

Abstract
Plexins are transmembrane high-affinity receptors for semaphorins, regulating cell guidance, motility and invasion. Functional evidences implicate semaphorin signals in cancer progression and metastasis. Yet, it is largely unknown whether plexin genes are genetically altered in human tumors. We performed a comprehensive gene copy analysis and mutational profiling of all nine members of the plexin gene family (plexinome), in melanomas and pancreatic ductal adenocarcinomas (PDACs), which are characterized by high metastatic potential and poor prognosis. Gene copy analysis detected amplification of PLXNA4 in melanomas, while copy number losses of multiple plexin genes were seen in PDACs. Somatic mutations were detected in PLXNA4, PLXNB3 and PLXNC1; providing the first evidence that these plexins are mutated in human cancer. Functional assays in cellular models revealed that some of these missense mutations result in loss of plexin function. For instance, c.1613G>A, p.R538H mutation in the extracellular domain of PLXNB3 prevented binding of the ligand Sema5A. Moreover, while PLXNA4 signaling can inhibit tumor cell migration, the mutated c.5206C>T, p.H1736Y allele had lost this activity. Our study is the first systematic analysis of the “plexinome” in human tumors, and indicates that multiple mutated plexins may be involved in cancer progression.
Keywords: PLXNA4, semaphorin, cancer, melanoma, pancreas
INTRODUCTION
Plexins are a family of transmembrane receptors (Tamagnone and Comoglio, 2000), consisting of nine members, divided into four subfamilies, A thru D (PLXNA1, A2, A3, A4; B1, B2, B3; C1; D1). In the human genome, the corresponding genes are located on chromosomes 1 (PLXNA2), 3 (PLXNs A1, B1, and D1), 7 (PLXNA4), 12 (PLXNC1), 22 (PLXNB2), and X (PLXNs A3 and B3) (see Supp. Table S1). Plexins are a part of the semaphorin gene superfamily, which includes the semaphorins and the receptor tyrosine kinases MET and RON. We have previously shown that plexins function as high-affinity receptors for semaphorins, either alone or in complex with the neuropilins (Tamagnone et al., 1999). Semaphorins are a large family of molecular signals controlling cell migration, axon guidance and the immune response (for a recent review, see (Zhou et al., 2008)).
Several studies show the involvement of semaphorins and neuropilins in cancer, either as putative onco-suppressor genes or as mediators of tumor invasion and metastasis (Tomizawa et al., 2001; Bielenberg et al., 2004; Christensen et al., 2005; Basile et al., 2006; Catalano et al., 2006; for a review, see Neufeld and Kessler, 2008). An additional link between plexins and human tumors is provided by their ability to associate and functionally activate tyrosine kinase receptors, such as MET, RON, HER2, and KDR (Giordano et al., 2002; Conrotto et al., 2004; Toyofuku et al., 2004; Swiercz et al., 2008). These data suggest that plexins might play a role in the invasive and metastatic potential of cancer cells. Scattered reports have described the expression of individual plexins in human tumors (Rieger et al., 2003; Roodink et al., 2005; Rody et al., 2007; Wong et al., 2007). On the other hand, whether and to what extent plexin genes may be genetically altered in cancer has been analyzed in few studies, limited to selected family members or tumour types (Wong et al., 2007; Jones et al., 2008). The availability of a reference human genome sequence coupled with advances in high-throughput DNA analysis has recently opened up new strategies for cancer gene identification. Thence, we have previously used post-genomic approaches to systematically analyze entire gene families in human cancer (Bardelli et al., 2003). Here we undertook a comprehensive genomic analysis of the plexin gene family (which we define as “plexinome”) in two cancer types – melanoma and pancreatic ductal adenocarcinomas (PDAC) - displaying high invasive metastatic potential. Metastatic melanoma has a poor prognosis with a median survival of 6–9 months (Balch et al., 2001). PDACs have a propensity for early metastasis and are highly aggressive and resistant to conventional and targeted therapeutic agents, resulting in a dismal 5-year survival rate of 3-5% (Hezel et al., 2006). Using a systematic genetic profiling approach, we detected gene copy number variations and somatic mutations in plexin genes in melanoma and PDACs. We experimentally verified that a subset of the missense mutations described here result in loss of plexin functions. Our study is the first comprehensive molecular profile of the human plexin gene family in cancer and provides evidence of the involvement of different plexins in melanoma and pancreatic cancers.
