and the corresponding chloride as being equipotent with the nitrile being much less lipophilic and more metabolically stable.93 34 (MIV-150) was developed as an NNRTI but is not suitable as an anti-viral therapy because of poor systemic absorption after oral administration.94 Exploiting the poor absorption, 34 is now being evaluated as a vaginal microbicide.95 Early pharmacokinetics and oral bioavailability of 14C-labeled 35 (IDX899) in humans are promising with further clinical studies in progress.96
36 (febuxostat) is a non-purine xanthine oxidase inhibitor used to treat gouty arthritis (Figure 11).97 X-ray crystallography suggests hydrogen bonding between the nitrile, a bound water, and amino acids, which positions 36 in the channel leading to the molybdenum active site.98 In the more potent inhibitor 37 (FYX-051) with better binding affinity,99 the nitrile has a direct hydrogen bond with an arginine residue.100
Figure 11.

Nitrile-Containing Xanthine Oxidase Inhibitors.
39 (alogliptin) is a non-covalent inhibitor of dipeptidyl peptidase IV (DPP IV) in phase III clinical trials for the treatment of diabetes (Figure 12, bottom right).101 39 exhibits a 10,000 times greater preference for DPP IV over the related peptidase DPP VIII and has therapeutic advantages over existing DPP IV inhibitors.102 The inhibitor design was based on docking the cyanoaryl ring of a substituted quinazolinone (38) into a hydrophobic pocket augmented with hydrogen bonding between the nitrile and an arginine residue.103 X-ray crystallography of a cocrystal structure of 38 in the active site of DPP IV is consistent with the design motif, clearly showing a hydrogen bond from arginine to the nitrile (Figure 12, top). Optimizations to minimize hERG activity and maximize the metabolic half-life led to 39 which presumably has analogous enzyme interactions.
Figure 12.

Nitrile-Containing DPP IV Inhibitors.
Unfortunately no information is available for the role of the nitrile in medications 40-47 (Figure 13). 40 (lodoxamide) and 41 (lodoxamide ethyl) are histamine inhibitors used as ocular anti-inflammatory agents.104 Structurally related 42 (TYB-2285) entered phase II clinical trials in Japan for asthma and atopic dermatitis.105 43 (strontium ranelate) is prescribed to promote bone formation106 through a dual-action mechanism of preventing bone resorption while stimulating bone formation.107 44 (ravuconazole) is a recently released broad spectrum anti-fungal agent108 for which molecular modeling suggests the cyanophenyl ring participates in π-stacking in the target lanosterol 14-demethylase.109 45 (isavuconazole) is a structural isomer in advanced clinical trials as a broad-spectrum azole anti-fungal agent.110 46 (NO-1886, ibrolipim) is a lipoprotein lipase activator that entered phase II trials in Japan for the potential treatment of hyperlipidemia.111 47 (epanolol) is a beta blocker used for angina pectoris.112 Although the exact role of the nitrile is unknown in these agents, a reasonable speculation is that the nitrile was installed to balance the electronics of the aromatic rings and/or to reduce potential for oxidative metabolism.
Figure 13.

Miscellaneous Arylnitrile-Containing Inhibitors.
α-Aryl Acetonitriles
α-Aryl acetonitrile drugs contain the nitrile on a quaternary carbon adjacent to an aromatic ring. Positioning the nitrile on a fully substituted carbon prevents oxidation at the nitrile-bearing carbon and thereby prevents cyanide release.113 48 (anastrazole), 49 (verapamil), and 50 (gallopamil) are widely prescribed and are among the best-studied nitrile-containing pharmaceuticals (Figure 14). The block-buster drug 48, marketed by Astra-Zeneca under the trade name Arimidex, is considered the drug of choice for treating oestrogen-dependent breast cancer.114 Docking of 48 into the human aromatase homology model reveals a potential hydrogen bonding interaction of the two nitriles with an adjacent serine residue.115
Figure 14.

Most Widely Prescribed Nitrile-Containing Pharmaceuticals.
49 is a calcium channel antagonist used as an antiarrhythmic agent to treat angina.116 49 relaxes blood vessels so the heart does not have to pump as hard and simultaneously increases the supply of blood and oxygen to the heart which reduces chest pain. 50 is a methoxy derivative of 48 having a ten-fold increase in potency.117 Molecular modeling suggests that the polar nitrile group of these inhibitors, which is required for activity,118 engages in a strong dipole interaction with the enzyme-bound calcium through the nitrile nitrogen.119
49 blocks the drug efflux pump P-glycoprotein and is often employed as a standard to gauge reversal of multidrug resistance (MDR). Strategies to modify verapamil into more potent and selective MDR agents highlight the necessity of the nitrile group.120
51 (cilomilast, Ariflo) was developed as a phosphodiesterase inhibitor (DPP4) for use as an anti-inflammatory and anti-asthmatic agent.121 51122 completed phase III trials with less than ideal results, leaving the drug's fate in question. Numerous analogs, particularly with more rigid scaffolds,123 have been developed to optimize activity and minimize nausea, diarrhea and headache symptoms associated with this class of inhibitors.124 A co-crystal structure with 51 bound in the phosphodiesterase reveals an interaction of the nitrile with methionine and leucine residues.125 As with many of these polar interactions, slight movement of the protein may allow intimate hydrogen bonding. 52 (levocabastine) is a selective second-generation H1-receptor antagonist used for allergic conjunctivitis.126 The precise interaction of 52 with the receptor is not known.
53 (piritramide) is a synthetic opioid, almost equipotent with morphine, which is prescribed for treating postoperative pain.127 Structurally related 54 (diphenoxylate, lomotil) is used to treat diarrhea.128 54 is rapidly hydrolyzed to the corresponding acid, difenoxin, which slows intestinal contractions and peristalsis, allowing the body to remove water and return the intestine to normal function.
Alkenenitrile-Containing Pharmaceuticals
Drugs containing the unsaturated nitrile functionality are conjugated either with additional electron withdrawing groups or heteroatoms (Figure 16). Often the nitrile is positioned adjacent to a hydrogen bond donor or acceptor implying an electronic role for the nitrile group. Conjugation of the nitrile with an additional electron withdrawing group facilitates Michael additions129 as in 55 (entacapone).
Figure 16.

Alkenenitrile-Containing Pharmaceuticals
55130 is a potent inhibitor of catechol-O-methyltransferase and used for treating Parkinson's disease by facilitating passage of dopaminergic agents across the blood-brain barrier.131 Molecular docking reveals a series of hydrogen bonds with the nitrocatechol ring but no specific interactions for the nitrile.132 56 (trilostane) is an inhibitor of 3β-hydroxysteroid dehydrogenase that was used to treat Cushing's syndrome in humans but is now licensed only for treating dogs.133 56 has been successfully used in treating post-menopausal women for breast cancer134 by inhibiting 3β-hydroxysteroid dehydrogenase.135 Molecular modeling and mutant studies demonstrate the necessity of the nitrile group and identifies an interaction with a serine residue as being critical.136
57 (lanoconazole)137 and 58 (luliconazole)138 are topical antifungal drugs developed and marketed in Japan. Both 57 and 58 inhibit sterol 14α-methylase in fungi137 and although marketed as a racemate the activity resides in the S-enantiomer. A π-interaction is proposed between the enzyme and the dichlorobenzene ring139 with the dithioalkenenitrile acting as a Michael acceptor.140 59 (nilvadipine) is a calcium channel blocker used to treat hypertension and cerebral artery occlusion,141 and recently entered trials to treat Alzheimer's disease.142 The role of the nitrile is unknown.
Celgene Corp.'s antitumor agent 60 (CC-5079) prevents tubulin polymerization but remains active against multi-drug resistant cells.143 Subsequent assays identified the Z-diastereomer as being more active with molecular modeling suggesting a key hydrogen bond between the nitrile nitrogen and a tyrosine hydroxyl group in tubulin.144 61 (levosimendan, Simdax) is a novel inodilator used to manage acute or chronic heart failure.145 As with many Ca2+ sensitization and K+ channel-mediators, the enzyme-inhibitor interactions are not fully resolved. Development of a related dinitrile inhibitor, 62 (KW-5092), suggests that the dinitrile creates a rigid, polar moiety comparable to a nitro group.146
N-Cyanoguanidine Containing Drugs
Several drugs and drug leads contain the N-cyano guanidine functionality.147 Guanidinium cations interact strongly with carboxylates through lateral (63) or terminal (64) ionic interactions (Figure 17).148 In the series of N-cyano guanidine, amidine, and formamidine functionalities the nitrile significantly changes the nitrogen basicity,149 attenuating the ionization and H-bond acceptor properties.150 The cyanoguanidine moiety can act as a bioisostere for cyanoamidine,151 sulfonylguanidine,152 thiourea,153 amide, or thioamide154 functionalities.
Figure 17.

Cyanoguanidine Binding Motifs
65 (cimetidine) is a well-studied histamine H2-receptor antagonist that inhibits the production of acid in the stomach (Figure 18).155 Originally used to treat heartburn and peptic ulcers, 65 is being examined for several dermatological diseases156 and as an anti-tumor agent.157 66 (pinacidil) modulates ATP-dependent potassium channels and is used to treat hypertension.158 67 (terbogrel), a thromboxane A2 synthase inhibitor, has a similar cyanoguanidine substitution and is a potential long-term antithrombotic therapy.159
Figure 18.

N-Cyanoguanidine-Containing Drugs and Drug Leads
68 (CHS-828) is an antineoplastic agent160 under active development by Gemin X Pharmaceuticals as GMX-1778.161 The mechanism by which 68 triggers apoptosis remains to be clarified. 69 (KR-31378) is a potassium ATP channel opener in preclinical trials to protect retinal ganglion cells in glaucoma.162
α-Amino Nitrile Drugs and Drug Leads
Several amino nitriles have been developed as reversible inhibitors of dipeptidyl peptidase (DPP IV) for treating diabetes.163 Proline peptidases cleave peptide bonds after a proline residue which, for the hydrolysis of the glucagon-like peptide 1, switches off insulin production to provide a therapeutic approach for type 2 diabetes. 70 (saxagliptin)164 was recently launched under the name Onglyza, and the structurally related 1165 is licensed in Europe. The dinitrile 71 (NVP-DPP728)166 is in phase II clinical development.
These inhibitors function through attack of a serine residue on the nitrile resulting in a strong, reversible inhibition with a slow off rate (Figure 19, 72 ⇆ 73).163 A similar covalent binding occurs in nitrile-based cysteine protease inhibitors167 for which high selectivity between proteases is possible.168 Co-crystallization of 70 in DPP IV shows hydrogen bonding between the nitrile nitrogen and asparagines suggesting a delicate choreography of inhibition in which the amino nitrile binds, becomes activated, and is attacked to form the covalent bond.169
Figure 19.

Amino Nitrile DPP IV Inhibitors
Several cathepsins, which are cysteine proteases, have been identified as viable drug targets. The search for cathepsin K inhibitors for the treatment of osteoporosis, uncovered a series of aminoacetonitrile inhibitors in which the nitrile participates in a reversible, covalent interaction with the active site cysteine residue (Figure 20).170 Subsequent refinement identified 74 (odanacatib)171 which is currently in phase III clinical trials.172 Significant effort is focused on inhibiting cathepsin S using amino nitrile inhibitors.173 Amino nitrile 75 is a potent, reversible inhibitor (IC50 = 9 nM) whose co-crystallization with cathepsin S shows formation of a reversible thioimidate formed by attack of cysteine, rather than serine, on the nitrile “warhead” (cf. 73, Figure 19).174
Figure 20.

Cathepsin Inhibitors
Conclusion
Structurally diverse nitrile-containing drugs are in use for a variety of medical treatments. These range from blockbuster drugs such as 48 to numerous candidates currently being pursued in clinical trials. Surveying the interactions of the nitrile within these pharmaceuticals and drug candidates reveals that the biological function of the nitrile group varies considerably. In some instances the nitrile merely polarizes the adjacent electron density whereas in other cases the nitrile is a key component for molecular recognition.
Recent advances in molecular recognition, through crystallography, NMR, and modeling, is providing an increased understanding of the interactions between small molecule inhibitors and their targets. By surveying a range of pharmaceuticals and clinical candidates, several roles of the nitrile group have been identified.
Carbonyl bioisostere. In several non-steroidal inhibitors arylnitriles function as a carbonyl equivalent. These inhibitors have the advantage of minimizing interference with other steroid receptors and improving bioavailability. The homology is evident in the overlay of progesterone and the nitrile-inhibitor 76 in the progesterone receptor site (Figure 21).175 The nitrile nitrogen occupies virtually the same position as the carbonyl oxygen of progesterone and engages in the same polar interactions.