MATERIALS AND METHODS
Samples and Cell Lines
Twenty-four human melanoma tumors and matched normals (Supp. Table S2) were obtained from the tumor bank maintained by Department of Experimental Oncology, Istituto Nazionale Tumori, Milan, Italy. Twelve human PDAC samples and respective normals (Supp. Table S2) were obtained from the Department of Pathology, Section of Anatomic Pathology, University of Verona, Italy. Genomic DNA was isolated as previously described (Miller et al., 1988; Balakrishnan et al., 2007). For PLXNA4 copy number analysis, we also analyzed a panel of tumor cell lines from ATCC and our batches (Supp. Table S3), as well as 34 lung primary tumors (from our tumor bank). DNA of glioblastoma multiforme samples was obtained from the Departments of Neurosurgery and Neuropathology at the Academic Medical Center (Amsterdam, The Netherlands). DNA of breast, lung and prostate carcinomas was obtained from the Clinical Research Center, Center of Excellence on Aging at the University-Foundation (Chieti, Italy). Matching of tumor samples containing mutations with their respective normals was verified by direct sequencing of 25 single nucleotide polymorphism (SNP) at 23 loci (data not shown).
Sequencing
Exon-specific PCR and sequencing primers were designed using Primer 3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi/) and synthesized by Invitrogen™, Life Technologies, Inc., Paisley, England) (Supp. Table S4). PCR primers were designed to amplify exons and the flanking intronic sequences (including splicing donor and acceptor regions). PCR amplicons were designed to be approximately 400 bp in length, with multiple overlapping amplimers for larger exons. The procedure to amplify individual exons and to detect somatic mutations by sequencing has been previously reported by our group (Balakrishnan et al., 2007). To warrant reliable and reproducible results, we analyzed in each reaction 10 ng of genomic DNA (in a 10 μl reaction). Lower amounts of DNA derived from dissection of paraffin-embedded samples of prostate carcinoma (2-5 ng per reaction) were also subjected to direct sequencing, resulting in detection of artefactual mutations, which were not confirmed when performing the reaction with higher DNA inputs. As a threshold for sequence quality control, the phred value of 22 was used throughout the study. The reference database for gene information, primary DNA sequences and reported SNPs was Ensembl (http://www.ensembl.org). The position of nucleotide mutations corresponds to that in the coding sequence of each gene, where position 1 is the A of ATG initiation codon. The GenBank reference sequence and version number for the genes studied are: PLXNA1 (NM_032242.2), PLXNA2 (NM_025179.3), PLXNA3 (NM_017514.3), PLXNA4 (NM_020911.1), PLXNB1 (NM_002673.3), PLXNB2 (NM_012401.2), PLXNB3 (NM_005393.1), PLXNC1 (NM_005761.1) and PLXND1 (NM_015103.2).
Copy number analysis
Quantitative real-time TaqMan PCR was performed to determine copy number of all the plexins in melanoma and PDAC tumors (details provided in Supp. Methods). The average plexin copy number was calculated from the differences in the threshold amplification cycles between PLXN and RNaseP. The diploid retinal pigmental epithelial cell line (RPE) was used as a normal control.
Molecular threading and structure predictions
The PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) was used to carry out structure based sequence alignments (mGenTHREADER). The potential impact of amino acid mutations was evaluated in the corresponding positions of the homologous protein structures identified from the PDB protein structure database (http://www.pdb.org/).
Ectopic expression in mammalian cells and Functional assays
cDNAs encoding human plexins were modified by site directed mutagenesis (Quickchange II XL kit, Stratagene) to introduce the nucleotide mutations identified in human tumors (further details in Supp. Methods). Ectopic expression of wild type or mutated plexins in COS and MDA-MB435 cells was achieved by DNA transfection. Semaphorin binding, cellular collapse and cell migration assays were performed as previously described (Tamagnone et al., 1999; Artigiani et al., 2004; Barberis et al., 2004); see Supp. Methods for details.
RESULTS
Gene Copy Number analysis of the Plexinome in human tumors
Oncogene activation in cancer is often a consequence of gene amplification, while inactivation of tumor suppressor genes is frequently due to either hemizygous deletion associated with mutation, or homozygous deletion. Thus, the identification of changes in gene copy number is a powerful tool for cancer genes discovery. In this study, we determined gene copy numbers of the entire plexin gene family (Supp. Table S1) in a panel of 24 melanomas and 12 PDAC samples for which the corresponding matched normals and clinical information were available (Supp. Table S2). For most genes minor or moderate copy number alterations were observed both in melanoma and in PDACs (Figure 1, A-C, and additional melanoma data in Supp. Figure S1). PLXNB1 and PLXNC1 displayed a general tendency towards gene copy losses, both in melanomas and PDACs. Due to the unavailability of appropriate sample material we were unable to perform FISH analysis to confirm copy number changes. The most striking observation was a many fold increase of PLXNA4 copy numbers in several melanoma samples (Figure 1, A and Supp. Figure S1). Thirteen of the 26 melanomas analyzed showed significant gains, and three had borderline gain values at this locus. PLXNA4 gene copy numbers gains varied from two to more than 11 folds. Interestingly, none of the melanomas showed PLXNA4 gene copies lower than normal, while this was often seen in PDACs. To determine whether PLXNA4 copy number may be significantly altered in other tumor types, an assorted panel of 75 tumor samples and cell lines was additionally analysed. Results indicated that lung primary tumors in addition to melanomas have increased PLXNA4 copy number (data not shown).
Figure 1. Plexin gene copy numbers in melanomas and PDACs.
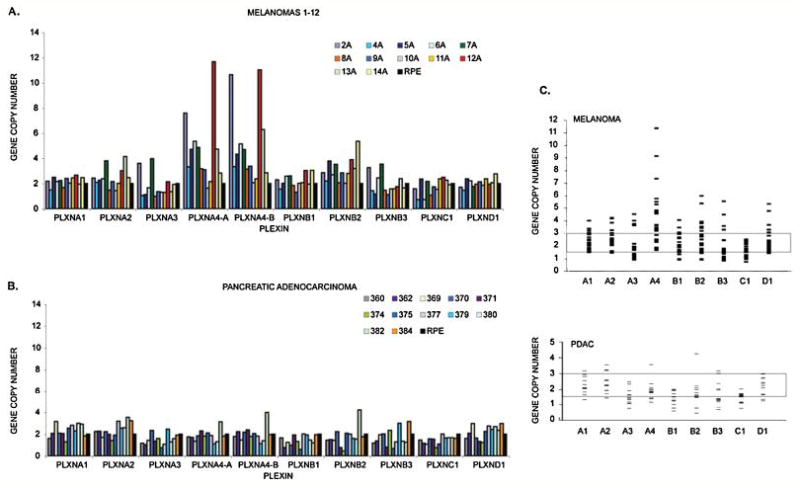
We performed Quantitative Real Time PCR with genomic DNA derived from twenty-four melanomas and twelve PDACs, using plexin-specific probes. The figure shows: A, copy number results for the first 12 melanomas (results for the next 12 melanomas are reported in detail in Supp. Figure S1) and B, the twelve PDACs. The nine plexins are indicated on the X-axis (A1 through D1). The Y-axis shows copy numbers after normalization with control DNA (RPE cells). Panel C summarizes the results of copy number analysis of the plexin gene family. Each small bar represents an individual case. Gene copy numbers below 1.5 and above 3 (highlighted range) are normally considered as aberrant.
In order to determine the outermost limits of the PLXNA4 containing amplicon observed in melanomas, the genomic DNAs were subjected to further analysis. In particular, we focused on PTPRZ1 and TRPV5, two genes located 10Mb upstream and 10Mb downstream of PLXNA4 locus, respectively. The BRAF oncogene, which is known to be frequently mutated in melanomas and is located about 8Mb downstream of the PLXNA4 locus, was also included in the analysis. Copy numbers of the PTPRZ1, PLXNA4, BRAF and TRPV5 genes were determined in the same samples in which we found PLXNA4 amplification (Supp. Figure S2). Copy numbers of PLXNA4 and TRPV5 varied concordantly in all the different samples. However, we noticed that alterations of PLXNA4 copy numbers did not always correlate with those of PTPRZ1, indicating that the latter may lie outside the limits of the amplicon. On this basis, it may be speculated that the implicated amplicon on 7q starts between PTPRZ1 and PLXNA4 and extends beyond TRPV5. Amplification of the BRAF gene was also often observed in melanomas samples.