Hydroxyl and carboxyl surrogate. The small, polar nitrile, is a strong hydrogen bond acceptor with a significant solvation shell. The combination allows the nitrile to function as hydroxyl and carboxyl surrogates. Hydrogen bonding is particularly common to the protein backbone, amino acid side chains, or water molecules enclosed within the binding domain. The relationship is exemplified by functional group replacements performed using the anti-cancer drug 79 (etoposide) as a lead structure.176 Replacing the glycoside with a nitrile 77 was significantly more efficacious than the acid 78.
The powerful electron-withdrawing nature of the CN unit allows non-specific dipole interactions with amino acids and metal ions. In cyanoguanidines, and related structures, nitrile substitution allows tuning of the guanidine basicity and hydrogen bonding properties.
Cyanoquinolines and cyanopyridines (81) can act as more potent azomethine-water (80) bioisosteres. Interchanging a water-bound quinazoline with a 3-cyano quinoline or pyridine effectively exchanges the mobile hydrogen-bonded pyrimidine-water complex for a direct hydrogen bond between the nitrile and the protein. Expulsion of water from the binding domain adds an additional entropic component to the binding affinity.
Carboxyl transition state analog. Several amino nitriles function as proline peptidases by reversibly forming covalently bound imino esters at the active site. Conversion of the nitrile to the imino ester appears choreographed through activation of the nitrile and addition of serine or cysteine.
Halogen bioisostere. The nitrile mimics the polarization of the halides and is often an excellent halogen bioisostere.177 Being smaller than bromine or iodine, the nitrile is capable of achieving better contact with amino acids lining an active site.
Improving ADME178-toxicology profiles. Computational properties and empirical rules such as Lipinski's rules179 are routinely employed to guide structure-based drug design. While a potent molecule is essential for drug discovery, ultimately ADME-Tox properties decide which molecule is advanced into clinical trials. During optimization, leads tend to increase in size and lipophilicity180 which can be offset by introducing the sterically insignificant nitrile group. Replacing a hydrogen with a nitrile can roughly lower cLogP181 by half an order of magnitude and nearly an order of magnitude reduction for LogD.182 A more dramatic decrease in lipophilicity by over a full order of magnitude for cLogP/LogD often occurs when replacing a halogen or methyl group by a nitrile.
Figure 21.

Nitrile Inhibitor 76 and Progesterone (10) Overlay
The development of 33 from 82 (capravirine) provides an excellent case study in modulating ADME properties through nitrile substitution.93 Refining 82 led to the truncated analog 83 that was evaluated and found to be less potent and more lipophilic. Consistent with the nitrile being a halogen bioisostere, interchanging the chlorine with a nitrile (83 → 84) reduced the lipophilicity by an order of magnitude and increased the lipophilic-ligand efficiency (LLE).183 Introduction of a second nitrile led to 33 with similar activity to 82, 141 mass units smaller, a decreased lipophilicity, and a much improved half-life in human liver microsomes (HLM); 7.5 minutes for 82 compared to 73 minutes for 33.
This review provides a comprehensive survey of the interactions between the nitrile group and a diverse range of bioactive receptors. Collating the nitrile-containing drugs and clinical candidates by their structural similarities, reveals at least seven different modes by which nitrile substituents accentuate binding to receptor targets. A greater understanding of these specific functions is likely to facilitate lead optimizations with nitrile-containing candidates and will, optimistically, increase the number of nitrile-containing pharmaceuticals.
Figure 15.

Bioactive Quatarnary Arylacetonitriles.
Figure 22.

Comparative Nitrile and Acid Etoposide Analogs.
Figure 21.

Cyanoquinolines and Cyanopyridines as Azomethine-Water Bioisosteres.
Figure 22.

ADME-Directed Evolution of Capravirine (82) into Lersivirine (33).
Acknowledgment
Financial support for nitrile-based research from the National Science Foundation (0808996 CHE) and the National Institutes of Health (2R15AI051352-03) is gratefully acknowledged. The Center for Advanced Studies, Ludwig Maximilians University (Munich) and the University of Göteborg are thanked for facilitating the writing of the review by hosting a sabbatical visit (FFF). Critical suggestions from Morten Grøtli and Robert Dancer are gratefully acknowledged.
Biographies
Fraser Fleming earned his B. Sc. (Hons.) at Massey University, New Zealand, in 1986 and then pursued a Ph. D. under the direction of Edward Piers at the University of British Columbia, Canada. After graduating in 1990 he moved to Oregon State University for postdoctoral research with James D. White and in 1992 joined the faculty at Duquesne University where he is a full professor. His research interests lie broadly in stereochemistry and the reactions of organometallics, particularly for the development of new reactions with nitriles.
Lihua Yao was born in Shanghai, China (1978) and obtained her B. Eng. in Chemical Engineering (2000) at Shanghai University of Engineeering Science. From 2000 until 2003, she was a research assistant in Shanghai Institute of Organic Chemistry, Chinese Academy Sciences, working on the development of organofluorine methodology. In 2007, she joined Duquesne University where she is currently working with Fraser Fleming on a transmissive olefination of alkenenitriles.
P. C. Ravikumar was born (1980) in Satyavedu, Andra Pradesh, India. He obtained his B.Sc. (2000) and M.Sc in Organic Chemistry (2002) from the University of Madras, India. He was awarded a research fellowship from the council for scientific and industrial research (CSIR), India for his doctoral research which he completed in 2006 at the Indian Institute of Science, Bangalore, under the guidance of Professor A. Srikrishna. He completed postdoctoral research with Fraser Fleming at Duquesne University (2007-2008) and with Professor Seth B. Herzon at Yale University (2009). He recently accepted an Assistant Professor position at the Indian Institute of Technology, Mandi, India, where he will be begin his independent academic career.
Lee Funk received his B.A. in Chemistry from Washington and Jefferson College, followed by a M.S. in Organic Chemistry from Duquesne University. From 2001 through 2008 he worked as a medicinal chemist for Pharmacia (Kalamazoo, MI), Pfizer (La Jolla, CA), and GlaxoSmithKline (Collegeville, PA). As a medicinal chemist, he worked in various therapeutic targets including HCV, oncology, and osteoporosis. In 2008, he transitioned to Mylan's legal Intellectual Property Department. He is currently working as a Senior Scientist performing prior art searches, mapping out patent landscapes, developing invalidity positions, supporting ongoing litigation, and working with R&D scientists to develop non-infringing API/formulations.
Brian C. Shook received a B.S. from the University of Pittsburgh, Johnstown (1996) and a Ph.D. in synthetic organic chemistry from Duquesne University in 2001 under the guidance of Fraser Fleming. His thesis work included organometallic additions to unsaturated nitriles and the stereoselective cyclizations of nitrile anions to cis- and trans-decalins. He then pursued a postdoctoral appointment with Robert K. Boeckman, Jr. at the University of Rochester where he completed the asymmetric total synthesis of bengamide B. In 2002, he joined the medicinal chemistry department at Johnson & Johnson where he has worked on programs for Parkinson's disease, pain, migraine, and diabetes.
Footnotes
Abbreviations: EGFR, epidermal growth factor receptor; vascular epidermal growth factor receptor-2, VEGFR-2; GABA, γ-aminobutyric acid; 5-HT1B, 5-hydroxytryptamine receptor 1B; NNRTI, non-nucleoside reverse transcriptase inhibitor; DPP IV, dipeptidyl peptidase IV; MDR, multidrug resistance; ADME, absorption, distribution, metabolism, and excretion.
References
- 1.a Murphy ST, Case HL, Ellsworth E, Hagen S, Huband M, Joannides T, Limberakis C, Marotti KR, Ottolini AM, Rauckhorst M, Starr J, Stier M, Taylor C, Zhu T, Blaser A, Denny WA, Lu GL, Smaill JB, Rivault F. The synthesis and biological evaluation of novel series of nitrile-containing fluoroquinolones as antibacterial agents. Bioorg. Med. Chem. Lett. 2007;17:2150–2155. doi: 10.1016/j.bmcl.2007.01.090. [DOI] [PubMed] [Google Scholar]; b Markus B, Kwon C-H. In vitro Metabolism of Aromatic Nitriles. J. Pharm. Sci-US. 1994;83:1729–1734. doi: 10.1002/jps.2600831216. [DOI] [PubMed] [Google Scholar]
- 2.MacFaul PA, Morley AD, Crawford JJ. A simple in vitro assay for assessing the reactivity of nitrile containing compounds. Bioorg. Med. Chem. Lett. 2009;19:1136–1138. doi: 10.1016/j.bmcl.2008.12.105. [DOI] [PubMed] [Google Scholar]
- 3.Oballa RM, Truchon J-F, Bayly CI, Chauret N, Day S, Crane S, Berthelette C. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg. Med. Chem. Lett. 2007;17:998–1002. doi: 10.1016/j.bmcl.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 4.For an example encountered during the development of Cathepsin Ka and Sb analogs see: Boyd MJ, Crane SN, Robichaud J, Scheigetz J, Black WC, Chauret N, Wang Q, Masse F, Oballa RM. Investigation of ketone warheads as alternatives to the nitrile for preparation of potent and selective cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 2009;19:675–679. doi: 10.1016/j.bmcl.2008.12.053. Patterson AW, Wood WJL, Hornsby M, Lesley S, Spraggon G, Ellman JA. Identification of Selective, Nonpeptidic Nitrile Inhibitors of Cathepsin S Using the Substrate Activity Screening Method. J. Med. Chem. 2006;49:6298–6307. doi: 10.1021/jm060701s.
- 5.For olprinone see: Kaneko K, Tadano K, Nakajima K, Sano Y, Yuzuriha T, Motoji N, Kawai N, Hasegawa S, Shigematsu A. Metabolic fate of loprinone hydrochloride. 1: Blood level, distribution, metabolism, and excretion after a single intravenous administration to rats. Yakubutsu Dotai. 1994;9:149–162. For lodoxamide see: Leong BKJ, Coombs JK, Petzold EN, Hanchar AJ. A dosimetric endotracheal nebulization technique for pulmonary metabolic disposition studies in laboratory animals. Inhalation Toxicology. 1988:37–51. For cimetidine see: Strong HA, Spino MJ. Chromatogr. 1987;422:301–308. doi: 10.1016/0378-4347(87)80467-x. Ziemniak JA, Wynn RJ, Aranda JV, Zarowitz BJ, Schentag JJ. The pharmacokinetics and metabolism of cimetidine in neonates. Dev. Pharmacol. Therap. 1984;7:30–38. doi: 10.1159/000457141.
- 6.For entacapone see: Wikberg T, Vuorela A. Metabolite profiles of two [14C]-labeled catechol O-methyltransferase inhibitors, nitecapone and entacapone, in rat and mouse urine and rat bile. Eur. J. Drug Metab. Ph. 1994;19:125–135. doi: 10.1007/BF03188833. Wikberg T, Vuorela A, Ottoila P, Taskinen J. Identification of major metabolites of the catechol-O-methyltransferase inhibitor entacapone in rats and humans. Drug Metab. Dispos. 1993;21:81–92. For cromakalim see: Kudoh S, Nakamura H. Direct determination of the antihypertensive agent Cromakalim and its major metabolites in human urine by high-performance liquid chromatography. J. Chromatogr. 1990;515:597–602. doi: 10.1016/s0021-9673(01)89359-6. Kudoh S, Okada H, Nakahira K, Nakamura H. Simultaneous determination of antihypertensive agent Cromakalim and its major metabolites in human urine by high-performance liquid chromatography. Anal. Sci. 1990;6:53–56. doi: 10.1016/s0021-9673(01)89359-6. For lersivirine see: Vourvahi M, Gleave M, Nedderman ANR, Hyland R, Gardner I, Howard M, Kempshall S, Collins C, LaBadie R. Excretion and metabolism of lersivirine (5-{[3,5-diethyl-1-(2-hydroxyethyl)(3,5-14C2)-1H-pyrazol-4-yl]oxy}benzene-1,3-dicarbonitrile), a next-generation non-nucleoside reverse transcriptase inhibitor, after administration of [14C]Lersivirine to healthy volunteers. Drug Metab. Dispos. 2010;38:789–800. doi: 10.1124/dmd.109.031252.
- 7.For levosimendan see: Koskinen M, Puttonen J, Pykalainen M, Vuorela A, Lotta T. Metabolism of OR-1896, a metabolite of levosimendan, in rats and humans. Xenobiotica. 2008;38:156–170. doi: 10.1080/00498250701744658. Sundberg S, Antila S, Scheinin H, Hayha M, Virtanen M, Lehtonen L. Integrated pharmacokinetics and pharmacodynamics of the novel calcium sensitizer levosimendan as assessed by systolic time intervals. Int. J. Clin. Pharm. Th. 1998;36:629–635.