Sequence analysis of the Plexinome in human PDAC and melanoma
The mutational status of the plexin gene family in melanoma and PDAC is presently unknown. We performed a systematic mutational profiling of the whole plexin gene family in the same panel of melanomas and PDAC samples subjected to gene copy number analysis. Exon specific primers were designed to amplify and sequence the entire coding region, and at least 15 intronic bases at both the 5’ and 3’ ends, including the splicing donor and acceptor sites (Supp. Table S4). Of the 269 exons amplified, 93% could be successfully sequenced.
A total of 11448 PCR products, spanning 5.09 Mb of tumour genomic DNA, were generated, of which 4.36 Mb were subjected to direct sequencing. Sequence data for each amplicon was evaluated for quality within the target region. Over 2300 nucleotide changes were identified during the initial screening. These included both synonymous (silent) and non-synonymous alterations, leading to amino acid changes. Changes previously reported as SNPs in public databases (see Footnote 1) were excluded from further analysis. To rule out PCR or sequencing artefacts, amplicons were independently re-amplified and resequenced in the corresponding tumors. All verified changes were re-sequenced in parallel with their matched normal DNA to distinguish between somatic mutations and SNPs not previously described. This led to the identification of seven novel somatic mutations in three plexins: six of them led to amino acid substitutions in the coding sequence (Table 1), while one was found in an intron-exon boundary sequence containing a splice donor site. Three missense somatic mutations were found in PLXNA4 in melanoma samples (c.5206C>T, p.H1736Y, shown in Figure 2; and c.1920G>C, p.K640N; c.3460G>A, p.E1154K, shown in Supp. Figure S3). A missense mutation was found in PLXNB3 (c.1613G>A, p.R538H, see Figure 2) and two missense mutations were found in PLXNC1 (c.1475A>T, p.N492I and c.2554G>A, p.E852K, Supp. Figure S4, A and B) in three different PDAC samples. A PLXNC1 mutation was furthermore detected in the intronic sequence following exon 2 in one melanoma sample (IVS2+1G>T, Supp. Figure S4, C). In silico analysis (with splice site predictor programs such as NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html)) revealed that this genetic alteration may affect mRNA splicing by loss of a potential donor site. All seven mutations were ruled out as previously reported changes or as SNPs (Riva and Kohane, 2004; and Footnote 1) and confirmed as being somatic by sequencing the matched normal genomic DNA.
Table 1.
Plexin mutations found in melanoma and PDAC
| GENE† | OMIM | NUCLEOTIDE | AMINO ACID | ZYGOSITY | PUTATIVE LOCATION IN PROTEIN | SAMPLE |
|---|---|---|---|---|---|---|
| PLXNA4 | 604280 | c.1920G>C | p.K640N | heterozygous | Sema domain | 11A (melanoma) |
| c.3460G>A | p.E1154K | heterozygous | Extracellular, Interdomain between PSI domains 2 and 3 | 16A (melanoma) | ||
| c.5206C>T | p.H1736Y | heterozygous | Cytoplasmic domain | 14A (melanoma) | ||
| PLXNB3 | 300214 | c.1613G>A | p. R538H | heterozygous | Extracellular, Interdomain between PSI domains 1 and 2 | 384 (PDAC) |
| PLXNC1 | 604259 | c.1204+1G>T | splice var. | heterozygous | Intronic, potentially affects splicing* | 18A* (melanoma) |
| c.1475A>T | p.N492I | heterozygous | Extracellular, PSI domain 1 | 370 (PDAC) | ||
| c.2554G>A | p.E852K | heterozygous | Extracellular, second IPT domain | 375 (PDAC) |
List of the seven mutations found in the coding sequence of PLXNA4, PLXNB3 and PLXNC1. The position of nucleotide mutations corresponds to that in the coding sequence of each gene, where position 1 is the A of ATG initiation codon. Zygosity of the mutations is shown.
PLXNC1 was also mutated in the splice donor site flanking exon 2 in melanoma sample 18A (c.1204+1G>T).
GenBank accession numbers for plexins with mutations are: PLXNA4 (NM_020911.1), PLXNB3 (NM_005393.1) and PLXNC1 (NM_005761.1).
Figure 2. Somatic mutations of plexin genes in melanoma and PDAC.
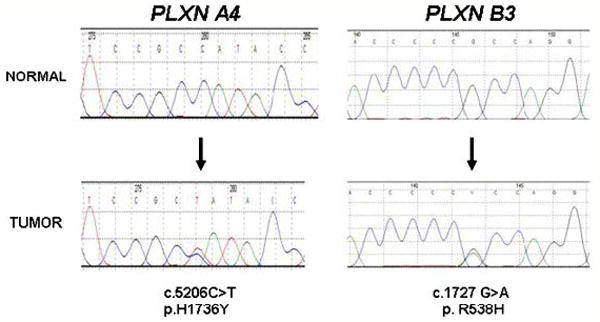
Chromatograms of two of the seven somatic mutations found in plexin genes in different melanomas and PDAC (other mutations are shown in Supp. Figure S3 and S4). PLXNA4 p.H1736Y mutation was found in melanoma sample 14A, while PLXNB3 p.R538H was identified in PDAC sample 384. In both cases, the lower chromatogram is from the tumor sample, and the upper chromatogram is from the corresponding normal. Arrows indicate the location of missense somatic mutations, and the nucleotide and amino acid alterations are indicated below the traces. Numbers above the sequences are part of the software output.
Mutational analysis of PLXNB1 in solid tumors
Wong et al. have recently reported that the intracellular domain of PLXNB1 is mutated at very high frequency (89%) in prostate cancers (Wong et al., 2007), indicating that this gene may play a major role in the progression of this cancer lineage. According to our results and those of Jones’ (Jones et al., 2008), PLXNB1 is not frequently mutated in melanoma and PDAC. When considered with the data of Wong and colleagues this suggests that the mutational profile of PLXNB1 may be tumor specific. To test this hypothesis we sequenced exons 23 and 27 of PLXNB1 (where most mutations had been detected) in a panel of common solid tumors including 15 primary prostate, 83 lung and 120 breast carcinomas and 120 glioblastoma multiforme samples. None of these displayed nucleotide changes in PLXNB1. The discrepancy between our results and those reported by Wong in primary prostate tumors is highly significant (Fisher’s exact test, p-value < 0.001). In an attempt to explain this inconsistency, we noted that 99% (79/80) of the changes reported by Wong were C>T/G>A or A>G/T>C and that multiple mutations of PLXNB1 were found in the same samples. Previous work has demonstrated that PCR errors due to deamination of cytosine or adenine occur frequently in low concentrated DNA, especially with DNA extracted from paraffin embedded tissue (Marchetti et al, 2006; Hofreiter et al., 2001; Akbari et al., 2005; Williams et al., 1999). To elucidate this issue, we repeated the PCR and sequencing approach using lower amounts of DNA extracted from prostate tumours as compared to our initial analysis. Under these conditions we detected A>G/T>C nucleotide changes potentially resulting in a number of non-synonymous amino acid changes (p.L1547F, p.T1750A, p.V1767A, p.V1769A, p.L1772P, p.T1802A; see Supp. Figure S5). Notably, one of the observed changes (p.T1802A) is identical to that described in (Wong et al., 2007). These changes, however, could not be confirmed when the samples were analyzed multiple times by PCR amplification and sequencing. Overall, our results indicate that the intracellular domain of PLXNB1 is not frequently mutated in prostate, lung and breast carcinomas and in glioblastoma multiforme.
Phylogenetic conservation and structural relevance of mutated residues
The six novel missense mutations in plexins that we have identified in human cancers are distributed in different domains of the receptors, both extracellular and cytoplasmic (Table 1, Figure 3). Many of them affect amino acid residues conserved throughout evolution and in other human plexin family members. These mutations fall into protein domains homologous to structures that have been solved by x-ray crystallography (McGuffin et al., 2000), thus allowing in silico evaluation of their potential impact on the structure and function of the corresponding receptors.