- 8.For N-dealkylation of verapamil see: Borlak J, Walles M, Elend M, Thum T, Preiss A, Levsen K. Verapamil: Identification of novel metabolites in cultures of primary human hepatocytes and human urine by LC-MSn and LC-NMR. Xenobiotica. 2003;33:655–676. doi: 10.1080/0049825031000093600. McIlhenny HM. Metabolism of [14C]-verapamil. J. Med. Chem. 1971;14:1178–1184. doi: 10.1021/jm00294a010. For N-dealkylation of zaleplon see: Horstkötter C, Schepmann D, Blaschke G. Separation and identification of zaleplon metabolites in human urine using capillary electrophoresis with laser-induced fluorescence detection and liquid chromatography-mass spectrometry. J. Chromatogr. A. 2003;1014:71–81. doi: 10.1016/s0021-9673(03)00564-8. Renwick AB, Mistry H, Ball SE, Walters DG, Kao J, Lake BG. Metabolism of zaleplon by human hepatic microsomal cytochrome P450 isoforms. Xenobiotica. 1998;28:337–348. doi: 10.1080/004982598239452. For N-dealkylation of citalopram see: Sindrup SH, Broesen K, Hansen MGJ, Aaes-Joergensen T, Overoe KF, Gram LF. Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Ther. Drug Monit. 1993;15:11–17. doi: 10.1097/00007691-199302000-00002. For N-dealkylation of gallopamil see: Weymann J, Buehler V, Hege HC, Mueller-Peltzer H, Schenk G, Stieren B, Hollmann M. Metabolism of the calcium antagonist gallopamil in man. Arzneimittel-Forschung. 1989;39:605–607.
- 9.For N-acetylation of alogliptin see: Christopher R, Covington P, Davenport M, Fleck P, Mekki QA, Wann ER, Karim A. Pharmacokinetics, pharmacodynamics, and tolerability of single increasing doses of the dipeptidyl peptidase-4 inhibitor alogliptin in healthy male subjects. Clin. Ther. 2008;30:513–527. doi: 10.1016/j.clinthera.2008.03.005.
- 10.For oxidation of trilostane: Suzuki M, Mori Y, Tsuboi M, Tatematsu A. Study on metabolites of trilostane, an inhibitor of adrenal steroidogenesis. J. Pharmacobio-Dynamics. 1980;3:S10, S-22. For oxidation and methylation of cimetidine: Waring RH, Mitchell SC, Blundell R, Smith RL. Novel aspects of the metabolism of cimetidine in the guinea pig. Biochem. Soc. Trans. 1986;14:98–99. Taylor DC, Cresswell PR. The metabolism of cimetidine in the rat, dog and man. Biochem. Soc. Trans. 1975;3:884–885. For oxidation of pinicidil: DeLong AF, Oldham SW, DeSante KA, Nell G, Henry DP. Disposition of [14C]pinacidil in humans. J. Pharm. Sci. 1988;77:153–156. doi: 10.1002/jps.2600770212. McBurney A, Farrow PR, Ainsworth S, Ward JW. Serum concentrations and urinary excretion of pinacidil and its major metabolite, pinacidil pyridine-N-oxide following i.v. and oral administration in healthy volunteers. Br. J. Clin. Pharmacol. 1985;19:91–94. doi: 10.1111/j.1365-2125.1985.tb02618.x. For oxidation of letrozole see: Rodriguez-Flores J, Salcedo AMC, Fernandez LM. Rapid quantitative analysis of letrozole, fluoxetine and their metabolites in biological and environmental samples by MEKC. Electrophoresis. 2009;30:624–632. doi: 10.1002/elps.200800495. Rodriguez F,J, Salcedo AMC, Villasenor Llerena MJ, Munoz Fernandez L. Micellar electrokinetic chromatographic method for the determination of letrozole, citalopram and their metabolites in human urine. J. Chromatogr. A. 2008;1185:281–290. doi: 10.1016/j.chroma.2008.01.067. (h) For oxidation of nilvadipine see: Terashita S, Tokuma Y, Fujiwara T, Shiokawa Y, Okumura K, Noguchi H. Metabolism of nilvadipine, a new dihydropyridine calcium antagonist, in rats and dogs. Xenobiotica. 1987;17:1415–1425. doi: 10.3109/00498258709044002.
- 11.a Wit JG, van Genderen H. Metabolism of the herbicide 2,6-dichlorobenzonitrile in rabbits and rats. Biochem. J. 1966;101:698–706. doi: 10.1042/bj1010698. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hutson DH, Hoadley EC, Griffiths MH, Donninger C. Mercapturic acid formation in the metabolism of 2-chloro-4-ethylamino-6-(1-methyl-1-cyanoethylamino)-s- triazine in the rat. J. Agr. Food Chem. 1970;18:507–512. doi: 10.1021/jf60169a035. [DOI] [PubMed] [Google Scholar]
- 12.Tanii H, Hashimoto K. Influence of ethanol on the in vivo and in vitro metabolism of nitriles in mice. Arch. Toxicol. 1986;58:171–176. doi: 10.1007/BF00340977. [DOI] [PubMed] [Google Scholar]
- 13.Singh PDA, Jackson JR, James SP. Metabolism of mandelonitrile in the rat. Biochem. Pharmacol. 1985;34:2207–2209. doi: 10.1016/0006-2952(85)90420-4. [DOI] [PubMed] [Google Scholar]
- 14.a Pryde David C., Henry Steven S., Meyers AI. Synthesis of 2-tetralones via a novel 1,2-carbonyl transposition of 1-tetralones. Tetrahedron Lett. 1996;37:3243–3246. [Google Scholar]; b Aiai M, Robert A, Baudy-Floc'h M, Le Grel P. First Sharpless epoxidation of electrophilic 2-cyano allylic alcohols. Tetrahedron Asym. 1995;6:2249–2252. [Google Scholar]; c Miyashita M, Suzuki T, Yoshikoshi A. Fluoride-promoted epoxidation of α,β-unsaturated compounds. Chem. Lett. 1987:285–288. [Google Scholar]
- 15.He H, Tran P, Yin H, Smith H, Batard Y, Wang L, Einolf H, Gu H, Mangold JB, Fischer V, Howard D. Absorption, metabolism, and excretion of [14C]vildagliptin, a novel dipeptidyl peptidase 4 inhibitor, in humans. Drug Metab. Dispos. 2009;37:536–544. doi: 10.1124/dmd.108.023010. [DOI] [PubMed] [Google Scholar]
- 16.Sakamoto K, Nakamura Y. Urinary metabolites of pinacidil. II. Species difference in the metabolism of pinacidil. Xenobiotica. 1993;23:649–656. doi: 10.3109/00498259309059402. Sakamoto K, Nakamura Y. Urinary metabolites of pinacidil: I. Isolation and identification of the metabolites in rat urine. Xenobiotica. 1993;23:391–400. doi: 10.3109/00498259309057027. For an example where nitrile hydrolysis may be present or an artifact of the analysis technique see reference 10h. An extensive metabolic profiling of lersivirine identified less than two percent of metabolites arising from nitrile hydrolysis.6e
- 17.Le Questel J-Y, Berthelot M, Laurence C. Hydrogen-bond acceptor properties of nitriles: a combined crystallographic and ab initio theoretical investigation. J. Phys. Org. Chem. 2000;13:347–358. The mean bond length of 1.14 Å was obtained by averaging 5059 nitriles located in the Cambridge Structural Database.
- 18.Sheppard WA. In: The Chemistry of the Cyano Group. Rappoport, editor. 1970. Ch. 5. [Google Scholar]
- 19.Based on a comparison of A values for nitrile and methyl groups: Eliel Ernest L., Wilen Samuel H., Mander Lewis N. Stereochemistry of Organic Compounds. Wiley, NY: 1994. pp. 696–697.
- 20.See for example the cocrystal structures of cathepsin K with bound inhibitor: Teno N, Miyake T, Ehara T, Irie O, Sakaki J, Ohmori O, Gunji H, Matsuura N, Masuya K, Hitomi Y, Nonomuraa K, Horiuchia M, Gohdaa K, Iwasakia A, Umemuraa I, Tadaa S, Kometania M, Iwasakia G, Cowan-Jacobb SW, Missbachb M, Lattmannb R, Betscharta C. Novel scaffold for cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 2007;17:6096–6100. doi: 10.1016/j.bmcl.2007.09.047.
- 21.The hydrogen bond acceptor properties of nitriles are variously described as “medium to weak”a and excellent.b Laurence C, Brameld KA, Graton J, Le Questel J-Y, Renault E. The pKBHX Database: Toward a Better Understanding of Hydrogen-Bond Basicity for Medicinal Chemists. J. Med. Chem. 2009;52:4073–4086. doi: 10.1021/jm801331y. Allerhand A, Schleyer P. von Rague Nitriles and isonitriles as proton acceptors in hydrogen bonding: correlation of ΔνOH with acceptor structure. J. Am. Chem. Soc. 1963;85:866–870.
- 22.a Recanatini Maurizio, Bisi Alessandra, Cavalli Andrea, Belluti Federica, Gobbi Silvia, Rampa Angela, Valenti Piero, Palzer Martina, Palusczak Anja, Hartmann Rolf W. A New class of nonsteroidal aromatase inhibitors: design and synthesis of chromone and xanthone derivatives and inhibition of the P450 enzymes aromatase and 17α-hydroxylase/C17,20-lyase. J. Med. Chem. 2001;44:672–680. doi: 10.1021/jm000955s. [DOI] [PubMed] [Google Scholar]; b Furet P, Batzl C, Bhatnagar A, Francotte E, Rihs G, Lang M. Aromatase inhibitors: synthesis, biological activity, and binding mode of azole-type compounds. J. Med. Chem. 1993;36:1393–1400. doi: 10.1021/jm00062a012. [DOI] [PubMed] [Google Scholar]
- 23.Modeling indicates that the nitrile mimics the carbonyl by interacting with amino acids at the active site, possibly through hydrogen bonds.22b
- 24.Kelloff GJ, Lubet RA, Lieberman R, Eisenhauer K, Steele VE, Crowell JA, Hawk ET, Boone CW, Sigman CC. Aromatase inhibitors as potential cancer chemopreventives. Cancer Epidem. Biomar. 1998;7:65–78. [PubMed] [Google Scholar]
- 25.Browne LJ, Gude C, Rodriguez H, Steele RE, Bhatnager A. Fadrozole hydrochloride: a potent, selective, nonsteroidal inhibitor of aromatase for the treatment of estrogen-dependent disease. J. Med. Chem. 1991;34:725–736. doi: 10.1021/jm00106a038. [DOI] [PubMed] [Google Scholar]
- 26.Bhatnagar AS. The discovery and mechanism of action of letrozole. Breast Cancer Res. Tr. 2007;105:7–17. doi: 10.1007/s10549-007-9696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong Y, Yu B, Sherman M, Yuan Y-C, Zhou D, Chen S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol. Endocrinol. 2007;21:401–414. doi: 10.1210/me.2006-0281. [DOI] [PubMed] [Google Scholar]
- 28.Ahokoshi O, Irjala K, Taalikka M, Manninen P, Halonen K, Kangas L, Salminen E, Huupponen R, Scheinin H. Pharmacokinetics of finrozole (MPV-2213ad), a novel selective aromatase inhibitor, in healthy men. Brit. J. Clin. Pharmacol. 2001;52:702–704. doi: 10.1046/j.1365-2125.2001.01488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roumen L, Peeters JW, Emmen JMA, Beugels IPE, Custers EMG, de Gooyer M, Plate R, Pieterse K, Hilbers PAJ, Smits JFM, Vekemans JAJ, Leysen D, Ottenheijm HCJ, Janssen HM, Hermans JJR. Synthesis, Biological Evaluation, and Molecular Modeling of 1-Benzyl-1H-imidazoles as Selective Inhibitors of Aldosterone Synthase (CYP11B2). J. Med. Chem. 2010;53:1712–1725. doi: 10.1021/jm901356d. [DOI] [PubMed] [Google Scholar]
- 30.Parkkinen T, Nevanen TK, Koivula A, Rouvinen J. Crystal Structures of an Enantioselective Fab-fragment in Free and Complex Forms. J. Mol. Biol. 2006;357:471–480. doi: 10.1016/j.jmb.2005.12.045. [DOI] [PubMed] [Google Scholar]
- 31.a Gao W, Bohl CE, Dalton JT. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005;105:3352–3370. doi: 10.1021/cr020456u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Singh SM, Gauthier S, Labrie F. Androgen receptor antagonists (antiandrogens): structure-activity relationships. Curr. Med. Chem. 2000;7:211–247. doi: 10.2174/0929867003375371. [DOI] [PubMed] [Google Scholar]
- 32.a Wellington K, Keam SJ. Spotlight on bicalutamide 150mg in the treatment of locally advanced prostate cancer. Drug Aging. 2007;24:169–171. doi: 10.2165/00002512-200724020-00008. [DOI] [PubMed] [Google Scholar]; b Fradet Y. Bicalutamide (Casodex) in the treatment of prostate cancer. Exp. Rev. Anticancer Ther. 2004;4:37–48. doi: 10.1586/14737140.4.1.37. [DOI] [PubMed] [Google Scholar]
- 33.Kirkovsky L, Mukherjee A, Yin D, Dalton JT, Miller DD. Chiral Nonsteroidal Affinity Ligands for the Androgen Receptor. 1. Bicalutamide Analogues Bearing Electrophilic Groups in the B Aromatic Ring. J. Med. Chem. 2000;43:581–590. doi: 10.1021/jm990027x. [DOI] [PubMed] [Google Scholar]
- 34.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. P. Natl. Acad. Sci. USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Dort ME, Jung Y-W. Synthesis and structure-activity studies of side-chain derivatized arylhydantoins for investigation as androgen receptor radioligands. Bioorg. Med. Chem. Lett. 2001;11:1045–1047. doi: 10.1016/s0960-894x(01)00146-9. [DOI] [PubMed] [Google Scholar]
- 36.Li JJ, Iula DM, Nguyen MN, Hu L-Y, Dettling D, Johnson TR, Du DY, Shanmugasundaram V, Van Camp JA, Wang Z, William GH, Wen-Song Y, Boys ML, Wade KJ, Drummond EM, Samas BM, Lefker BA, Hoge GS, Lovdahl MJ, Asbill J, Carroll M, Meade MA, Ciotti SM, Krieger-Burke T. Rational Design and Synthesis of 4-((1R,2R)-2-Hydroxycyclohexyl)-2(trifluoromethyl)benzonitrile (PF-998425), a Novel, Nonsteroidal Androgen Receptor Antagonist Devoid of Phototoxicity for Dermatological Indications. J. Med. Chem. 2008;51:7010–7014. doi: 10.1021/jm8009316. [DOI] [PubMed] [Google Scholar]
- 37.Kern JC, Terefenko E, Trybulski E, Berrodin TJ, Cohen J, Winneker RC, Yudt MR, Zhang Z, Zhu Y, Zhang P. 5-Aryl indanones and derivatives as nonsteroidal progesterone receptor modulators. Bioorg. Med. Chem. Lett. 2009;19:6666–6669. doi: 10.1016/j.bmcl.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 38.a Wang L. Andrew Fensome receives Robertson Award. Chem. Eng. News. 2007;85:40. [Google Scholar]; b Fensome A, Bender R, Chopra R, Cohen J, Collins MA, Hudak V, Malakian K, Lockhead S, Olland A, Svenson K, Terefenko EA, Unwalla RJ, Wilhelm JM, Wolfrom S, Zhu Y, Winneker RC, Wrobel J. Synthesis and structure-activity relationship of novel 6-aryl-1,4-dihydrobenzo[d][1,3]oxazine-2-thiones as progesterone receptor modulators leading to the potent and selective nonsteroidal progesterone receptor agonist Tanaproget. J. Med. Chem. 2005;48:5092–5095. doi: 10.1021/jm050358b. [DOI] [PubMed] [Google Scholar]
- 39.Zhou H-B, Lee JH, Mayne CG, Carlson KE, Katzenellenbogen JA. Imaging Progesterone Receptor in Breast Tumors: Synthesis and Receptor Binding Affinity of Fluoroalkyl-Substituted Analogues of Tanaproget. J. Med. Chem. 2010;53:3349–3360. doi: 10.1021/jm100052k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shivakumar L, Lancet JE. The role of farnesyl transferase inhibitors in the treatment of patients with leukemia and myelodysplastic syndrome. Clinical Leukemia. 2006;1:80–83. [Google Scholar]
- 41.a Bailey HH, Alberti DB, Thomas JP, Mulkerin DL, Binger KA, Gottardis MM, Martell RE, Wilding G. Phase I Trial of Weekly Paclitaxel and BMS-214662 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2007;13:3623–3629. doi: 10.1158/1078-0432.CCR-07-0158. [DOI] [PubMed] [Google Scholar]; b Caponigro F, Casale M, Bryce J. Farnesyl transferase inhibitors in clinical development. Expert Opin. Invest. Drugs. 2003;12:943–954. doi: 10.1517/13543784.12.6.943. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen HG, Holyoake TL. Characterization of cancer stem cells in chronic myeloid leukaemia. Biochem. Soc. T. 2007;35:1347–1351. doi: 10.1042/BST0351347. [DOI] [PubMed] [Google Scholar]
- 43.Reid TS, Beese LS. Crystal structures of the anticancer clinical candidates R115777 (Tipifarnib) and BMS-214662 complexed with protein farnesyltransferase suggest a mechanism of FTI selectivity. Biochemistry. 2004;43:6877–6884. doi: 10.1021/bi049723b. [DOI] [PubMed] [Google Scholar]
- 44.Hunt JT, Ding CZ, Batorsky R, Bednarz M, Bhide R, Cho Y, Chong S, Chao S, Gullo-Brown J, Guo P, Kim SK, Lee FYF, Leftheris K, Miller A, Mitt T, Patel M, Penhallow BA, Ricca C, Rose WC, Schmidt RC, Slusarchyk WA, Vite G, Manne V. Discovery of (R)-7-Cyano-2,3,4,5-tetrahydro-1-(1H-imidazol-4-ylmethyl)-3- (phenylmethyl)-4-(2-thienylsulfonyl)-1H-1,4-benzodiazepine (BMS-214662), a Farnesyltransferase Inhibitor with Potent Preclinical Antitumor Activity. J. Med. Chem. 2000;43:3587–3595. doi: 10.1021/jm000248z. [DOI] [PubMed] [Google Scholar]
- 45.Martin NE, Brunner TB, Kiel KD, DeLaney TF, Regine WF, Mohiuddin M, Rosato EF, Haller DG, Stevenson JP, Smith D, Pramanik B, Tepper J, Tanaka WK, Morrison B, Deutsch P, Gupta AK, Muschel RJ, McKenna WG, Bernhard EJ, Hahn SM. A Phase I trial of the dual farnesyltransferase and geranylgeranyltransferase inhibitor L-778,123 and radiotherapy for locally advanced pancreatic cancer. Clin. Cancer Res. 2004;10:5447–5454. doi: 10.1158/1078-0432.CCR-04-0248. [DOI] [PubMed] [Google Scholar]
- 46.Reid TS, Long SB, Beese LS. Crystallographic Analysis Reveals that Anticancer Clinical Candidate L-778,123 Inhibits Protein Farnesyltransferase and Geranylgeranyltransferase-I by Different Binding Modes. Biochemistry. 2004;43:9000–9008. doi: 10.1021/bi049280b. [DOI] [PubMed] [Google Scholar]
- 47.Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR, Awada A, Ranade A, Jiao S, Schwartz G, Abbas R, Powell C, Turnbull K, Vermette J, Zacharchuk C, Badwe R. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J. Clin. Oncol. 2010;28:1301–1307. doi: 10.1200/JCO.2009.25.8707. [DOI] [PubMed] [Google Scholar]
- 48.Bose P, Ozer H. Neratinib: an oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin. Invest. Drugs. 2009;18:1735–1751. doi: 10.1517/13543780903305428. [DOI] [PubMed] [Google Scholar]
- 49.Erlichman C, Hidalgo M, Boni JP. Phase I study of EKB-569, an irreversible inhibitor of the epidermal growth factor receptor, in patients with advanced solid tumors. J. Clin. Oncol. 2006;24:2252–2260. doi: 10.1200/JCO.2005.01.8960. [DOI] [PubMed] [Google Scholar]
- 50.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. P. Natl. Acad. Sci. USA. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. P. Natl. Acad. Sci. USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wissner A, Fraser HL, Ingalls CL, Dushin RG, Floyd MB, Cheung K, Nittoli T, Ravi MR, Tan X, Loganzo F. Dual irreversible kinase inhibitors: Quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg. Med. Chem. 2007;15:3635–3648. doi: 10.1016/j.bmc.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 53.a Morphy R. Selectively Nonselective Kinase Inhibition: Striking the Right Balance. J. Med. Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]; b Keller G, Schafhause P, Brummendorf TH. Bosutinib. Recent Res. Cancer. 2010;184:119–127. doi: 10.1007/978-3-642-01222-8_9. [DOI] [PubMed] [Google Scholar]
- 54.Gunby RH, Ahmed S, Sottocornola R, Gasser M, Redaelli S, Mologni L, Tartari CJ, Belloni V, Gambacorti-Passerini C, Scapozza L. Structural Insights into the ATP Binding Pocket of the Anaplastic Lymphoma Kinase by Site-Directed Mutagenesis, Inhibitor Binding Analysis, and Homology Modeling. J. Med. Chem. 2006;49:5759–5768. doi: 10.1021/jm060380k. [DOI] [PubMed] [Google Scholar]
- 55.a Tsou H-R, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, Rabindran SK, Shen R, Shi X, Wang YF, Upeslacis J, Wissner A. Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3-carbonitriles as Orally Active, Irreversible Inhibitors of Human Epidermal Growth Factor Receptor-2 Kinase Activity. J. Med. Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]; b Wissner A, Berger DM, Boschelli DH, Floyd MB, Jr., Greenberger LM, Gruber BC, Johnson BD, Mamuya N, Nilakantan R, Reich MF, Shen R, Tsou H-R, Upeslacis E, Wang YF, Wu B, Ye F, Zhang N. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J. Med. Chem. 2000;43:3244–3256. doi: 10.1021/jm000206a. [DOI] [PubMed] [Google Scholar]
- 56.Boschelli DH. 4-Anilino-3-quinolinecarbonitriles: an emerging class of kinase inhibitors - an update. Med. Chem. Rev. 2004;1:457–463. doi: 10.2174/1568026023393354. [DOI] [PubMed] [Google Scholar]
- 57.a Wissner A, Floyd MB, Rabindran SK, Nilakantan R, Greenberger LM, Shen R, Wang Y-F, Tsou H-R. Syntheses and EGFR and HER-2 kinase inhibitory activities of 4-anilinoquinoline-3-carbonitriles: analogues of three important 4-anilinoquinazolines currently undergoing clinical evaluation as therapeutic antitumor agents. Bioorg. Med. Chem. Lett. 2002;12:2893–2897. doi: 10.1016/s0960-894x(02)00598-x. [DOI] [PubMed] [Google Scholar]; b Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discfani CM. Combinatorial chemoprevention of intestinal neoplasia. Nat. Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- 58.Chen JM, Xu SL, Wawrzak Z, Basarab GS, Jordan DB. Structure-Based Design of Potent Inhibitors of Scytalone Dehydratase: Displacement of a Water Molecule from the Active Site. Biochemistry. 1998;37:17735–17744. doi: 10.1021/bi981848r. [DOI] [PubMed] [Google Scholar]
- 59.Bayram M, De Luca L, Massie MB, Gheorghiade M. Reassessment of dobutamine, dopamine, and milrinone in the management of acute heart failure syndromes. Am. J. Cardiol. 2005;96:47G–58G. doi: 10.1016/j.amjcard.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 60.Hannon JD, Housmans PR. Inotropic therapy. Advances in Cardiovascular Pharmacology. 2008:1–15. [Google Scholar]
- 61.Mizushige K, Ueda T, Yukiiri K, Suzuki H. Olprinone: a phosphodiesterase III inhibitor with positive inotropic and vasodilator effects. Cardiovasc. Drug Rev. 2002;20:163–174. doi: 10.1111/j.1527-3466.2002.tb00085.x. [DOI] [PubMed] [Google Scholar]
- 62.a Wojtczak A, Luft JR, Cody V. Structural aspects of inotropic bipyridine binding. Crystal structure determination to 1.9 Å of the human serum transthyretin-milrinone complex. J. Biol. Chem. 1993;268:6202–6206. doi: 10.2210/pdb1tlm/pdb. [DOI] [PubMed] [Google Scholar]; b Cody V, Wojtczak A, Ciszak E, Luft J. Gordon A, Gross J, Hennemann G, editors. Differences in inhibitor and substrate binding in transthyretin crystal complexes. Prog. Thyroid Res., Proc. Int. Thyroid Conf. 1991:793–796. [Google Scholar]
- 63.Cody V, Wojtczak A, Davis FB, Davis PJ, Blas SD. Structure-Activity Relationships of Milrinone Analogs Determined in Vitro in a Rabbit Heart Membrane Ca2+-ATPase Model. J. Med. Chem. 1995;38:1990–1997. doi: 10.1021/jm00011a018. [DOI] [PubMed] [Google Scholar]
- 64.Thornber CW. Isosterism and molecular modification in drug design. Chem. Soc. Rev. 1979;8:563–580. [Google Scholar]
- 65.Matthews DP, McCarthy JR, Whitten JP, Kastner PR, Barney CL, Marshall FN, Ertel MA, Burkhard T, Shea PJ, Kariya T. Synthesis and cardiotonic activity of novel biimidazoles. J. Med. Chem. 1990;33:317–327. doi: 10.1021/jm00163a052. [DOI] [PubMed] [Google Scholar]
- 66.Bell AS, Campbell SF, Morris DS, Roberts DA, Stefaniak MH. 2(1H)-Quinolinones with cardiac stimulant activity. 3. Synthesis and biological properties of 6-imidazol-1-yl derivatives. J. Med. Chem. 1989;32:1552–1558. doi: 10.1021/jm00127a025. [DOI] [PubMed] [Google Scholar]
- 67.Cecchetti V, Tabarrini O, Sabatini S. From cromakalim to different structural classes of KATP channel openers. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2006;6:1049–1068. doi: 10.2174/156802606777323683. [DOI] [PubMed] [Google Scholar]; b Salamon E, Mannhold R, Weber H, Lemoine H, Frank W. 6-Sulfonylchromenes as Highly Potent KATP-Channel Openers. J. Med. Chem. 2002;45:1086–1089. doi: 10.1021/jm010999g. [DOI] [PubMed] [Google Scholar]
- 68.Ding CZ, Rovnyak GC, Misra RN, Grover GJ, Miller AV, Ahmed SZ, Kelly Y, Normandin DE, Sleph PG, Atwal KS. Cardioselective Antiischemic ATP-Sensitive Potassium Channel (KATP) Openers. 6. Effect of Modifications at C6 of Benzopyranyl Cyanoguanidines. J. Med. Chem. 1999;42:3711–3717. doi: 10.1021/jm990196h. [DOI] [PubMed] [Google Scholar]; b Atwal KS, Grover GJ, Ahmed SZ, Ferrara FN, Harper TW, Kim KS, Sleph PG, Dzwonczyk S, Russell AD, Moreland S, McCullough JR, Normandin DE. Cardioselective anti-ischemic ATP-sensitive potassium channel openers. J. Med. Chem. 1993;36:3971–3974. doi: 10.1021/jm00076a027. [DOI] [PubMed] [Google Scholar]
- 69.a Gaspar T, Snipes JA, Busija AR, Kis B, Domoki F, Bari F, Busija DW. ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095. J. Cerebr. Blood F. Met. 2008;28:1090–1103. doi: 10.1038/sj.jcbfm.9600611. [DOI] [PubMed] [Google Scholar]; b Rovnyak GC, Ahmed SZ, Ding CZ, Dzwonczyk S, Ferrara FN, Humphreys WG, Grover GJ, Santafianos D, Atwal KS, Baird AJ, McLaughlin LG, Normandin DE, Sleph PG, Traeger SC. Cardioselective Antiischemic ATP-Sensitive Potassium Channel (KATP) Openers. 5. Identification of 4-(N-Aryl)-Substituted Benzopyran Derivatives with High Selectivity. J. Med. Chem. 1997;40:24–34. doi: 10.1021/jm9605905. [DOI] [PubMed] [Google Scholar]
- 70.b Horino H, Mimura T, Kagechika K, Ohta M, Kubo H, Kitagawa M. Synthesis and antihypertensive activity of 4-(diazabicyclo[4.1.0]heptenyloxy)benzopyran derivatives and their analogs. Chem. Pharm. Bull. 1998;46:602–609. doi: 10.1248/cpb.46.602. [DOI] [PubMed] [Google Scholar]; c Bergmann R, Gericke R. Synthesis and antihypertensive activity of 4-(1,2-dihydro-2-oxo-1-pyridyl)-2H-1-benzopyrans and related compounds, new potassium channel activators. J. Med. Chem. 1990;33:492–504. doi: 10.1021/jm00164a005. [DOI] [PubMed] [Google Scholar]
- 71.Atwal KS, Grover GJ, Lodge NJ, Normandin DE, Traeger SC, Sleph PG, Cohen RB, Bryson CC, Dickinson KEJ. Binding of ATP-Sensitive Potassium Channel (KATP) Openers to Cardiac Membranes: Correlation of Binding Affinities with Cardioprotective and Smooth Muscle Relaxing Potencies. J. Med. Chem. 1998;41:271–275. doi: 10.1021/jm970762d. [DOI] [PubMed] [Google Scholar]
- 72.Sanchez C. The pharmacology of citalopram enantiomers: the antagonism by R-citalopram on the effect of S-citalopram. Basic Clin. Pharmacol. Toxicol. 2006;99:91–95. doi: 10.1111/j.1742-7843.2006.pto_295.x. [DOI] [PubMed] [Google Scholar]
- 73.Koldso H, Severinsen K, Tran TT, Celik L, Jensen HH, Wiborg O, Schiott B, Sinning S. The Two Enantiomers of Citalopram Bind to the Human Serotonin Transporter in Reversed Orientations. J. Am. Chem. Soc. 2010;132:1311–1322. doi: 10.1021/ja906923j. [DOI] [PubMed] [Google Scholar]
- 74.Puchler K, Sasahara K, Plenker A, Nitanai T, Witte PU. The pharmacokinetic profile of RS-8359. Int. Clin. Psychopharmacol. 1997;12:S11–S16. doi: 10.1097/00004850-199709005-00003. [DOI] [PubMed] [Google Scholar]
- 75.Takasaki W, Yamamura M, Nozaki A, Nitanai T, Sasahara K, Itoh K, Tanaka Y. Stereoselective pharmacokinetics of RS-8359, a selective and reversible MAO-A inhibitor, by species-dependent drug-metabolizing enzymes. Chirality. 2005;17:135–141. doi: 10.1002/chir.20124. [DOI] [PubMed] [Google Scholar]
- 76.Itoh K, Hoshino K, Endo A, Asakawa T, Yamakami K, Noji C, Kosaka T, Tanaka Y. Chiral inversion of RS-8359: a selective and reversible MAO-A inhibitor via oxido-reduction of keto-alcohol. Chirality. 2006;18:698–706. doi: 10.1002/chir.20309. [DOI] [PubMed] [Google Scholar]
- 77.Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin. Invest. Drugs. 2009;18:1753–1764. doi: 10.1517/13543780903286396. [DOI] [PubMed] [Google Scholar]
- 78.Heinrich T, Boettcher H, Gericke R, Bartoszyk GD, Anzali S, Seyfried CA, Greiner HE, van Amsterdam C. Synthesis and Structure-Activity Relationship in a Class of Indolebutylpiperazines as Dual 5-HT1A Receptor Agonists and Serotonin Reuptake Inhibitors. J. Med. Chem. 2004;47:4684–4692. doi: 10.1021/jm040793q. [DOI] [PubMed] [Google Scholar]
- 79.Bromidge SM, Brown F, Cassidy F, Clark MSG, Dabbs S, Hadley MS, Hawkins J, Loudon JM, Naylor CB, Orlek BS, Riley GJ. Design of [R-(Z)]-(+)-α-(Methoxyimino)-1-azabicyclo[2.2.2]octane-3-acetonitrile (SB 202026), a Functionally Selective Azabicyclic Muscarinic M1 Agonist Incorporating the N-Methoxyimidoyl Nitrile Group as a Novel Ester Bioisostere. J. Med. Chem. 1997;40:4265–4280. doi: 10.1021/jm9702903. [DOI] [PubMed] [Google Scholar]
- 80.Vialla A. Propericiazine (Neuleptil), chemistry, and pharmacology. Encephale. 1969;58:3–7. [Google Scholar]
- 81.a Favre J-D, Allain H, Aubin H-J, Frija-Orvoen E, Gillet C, Lejoyeux M, Payen A, Weber M, Garcia-Acosta S, Kermadi I, Dib M. Double-blind study of cyamemazine and diazepam in the alcohol withdrawal syndrome. Hum. Psychopharmacol. Clin. Exp. 2005;20:511–519. doi: 10.1002/hup.718. [DOI] [PubMed] [Google Scholar]; b Lemoine P, Kermadi I, Garcia-Acosta S, Garay RP, Dib M. Double-blind, comparative study of cyamemazine vs. bromazepam in the benzodiazepine withdrawal syndrome. Prog. Neuropsychopharmacol. Biol. Psych. 2006;30:131–137. doi: 10.1016/j.pnpbp.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 82.Patat A, Paty I, Hindmarch I. Pharmacodynamic profile of zaleplon, a new non-benzodiazepine hypnotic agent. Hum. Psychopharm. 2001;16:369–392. doi: 10.1002/hup.310. [DOI] [PubMed] [Google Scholar]
- 83.Sanger DJ, Morel E, Perrault G. Comparison of the pharmacological profiles of the hypnotic drugs, zaleplon and zolpidem. Eur. J. Pharmacol. 1996;313:35–42. doi: 10.1016/0014-2999(96)00510-9. [DOI] [PubMed] [Google Scholar]
- 84.Munoz-Islas E, Gupta S, Jimenez-Mena LR, Lozano-Cuenca J, Sanchez-Lopez A, Centurion D, Mehrotra S, MaassenVanDenBrink A, Villalon CM. Donitriptan, but not sumatriptan, inhibits capsaicin-induced canine external carotid vasodilatation via 5-HT1B rather than 5-HT1D receptors. Br. J. Pharmacol. 2006;149:82–91. doi: 10.1038/sj.bjp.0706839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Letienne R, Verscheure Y, Perez M, Le Grand B, Colpaert FC, John GW. Donitriptan selectively decreases jugular venous oxygen saturation in the anesthetized pig: Further insights into its mechanism of action relevant to headache relief. J. Pharmacol. Exp. Ther. 2003;305:749–754. doi: 10.1124/jpet.102.047225. [DOI] [PubMed] [Google Scholar]
- 86.Mehellou Y, De Clercq E. Twenty-Six Years of Anti-HIV Drug Discovery: Where Do We Stand and Where Do We Go? J. Med. Chem. 2010;53:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 87.Hughes CA, Robinson L, Tseng A, MacArthur RD. New antiretroviral drugs: a review of the efficacy, safety, pharmacokinetics, and resistance profile of tipranavir, darunavir, etravirine, rilpivirine, maraviroc, and raltegravir. Expert Opinion Pharmacother. 2009;10:2445–2466. doi: 10.1517/14656560903176446. [DOI] [PubMed] [Google Scholar]
- 88.a Nuttall JP, Thake DC, Lewis MG, Ferkany JW, Romano JW, Mitchnick MA. Concentrations of dapivirine in the rhesus macaque and rabbit following once daily intravaginal administration of a gel formulation of [14C]dapivirine for 7 days. Antimicrob. Agents Chemother. 2008;52:909–914. doi: 10.1128/AAC.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Van Herrewege Y, Michiels J, Van Roey J, Fransen K, Kestens L, Balzarini J, Lewi P, Vanham G, Janssen P. In vitro evaluation of nonnucleoside reverse transcriptase inhibitors UC-781 and TMC120-R147681 as human immunodeficiency virus microbicides. Antimicrob. Agents Chemother. 2004;48:337–339. doi: 10.1128/AAC.48.1.337-339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Clercq E. Emerging anti-HIV drugs. Expert Opin. Emerging Drugs. 2005;10:241–274. doi: 10.1517/14728214.10.2.241. [DOI] [PubMed] [Google Scholar]
- 90.Das K, Lewi PJ, Hughes SH, Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Bio. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 91.Das K, Clark AD, Jr., Lewi PJ, Heeres J, De Jonge MR, Koymans LMH, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, de Béthune MP, Boyer PL, Clark P, Hughes SH, Janssen PAJ, Arnold E. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (Etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 92.a Faetkenheuer G, Staszewski S, Plettenburg A, Hackman F, Layton G, McFadyen L, Davis J, Jenkins TM. Activity, pharmacokinetics and safety of lersivirine (UK-453,061), a next-generation nonnucleoside reverse transcriptase inhibitor, during 7-day monotherapy in HIV-1-infected patients. AIDS. 2009;23:2115–2122. doi: 10.1097/QAD.0b013e32832fef5b. [DOI] [PubMed] [Google Scholar]; b Abdel-Malak M, Gallati C, Mousa SA. The rise of second-generation non-nucleoside reverse transcriptase inhibitors: etravirine, rilpivirine, UK-453061 and RDEA-806. Drugs Fut. 2008;33:691–699. [Google Scholar]
- 93.a Mowbray CE, Corbau R, Hawes M, Jones LH, Mills JE, Perros M, Selby MD, Stupple PA, Webster R, Wood A. Pyrazole NNRTIs 3: Optimisation of physicochemical properties. Bioorg. Med. Chem. Lett. 2009;19:5603–5606. doi: 10.1016/j.bmcl.2009.08.043. [DOI] [PubMed] [Google Scholar]; b Mowbray CE, Burt C, Corbau R, Gayton S, Hawes M, Perros M, Tran I, Price DA, Quinton FJ, Selby MD, Stupple PA, Webster R, Wood A. Pyrazole NNRTIs 4: Selection of UK-453,061 (lersivirine) as a Development Candidate. Bioorg. Med. Chem. Lett. 2009;19:5857–5860. doi: 10.1016/j.bmcl.2009.08.080. [DOI] [PubMed] [Google Scholar]
- 94.Hoegberg M, Sahlberg C, Engelhardt P, Noreen R, Kangasmetsae J, Johansson NG, Oeberg B, Vrang L, Zhang H, Sahlberg B-L, Unge T, Lövgren S, Fridborg K, Bäckbro K. Urea-PETT Compounds as a New Class of HIV-1 Reverse Transcriptase Inhibitors. 3. Synthesis and Further Structure-Activity Relationship Studies of PETT Analogues. J. Med. Chem. 1999;42:4150–4160. doi: 10.1021/jm990095j. [DOI] [PubMed] [Google Scholar]
- 95.a Crostarosa F, Aravantinou M, Akpogheneta OJ, Jasny E, Shaw A, Kenney J, Piatak M, Jr., Lifson JD, Teitelbaum A, Hu L, Chudolij A, Zydowsky TM, Blanchard J, Gettie A, Robbiani M. A macaque model to study vaginal HSV-2/Immunodeficiency virus co-infection and the impact of HSV-2 on microbicide efficacy. PLoS One. 2009;4:e8060, 1–14. doi: 10.1371/journal.pone.0008060. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fernandez-Romero JA, Thorn M, Turville SG, Titchen K, Sudol K, Li J, Miller T, Robbiani M, Maguire RA, Buckheit RW, Jr., Hartman TL, Phillips DM. Carrageenan/MIV-150 (PC-815), a Combination Microbicide. Sex Transm. Dis. 2006;34:9–14. doi: 10.1097/01.olq.0000223287.46097.4b. [DOI] [PubMed] [Google Scholar]
- 96.Zhou X-J, Garner RC, Nicholson S, Kissling CJ, Mayers D. Microdose pharmacokinetics of IDX899 and IDX989, candidate HIV-1 non-nucleoside reverse transcriptase inhibitors, following oral and intravenous administration in healthy male subjects. J. Clin. Pharmacol. 2009;49:1408–1416. doi: 10.1177/0091270009343698. [DOI] [PubMed] [Google Scholar]
- 97.Pascual E, Sivera F, Yasothan U, Kirkpatrick P. Febuxostat. Nat. Rev. Drug Discovery. 2009;8:191–192. doi: 10.1038/nrd2831. [DOI] [PubMed] [Google Scholar]
- 98.Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An Extremely Potent Inhibitor of Xanthine Oxidoreductase. J. Biol. Chem. 2003;278:1848–1855. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- 99.Sato T, Ashizawa N, Matsumoto K, Iwanaga T, Nakamura H, Inoue T, Nagata O. Discovery of 3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole, FYX-051 - a xanthine oxidoreductase inhibitor for the treatment of hyperuricemia. Bioorg. Med. Chem. Lett. 2009;19:6225–6229. doi: 10.1016/j.bmcl.2009.08.091. [DOI] [PubMed] [Google Scholar]
- 100.Okamoto K, Matsumoto K, Hille R, Eger BT, Pai EF, Nishino T. The crystal structure of xanthine oxidoreductase during catalysis: Implications for reaction mechanism and enzyme inhibition. P. Natl. Acad. Sci. USA. 2004;101:7931–7936. doi: 10.1073/pnas.0400973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pratley RE. Alogliptin: a new, highly selective dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2009;10:503–512. doi: 10.1517/14656560802694713. [DOI] [PubMed] [Google Scholar]
- 102.a Andukuri R, Drincic A, Rendell M. Alogliptin: a new addition to the class of DPP-4 inhibitors. Diabetes, Metabolic Syndrome and Obesity. 2009;2:117–126. [PMC free article] [PubMed] [Google Scholar]; b Lee B, Shi L, Kassel DB, Asakawa T, Takeuchi K, Christopher RJ. Pharmacokinetic, pharmacodynamic, and efficacy profiles of alogliptin, a novel inhibitor of dipeptidyl peptidase-4, in rats, dogs, and monkeys. Eur. J. Pharmacol. 2008;589:306–314. doi: 10.1016/j.ejphar.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 103.Feng J, Zhang Z, Wallace MB, Stafford JA, Kaldor SW, Kassel DB, Navre M, Shi L, Skene RJ, Asakawa T, Takeuchi K, Xu R, Webb DR, Gwaltney SL., II Discovery of Alogliptin: A Potent, Selective, Bioavailable, and Efficacious Inhibitor of Dipeptidyl Peptidase IV. J. Med. Chem. 2007;50:2297–2300. doi: 10.1021/jm070104l. [DOI] [PubMed] [Google Scholar]
- 104.Johnson HG. New anti-allergy drugs - the lodoxamides. Trends Pharmacol. Sci. 1980;1:343–345. [Google Scholar]
- 105.a Norman P. TYB-2285 (Toyobo). Curr. Opin. Anti-Inflam. Immunomodul. Invest. Drugs. 2000;2:113–116. [Google Scholar]; b Ban M, Taguchi H, Katsushima T, Takahashi M, Shinoda K, Watanabe A, Tominaga T. Novel antiallergic and antiinflammatory agents. Part II: synthesis and pharmacology of TYB-2285 and its related compounds. Bioorg. Med. Chem. 1998;6:1077–1087. doi: 10.1016/s0968-0896(98)00066-2. [DOI] [PubMed] [Google Scholar]
- 106.Ortolani S. Strontium ranelate: a new first-line treatment for patients with postmenopausal osteoporosis. Postmenopausal Osteoporosis. 2006:247–253. [Google Scholar]
- 107.Maimoun L, Brennan TC, Badoud I, Dubois-Ferriere V, Rizzoli R, Ammann P. Strontium ranelate improves implant osseointegration. Bone. 2010;46:1436–1441. doi: 10.1016/j.bone.2010.01.379. (b) There has been some question as to whether this dual action mechanism operates: Blake GM, Compston JE, Fogelman I. Could strontium ranelate have a synertistic role in the treatment of osteoporosis? J. Bone Miner. Res. 2009;24:1354–1357. doi: 10.1359/jbmr.090601.
- 108.a Pasqualotto AC, Thiele KO, Goldani LZ. Novel triazole antifungal drugs: Focus on isavuconazole, ravuconazole and albaconazole. Curr. Opin. Investig. Drugs. 2010;11:165–174. [PubMed] [Google Scholar]; b Petrikkos G, Skiada A. Recent advances in antifungal chemotherapy. Int. J. Antimicrob. Agents. 2007;30:108–117. doi: 10.1016/j.ijantimicag.2007.03.009. [DOI] [PubMed] [Google Scholar]; c Yamazumi T, Pfaller MA, Messer SA, Houston A, Hollis RJ, Jones RN. In vitro activities of ravuconazole (BMS-207147) against 541 clinical isolates of Cryptococcus neoformans. Antimicrob. Agents Chemother. 2000;44:2883–2886. doi: 10.1128/aac.44.10.2883-2886.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sheng C, Miao Z, Ji H, Yao J, Wang W, Che X, Dong G, Lu J, Guo W, Zhang W. Three-dimensional model of lanosterol 14α-demethylase from Cryptococcus neoformans: active-site characterization and insights into azole binding. Antimicrob. Agents Chemother. 2009;53:3487–3495. doi: 10.1128/AAC.01630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.a Schmitt-Hoffmann A, Roos B, Spickermann J, Heep M, Peterfai E, Edwards DJ, Stoeckel K. Effect of mild and moderate liver disease on the pharmacokinetics of isavuconazole after intravenous and oral administration of a single dose of the prodrug BAL8557. Antimicrob. Agents Chemother. 2009;53:4885–4890. doi: 10.1128/AAC.00319-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Verweij PE, Gonzalez GM, Wiederhold NP, Lass-Florl C, Warn P, Heep M, Ghannoum MA, Guinea J. In vitro antifungal activity of isavuconazole against 345 mucorales isolates collected at study centers in eight countries. J. Chemotherapy. 2009;21:272–281. doi: 10.1179/joc.2009.21.3.272. [DOI] [PubMed] [Google Scholar]
- 111.Yin W, Tsutsumi K. Lipoprotein lipase activator NO-1886. Cardiovasc. Drug Rev. 2003;21:133–142. doi: 10.1111/j.1527-3466.2003.tb00111.x. [DOI] [PubMed] [Google Scholar]
- 112.a Blake P. An overview of clinical trial experience with epanolol. Drugs. 1989;38:81–85. doi: 10.2165/00003495-198900382-00022. [DOI] [PubMed] [Google Scholar]; b Harry JD. Clinical pharmacology of epanolol. Pharmacodynamic aspects. Drugs. 1989;38:18–27. doi: 10.2165/00003495-198900382-00006. [DOI] [PubMed] [Google Scholar]
- 113.Tanii H, Hashimoto K. Structure-toxicity relationship of aliphatic nitriles. Toxicol. Lett. 1984;22:267–272. doi: 10.1016/0378-4274(84)90077-8. Ahmed AE, Trieff NM. Aliphatic nitriles: metabolism and toxicity. Prog. Drug Metab. 1983;7:229–294. For recent AMES analyses of several alkylnitriles see: Wu J-C, Hseu YC, Chen C-H, Wang S-H, Chen SC. Comparative investigations of genotoxic activity of five nitriles in the comet assay and the Ames test. J. Hazard. Mater. 2009;169:492–497. doi: 10.1016/j.jhazmat.2009.03.121.
- 114.a Milani M, Jha G, Potter DA. Anastrozole use in early stage breast cancer of postmenopausal women. Clin. Med. Ther. 2009;1:141–156. doi: 10.4137/cmt.s9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gangadhara S, Bertelli G. Long-term efficacy and safety of anastrozole for adjuvant treatment of early breast cancer in postmenopausal women. Ther. Clin. Risk Man. 2009;5:291–300. doi: 10.2147/tcrm.s3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.a Jackson T, Woo LWL, Trusselle MN, Purohit A, Reed MJ, Potter BVL. Non-steroidal aromatase inhibitors based on a biphenyl scaffold: synthesis, in vitro SAR, and molecular modeling. ChemMedChem. 2008;3:603–618. doi: 10.1002/cmdc.200700266. [DOI] [PubMed] [Google Scholar]; b Jackson T, Woo LWL, Trusselle MN, Chander SK, Purohit A, Reed MJ, Potter BVL. Dual aromatase-sulfatase inhibitors based on the anastrozole template: Synthesis, in vitro SAR, molecular modelling and in vivo activity. Org. Biomol. Chem. 2007;5:2940–2952. [PubMed] [Google Scholar]
- 116.a Cooper-DeHoff RM, Handberg EM, Mancia G, Zhou Q, Champion A, Legler UF, Pepine CJ. INVEST revisited: review of findings from the International Verapamil SR-Trandolapril Study. Expert Rev. Cardiovasc. Ther. 2009;7:1329–1340. doi: 10.1586/erc.09.102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sica DA, Prisant LM. Pharmacologic and therapeutic considerations in hypertension therapy with calcium channel blockers: focus on verapamil. J. Clin. Hypertens. 2007;9:1–22. [Google Scholar]
- 117.Brogden RN, Benfield P. Gallopamil. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in ischaemic heart disease. Drugs. 1994;47:93–115. doi: 10.2165/00003495-199447010-00007. [DOI] [PubMed] [Google Scholar]
- 118.Mannhold R, Holtje HD, Koke V. Importance of nitrile substitution for the calcium antagonistic action of verapamil. Arch. Pharm. 1986;319:990–998. doi: 10.1002/ardp.19863191107. [DOI] [PubMed] [Google Scholar]
- 119.Cheng RCK, Tikhonov DB, Zhorov BS. Structural model for phenylalkylamine binding to L-type calcium channels. J. Biol. Chem. 2009;284:28332–28342. doi: 10.1074/jbc.M109.027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.a Berger D, Citarella R, Dutia M, Greenberger L, Hallett W, Paul R, Powell D. Novel Multidrug Resistance Reversal Agents. J. Med. Chem. 1999;42:2145–2161. doi: 10.1021/jm9804477. [DOI] [PubMed] [Google Scholar]; b Teodori E, Dei S, Quidu P, Budriesi R, Chiarini A, Garnier-Suillerot A, Gualtieri F, Manetti D, Romanelli MN, Scapecchi S. Design, Synthesis, and in Vitro Activity of Catamphiphilic Reverters of Multidrug Resistance: Discovery of a Selective, Highly Efficacious Chemosensitizer with Potency in the Nanomolar Range. J. Med. Chem. 1999;42:1687–1697. doi: 10.1021/jm980440p. [DOI] [PubMed] [Google Scholar]
- 121.Rennard S, Knobil K, Rabe KF, Morris A, Schachter N, Locantore N, Canonica WG, Zhu Y, Barnhart F. The efficacy and safety of cilomilast in COPD. Drugs. 2008;68:3–57. doi: 10.2165/0003495-200868002-00002. [DOI] [PubMed] [Google Scholar]
- 122.Christensen SB, Guider A, Forster CJ, Gleason JG, Bender PE, Karpinski JM, DeWolf WE, Jr., Barnette MS, Underwood DC, Griswold DE, Cieslinski LB, Burman M, Bochnowicz S, Osborn RR, Manning CD, Grous M, Hillegas LM, O'Leary Bartus J, Ryan MD, Eggleston DS, Haltiwanger RC, Torphy TJ. 1,4-Cyclohexanecarboxylates: Potent and Selective Inhibitors of Phosophodiesterase 4 for the Treatment of Asthma. J. Med. Chem. 1998;41:821–835. doi: 10.1021/jm970090r. [DOI] [PubMed] [Google Scholar]
- 123.Ochiai H, Ohtani T, Ishida A, Kishikawa K, Obata T, Nakai H, Toda M. Orally active PDE4 inhibitors with therapeutic potential. Bioorg. Med. Chem. Lett. 2004;14:1323–1327. doi: 10.1016/j.bmcl.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 124.Kodimuthali A, Jabaris SSL, Pal M. Recent Advances on Phosphodiesterase 4 Inhibitors for the Treatment of Asthma and Chronic Obstructive Pulmonary Disease. J. Med. Chem. 2008;51:5471–5489. doi: 10.1021/jm800582j. [DOI] [PubMed] [Google Scholar]
- 125.Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillette S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim S-H, Schlessinger J, Zhang KYJ. Structural Basis for the Activity of Drugs that Inhibit Phosphodiesterases. Structure. 2004;12:2233–2247. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 126.Noble S, McTavish D. Levocabastine: An update of its pharmacology, clinical efficacy and tolerability in the topical treatment of allergic rhinitis and conjunctivitis. Drugs. 1995;50:1032–1049. doi: 10.2165/00003495-199550060-00009. [DOI] [PubMed] [Google Scholar]
- 127.a Brack A, Bottiger BW, Schafer M. New insights in postoperative pain therapy. Anasth. Intensiv. Notf. 2004;39:157–164. doi: 10.1055/s-2004-814314. [DOI] [PubMed] [Google Scholar]; b Bouillon T, Kietzmann D, Port R, Meineke I, Hoeft A. Population pharmacokinetics of piritramide in surgical patients. Anesthesiol. 1999;90:7–15. doi: 10.1097/00000542-199901000-00004. [DOI] [PubMed] [Google Scholar]; c Freye E. Postoperative pain treatment. Anaesthesiol. Reanim. 1991;16:379–392. [PubMed] [Google Scholar]
- 128.Jackson LS, Stafford JEH. The evaluation and application of a radioimmunoassay for the measurement of diphenoxylic acid, the major metabolite of diphenoxylate hydrochloride (Lomotil), in human plasma. J. Pharmacol. Methods. 1987;18:189–197. doi: 10.1016/0160-5402(87)90069-6. [DOI] [PubMed] [Google Scholar]
- 129.Fleming FF, Wang Q. Unsaturated Nitriles: Conjugate Additions of Carbon Nucleophiles to a Recalcitrant Class of Acceptors. Chem. Rev. 2003;103:2035–2078. doi: 10.1021/cr020045d. [DOI] [PubMed] [Google Scholar]
- 130.Seeberger LC, Hauser RA. Levodopa/carbidopa/entacapone in Parkinson's disease. Expert Rev. Neurother. 2009;9:929–940. doi: 10.1586/ern.09.64. [DOI] [PubMed] [Google Scholar]
- 131.a Lees AJ. Evidence-based efficacy comparison of tolcapone and entacapone as adjunctive therapy in Parkinson's disease. CNS Neurosci. Ther. 2008;14:83–93. doi: 10.1111/j.1527-3458.2007.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gordin A, Brooks DJ. Clinical pharmacology and therapeutic use of COMT inhibition in Parkinson's disease. J. Neurol. 2007;254:IV/37–IV/48. [Google Scholar]
- 132.Tervoa AJ, Nyrönenb TH, Rönkköa T, Posoa A. A structure-activity relationship study of catechol-O-methyltransferase inhibitors combining molecular docking and 3D QSAR methods. J. Comput. Aid. Mol. Des. 2003;17:797–810. doi: 10.1023/b:jcam.0000021831.47952.a7. For cocrystalization of COMT with a related inhibitor see: Rodrigues ML, Archer M, Bonifacio MJ, Soares-da-Silva P, Carrondo MA. Crystallization and preliminary crystallographic characterization of catechol-O-methyltransferase in complex with its cosubstrate and an inhibitor. Acta Crystallogr. 2001;D57:906–908. doi: 10.1107/s0907444901006539. Bonifacio MJ, Archer M, Rodrigues ML, Matias PM, Learmonth DA, Carrondo MA, Soares-da-Silva P. Kinetics and crystal structure of catechol-O-methyltransferase complex with co-substrate and a novel inhibitor with potential therapeutic application. Mol. Pharmacol. 2002;62:795–805. doi: 10.1124/mol.62.4.795.
- 133.Reine NJ. Medical management of pituitary-dependent hyperadrenocorticism: mitotane versus trilostane. Clin. Tech. Small An. P. 2007;22:18–25. doi: 10.1053/j.ctsap.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 134.Puddefoot JR, Barker S, Glover HR, Malouitre SDM, Vinson GP. Noncompetitive steroid inhibition of oestrogen receptor functions. Int. J. Cancer. 2002;101:17–22. doi: 10.1002/ijc.10547. [DOI] [PubMed] [Google Scholar]
- 135.Huq F. Molecular modelling analysis of the metabolism of trilostane. Pure Appl. Chem. 2007;2:353–358. [Google Scholar]
- 136.Thomas JL, Mack VL, Glow JA, Moshkelani D, Terrell JR, Bucholtz KM. Structure/function of the inhibition of human 3β-hydroxysteroid dehydrogenase type 1 and type 2 by trilostane. J. Steroid Biochem. 2008;111:66–73. doi: 10.1016/j.jsbmb.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Niwano Y, Ohmi T, Seo A, Kodama H, Koga H, Sakai A. Lanoconazole and its related optically active compound NND-502: Novel antifungal imidazoles with a ketene dithioacetal structure. Curr. Med. Chem. Anti-Infect. Agents. 2003;2:147–160. [Google Scholar]
- 138.Koga H, Nanjoh Y, Makimura K, Tsuboi R. In vitro antifungal activities of luliconazole, a new topical imidazole. Med. Mycol. 2009;47:640–647. doi: 10.1080/13693780802541518. [DOI] [PubMed] [Google Scholar]
- 139.Niwano Y, Koga H, Kodama H, Kanai K, Miyazaki T, Yamaguchi H. Inhibition of sterol 14α-demethylation of Candida albicans with NND-502, a novel optically active imidazole antimycotic agent. Med. Mycol. 1999;37:351–355. doi: 10.1046/j.1365-280x.1999.00243.x. [DOI] [PubMed] [Google Scholar]
- 140.Huq F. A molecular modelling analysis of luliconazole, lanconazole, and bifonazole. J. Pharmacol. Toxicol. 2007;2:567–573. [Google Scholar]
- 141.a Ogasawara K, Noda A, Yasuda S, Kobayashi M, Yukawa H, Ogawa A. Effect of calcium antagonist on cerebral blood flow and oxygen metabolism in patients with hypertension and chronic major cerebral artery occlusion: a positron emission tomography study. Nucl. Med. Commun. 2003;24:71–76. doi: 10.1097/00006231-200301000-00017. [DOI] [PubMed] [Google Scholar]; b Rosenthal J. Nilvadipine: profile of a new calcium antagonist. An overview. J. Cardiovasc. Pharm. 1994;24:S92–S107. [PubMed] [Google Scholar]
- 142.a Iwasaki K, Egashira N, Takagaki Y, Yoshimitsu Y, Hatip-Al-Khatib I, Mishima K, Fujiwara M. Nilvadipine prevents the impairment of spatial memory induced by cerebral ischemia combined with β-amyloid in rats. Biol. Pharm. Bull. 2007;30:698–701. doi: 10.1248/bpb.30.698. [DOI] [PubMed] [Google Scholar]; b Paris D, Quadros A, Humphrey J, Patel N, Crescentini R, Crawford F, Mullan M. Nilvadipine antagonizes both Aβ vasoactivity in isolated arteries, and the reduced cerebral blood flow in APPsw transgenic mice. Brain Res. 2004;999:53–61. doi: 10.1016/j.brainres.2003.11.061. [DOI] [PubMed] [Google Scholar]
- 143.Zhang L-H, Wu L, Raymon HK, Chen RS, Corral L, Shirley MA, Narla RK, Gamez J, Muller GW, Stirling DI, Bartlett JB, Schafer PH, Payvandi F. The Synthetic Compound CC-5079 Is a Potent Inhibitor of Tubulin Polymerization and Tumor Necrosis Factor-α Production with Antitumor Activity. Cancer Res. 2006;66:951–959. doi: 10.1158/0008-5472.CAN-05-2083. [DOI] [PubMed] [Google Scholar]
- 144.Fang Z, Song Y, Sarkar T, Hamel E, Fogler WE, Agoston GE, Fanwick PE, Cushman M. Stereoselective Synthesis of 3,3-Diarylacrylonitriles as Tubulin Polymerization Inhibitors. J. Org. Chem. 2008;73:4241–4244. doi: 10.1021/jo800428b. [DOI] [PubMed] [Google Scholar]
- 145.a Nieminen MS, Pollesello P, Vajda G, Papp Z. Effects of Levosimendan on the Energy Balance: Preclinical and Clinical Evidence. J. Cardiovasc. Pharmacol. 2009;53:302–310. doi: 10.1097/FJC.0b013e31819c9a17. [DOI] [PubMed] [Google Scholar]; b Gupta S, Garg S, Whig J. Levosimendan. J. Anaesth. Clin. Pharmacol. 2009;25:79–82. [Google Scholar]
- 146.Sasho S, Obase H, Ichikawa S, Kitazawa T, Nonaka H, Yoshizaki R, Ishii A, Shuto K. Synthesis of 2-imidazolidinylidene propanedinitrile derivatives as stimulators of gastrointestinal motility. 1. J. Med. Chem. 1993;36:572–579. [Google Scholar]
- 147.a King BF. Novel P2X7 receptor antagonists ease the pain. Br. J. Pharmacol. 2007;151:565–567. doi: 10.1038/sj.bjp.0707266. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen B-C, Quinlan SL, Reid JG, Jass PA, Robinson TP, Early WA, Delaney EJ, Humora MJ, Madding GD, Venit JJ, Winter WJ. Regio- and stereoselective ring opening of epoxide with cyanoguanidine dianions, a facile synthesis of the KATP opener BMS-180448. Tetrahedron Asym. 1998;9:1337–1340. [Google Scholar]; c Toombs CF, Shebuski RJ. U-89232 [BMS-189365], a novel antiischemic agent derived from cromakalim. Cardiovasc. Drug Rev. 1994;12:303–316. [Google Scholar]
- 148.Donetti A, Cereda E, Bellora E, Gallazzi A, Bazzano C, Vanoni P, Del Soldato P, Micheletti R, Pagani F, Giachetti A. (Imidazolylphenyl)formamidines. A structurally novel class of potent histamine H2 receptor antagonists. J. Med. Chem. 1984;27:380–386. doi: 10.1021/jm00369a025. [DOI] [PubMed] [Google Scholar]
- 149.Haaksma EEJ, Rademaker B, Kramer K, Eriks JC, Bast A, Timmerman H. Studies on the active molecular species of the H2-receptor antagonists cimetidine and mifentidine. J. Med. Chem. 1987;30:208–211. doi: 10.1021/jm00384a036. [DOI] [PubMed] [Google Scholar]
- 150.For leading references see: Makowski M, Raczynska ED, Chmurzynski L. Ab Initio Study of Possible and Preferred Basic Site(s) in Polyfunctional N1,N1-Dimethyl-N2-cyanoformamidine. J. Phys. Chem. A. 2001;105:869–874.
- 151.Yanagisawa I, Hirata Y, Ishii Y. Histamine H2 receptor antagonists. 1. Synthesis of N-cyano and N-carbamoyl amidine derivatives and their biological activities. J. Med. Chem. 1984;27:849–857. doi: 10.1021/jm00373a007. [DOI] [PubMed] [Google Scholar]
- 152.Eda M, Takemoto T, Ono S, Okada T, Kosaka K, Gohda M, Matzno S, Nakamura N, Fukaya C. Novel Potassium-Channel Openers: Preparation and Pharmacological Evaluation of Racemic and Optically Active N-(6-Amino-3-pyridyl)-N'-bicycloalkyl-N”-cyanoguanidine Derivatives. J. Med. Chem. 1994;37:1983–1990. doi: 10.1021/jm00039a011. [DOI] [PubMed] [Google Scholar]
- 153.Takemoto T, Eda M, Okada T, Sakashita H, Matzno S, Gohda M, Ebisu H, Nakamura N, Fukaya C, Hihara C, Eiraku M, Yamanouchi K, Yokoyama K. Novel potassium channel openers: synthesis and pharmacological evaluation of new N-(substituted-3-pyridyl)-N'-alkylthioureas and related compounds. J. Med. Chem. 1994;37:18–35. doi: 10.1021/jm00027a003. [DOI] [PubMed] [Google Scholar]
- 154.Nakajima T, Kashiwabara T, Izawa T, Nakajima S. Structure-activity studies of N-cyano-3-pyridinecarboxamidines and their amide and thioamide congeners. Bioorg. Med. Chem. Lett. 1994;4:2485–2488. [Google Scholar]
- 155.Ganellin CR. Discovery of the antiulcer drug Tagamet. Drug Discovery and Development. 2006;1:295–311. [Google Scholar]
- 156.Scheinfeld N. Cimetidine: a review of the recent developments and reports in cutaneous medicine. Dermatology. 2003;9(2):4. [PubMed] [Google Scholar]
- 157.Lefranc F, Yeaton P, Brotchi J, Kiss R. Cimetidine, an unexpected anti-tumor agent, and its potential for the treatment of glioblastoma (review). Int. J. Oncol. 2006;28:1021–1030. [PubMed] [Google Scholar]
- 158.Friedel HA, Brogden RN. Pinacidil. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the treatment of hypertension. Drugs. 1990;39:929–967. doi: 10.2165/00003495-199039060-00008. [DOI] [PubMed] [Google Scholar]
- 159.a Guth BD, Narjes H, Schubert H-D, Tanswell P, Riedel A, Nehmiz G. Pharmacokinetics and pharmacodynamics of terbogrel, a combined thromboxane A2 receptor and synthase inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 2004;58:40–51. doi: 10.1111/j.1365-2125.2004.02083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Soyka R, Guth BD, Weisenberger HM, Luger P, Mueller TH. Guanidine Derivatives as Combined Thromboxane A2 Receptor Antagonists and Synthase Inhibitors. J. Med. Chem. 1999;42:1235–1249. doi: 10.1021/jm9707941. [DOI] [PubMed] [Google Scholar]
- 160.a Olesen UH, Christensen MK, Bjoerkling F, Jaeaettelae M, Jensen PB, Sehested M, Nielsen SJ. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 2008;367:799–804. doi: 10.1016/j.bbrc.2008.01.019. [DOI] [PubMed] [Google Scholar]; b Johanson V, Arvidsson Y, Koelby L, Bernhardt P, Swaerd C, Nilsson O, Ahlman H. Antitumoural Effects of the Pyridyl Cyanoguanidine CHS 828 on Three Different Types of Neuroendocrine Tumours Xenografted to Nude Mice. Neuroendocrinology. 2005;82:171–176. doi: 10.1159/000091754. [DOI] [PubMed] [Google Scholar]
- 161.Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, Bernier C, Branchaud S, Chan H, Dairi K, Gilbert K, Goulet D, Gratton M-O, Isakau H, Jang A, Khadir A, Koch E, Lavoie M, Lawless M, Nguyen M, Paquette D, Turcotte E, Berger A, Mitchell M, Shore GC, Beauparlant P. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell Biol. 2009;29:5872–5888. doi: 10.1128/MCB.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.a Choi A, Choi J-S, Yoon Y-J, Kim K-A, Joo CK. KR-31378, a potassium-channel opener, induces the protection of retinal ganglion cells in rat retinal ischemic models. J. Pharmacol. Sci. 2009;109:511–517. doi: 10.1254/jphs.fp0072067. [DOI] [PubMed] [Google Scholar]; b Jeong TC, Ji-Young J, Young JH, Ha LD, Sun-Ok K, Hong L, Eun YS, Suk LH. Effects of a new neuroprotective agent KR-31378 on liver cytochrome P450s in male Sprague Dawley rats. Arch. Pharm. Res. 2003;26:800–804. doi: 10.1007/BF02980023. [DOI] [PubMed] [Google Scholar]
- 163.a Khun B, Hennig M, Mattei P. Molecular recognition of ligands in dipeptidyl peptidase IV. Curr. Top. Med. Chem. 2007;7:609–619. doi: 10.2174/156802607780091064. [DOI] [PubMed] [Google Scholar]; a Peters J-U. 11 years of cyanopyrrolidines as DPP-IV inhibitors. Curr. Top. Med. Chem. 2007;7:579–595. doi: 10.2174/156802607780091000. [DOI] [PubMed] [Google Scholar]; b Qiao L, Baumann CA, Crysler CS, Ninan NS, Abad MC, Spurlino JC, DesJarlais RL, Kervinen J, Neeper MP, Bayoumy SS, Williams R, Deckman IC, Dasgupta M, Reed RL, Huebert ND, Tomczuk BE, Moriarty KJ. Discovery, SAR, and X-ray structure of novel biaryl-based dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett. 2006;16:123–128. doi: 10.1016/j.bmcl.2005.09.037. [DOI] [PubMed] [Google Scholar]; c Hughes TE, Mone MD, Russell ME, Weldon SC, Villhauer EB. NVP-DPP728 (1-[[[2-[(5-Cyanopyridin-2-yl)amino]ethyl]amino]acetyl]-2-cyano-(S)-pyrrolidine), a Slow-Binding Inhibitor of Dipeptidyl Peptidase IV. Biochemistry. 1999;38:11597–11603. doi: 10.1021/bi990852f. [DOI] [PubMed] [Google Scholar]
- 164.a Dhillon S, Weber J. Saxagliptin. Drugs. 2009;69:2103–2114. doi: 10.2165/11201170-000000000-00000. [DOI] [PubMed] [Google Scholar]; b Deacon CF, Holst JJ. Saxagliptin: a new dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes. Adv. Ther. 2009;26:488–499. doi: 10.1007/s12325-009-0030-9. [DOI] [PubMed] [Google Scholar]
- 165.a Johnson JT, Golden KL, Braceras R. An update of recent trials with vildagliptin, a dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes. Journal of Pharmacy Technology. 2009;25:235–243. [Google Scholar]; b Mathieu C, Degrande E. Vildagliptin: a new oral treatment for type 2 diabetes mellitus. Vascular Health and Risk Management. 2008;4:1349–1360. doi: 10.2147/vhrm.s3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Willand N, Joossens J, Gesquiere J-C, Tartar AL, Evans DM, Roe MB. Solid and solution phase syntheses of the 2-cyanopyrrolidide DPP-IV inhibitor NVP-DPP728. Tetrahedron. 2002;58:5741–5746. [Google Scholar]
- 167.Babine RE, Bender SL. Molecular Recognition of Protein-Ligand Complexes: Applications to Drug Design. Chem. Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 168.Loeser R, Schilling K, Dimmig E, Guetschow M. Interaction of Papain-like Cysteine Proteases with Dipeptide-Derived Nitriles. J. Med. Chem. 2005;48:7688–7707. doi: 10.1021/jm050686b. [DOI] [PubMed] [Google Scholar]
- 169.Metzler WJ, Yanchunas J, Weigelt C, Kish K, Klei HE, Xie D, Zhang Y, Corbett M, Tamura JK, He B, Hamann LG, Kirby MS, Marcinkeviciene J. Involvement of DPP-IV catalytic residues in enzyme-saxagliptin complex formation. Protein Sci. 2008;17:240–250. doi: 10.1110/ps.073253208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li CS, Deschenes D, Desmarais S, Falgueyret J-P, Gauthier JY, Kimmel DB, Leger S, Masse F, McGrath ME, McKay DJ, Percivala MD, Riendeaua D, Rodan SB, Thériena M, Truong VL, Wesolowski G, Zamboni R, Black WC. Identification of a potent and selective non-basic cathepsin K inhibitor. Bioorg. Med. Chem. Lett. 2006;16:1985–1989. doi: 10.1016/j.bmcl.2005.12.071. [DOI] [PubMed] [Google Scholar]
- 171.Gauthier JY, Chauret N, Cromlish W, Desmarais S, Duong Le T., Falgueyret J-P, Kimmel DB, Lamontagne S, Leger S, LeRiche T, Lia CS, Masséa F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmerc JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Thérien M, Truong VL, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008;18:923–928. doi: 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- 172.Opar A. Late-stage osteoporosis drugs illustrate challenges in the field. Nat. Rev. Drug Discov. 2009;8:757–759. doi: 10.1038/nrd3015. [DOI] [PubMed] [Google Scholar]
- 173.Frizler M, Stirnberg M, Sisay MT, Guetschow M. Development of nitrile-based peptidic inhibitors of cysteine cathepsins. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2010;10:294–322. doi: 10.2174/156802610790725452. [DOI] [PubMed] [Google Scholar]
- 174.Ward YD, Thomson DS, Frye LL, Cywin CL, Morwick T, Emmanuel MJ, Zindell R, McNeil D, Bekkali Y, Giradot M, Hrapchak M, DeTuri M, Crane K, White D, Pav S, Wang Y, Hao M-H, Grygon CA, Labadia ME, Freeman DM, Davidson W, Hopkins JL, Brown ML, Spero DM. Design and synthesis of dipeptide nitriles as reversible and potent cathepsin S inhibitors. J. Med. Chem. 2002;45:5471–5482. doi: 10.1021/jm020209i. [DOI] [PubMed] [Google Scholar]
- 175.Wang Y, Duraiswami C, Madauss KP, Tran TB, Williams SP, Deng S-J, Graybill TL, Hammond M, Jones DG, Grygielko ET, Bray JD, Thompson SK. 2-Amino-9-aryl-3-cyano-4-methyl-7-oxo-6,7,8,9-tetrahydropyrido[2',3':4,5]thieno[2,3-b]pyridine derivatives as selective progesterone receptor agonists. Bioorg. Med. Chem. Lett. 2009;19:4916–4919. doi: 10.1016/j.bmcl.2009.07.100. [DOI] [PubMed] [Google Scholar]
- 176.Chen ZX, Ma WY, Zhang CN. Stereoselective synthesis and anticancer activities of new podophyllotoxin derivatives: 4-β-cyano-4-deoxy-4'-demethylepipodophyllotoxin and 4-β-carboxyl-4-deoxy-4'-demethylepipodophyllotoxin. Chin. Chem. Lett. 2000;11:505–508. [Google Scholar]
- 177.Nogrady T, Weaver DF. Medicinal Chemistry: a Molecular and Biochemical Approach. Oxford university Press; US: 2005. p. 22. [Google Scholar]
- 178.ADME is an common acronym describing the pharamacokinetics and pharmacology of an active pharmaceutical ingredient within an organism: absorption, distribution, metabolism, and excretion.
- 179.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 180.a Keserü GM, Makara GM. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery. 2009;8:203–212. doi: 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]; b Oprea TI, Davis AM, Teague SJ, Leeson PDJ. Is There a Difference between Leads and Drugs? A Historical Perspective. Chem. Inf. Comput. Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 181.LogP is the ratio of an unionized drug distributed between octanol and water phases at equilibrium. High logP ratios correlate with increased lipophilicity.
- 182.LogD is the 1-octanol-water coefficient at different pH values.
- 183.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Disc. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]