Figure 3. Schematic representation of plexin domain structure and mutations.
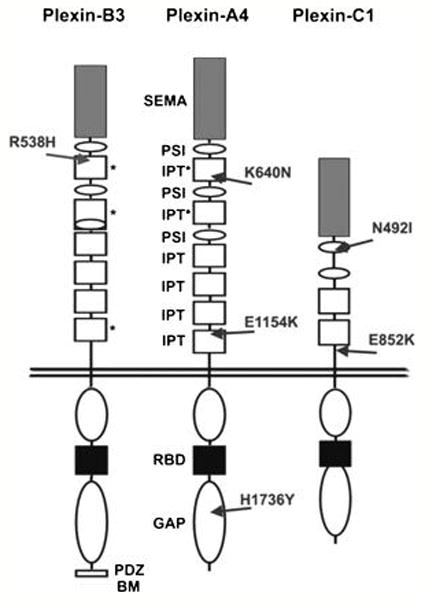
The location of novel missense mutations found in human cancer samples are indicated by arrows. In the extracellular portions, grey boxes with “SEMA” indicate semaphorin domains, white ovals with “PSI” indicate Plexin-Semaphorin-Integrin domains (also known as MRS motifs), and white boxes with “IPT” indicate Integrin-Plexin-Transcription-factor domains. Previously unidentified IPT-like domains described in this work are marked by asterisks. In the cytoplasmic portion of the receptors, white ovals indicate the conserved GAP-like regions, the black boxes indicate the Rac-Rnd GTP-ase Binding Domains (RBD), while the C-terminus of PlexinB3 includes a PDZ-domain Binding Motif.
The PLXNB3 p.R538H mutation is located in the extracellular domain of this receptor, immediately after the first PSI repeat (a protein domain found in Plexins, Semaphorins and Integrins) (Shi et al., 2005). We found that this region, previously described as non-structured, actually bears highly significant structural homology to an ‘Ig-like, Plexins, Transcription factors’ (IPT) domain (pdb id 1ttu, 21% sequence identity, p < 0.002). IPT domains are found in plexins, integrins and transcription factors; however, the presence of IPT domains between PSI motifs of plexins had not been recognized previously. Residue R538 in this new IPT of PLXNB3 is located at the beginning of beta-strand 2, and is conserved in other homologous IPT domains (in plexins and other proteins), suggesting its functional relevance. K640N mutation in PLXNA4 is similarly located between the first and second PSI repeat within a putative IPT domain. The p.E1154K mutation in PLXNA4, maps to the beginning of the last IPT domain (close to the transmembrane domain). It is noteworthy that the Glu to Lys mutation not only reverses the charge of the amino acid, but may also create a glycation site at residue 1154 as predicted by the NetGlycate1.0 server (http://www.cbs.dtu.dk/services/NetGlycate), thereby potentially affecting the conformation of the extracellular domain. The p.H1736Y mutation in PLXNA4 occurs in the cytoplasmic region of the receptor. This residue is conserved in all plexins and is located in the C-terminal domain homologous to Ras-GAP proteins (Rohm et al., 2000) (e.g. neurofibromin, pdb id 1nf1 with sequence identity 11%, p < 10-8). The equivalent position in the homologous crystal structure is at the solvent exposed end of an alpha-helix. This residue lies within 10 residues of the catalytic Arg finger in the GAP domain, but it is probably not part of the binding interface with Ras. The mutation to an uncharged residue in PLXNA4, however, may alter the protein conformation and/or binding affinity towards other protein domains or signaling molecules. Moreover, the mutation to Tyr may create a possible phosphorylation site, which would induce charge reversal, compared to His. Interestingly, the corresponding residue in the homologous domains of SynGAP and R-RasGAP is a tyrosine. The missense mutations found in PlexinC1 affect amino acid residues that do not display high level of homology among various family members: p.N492I is located at the C-terminus of the first conserved PSI repeat; whereas p.E852K, is located in a negatively-charged segment of the polypeptide chain flanking the last IPT domain and features a major shift in polarity, which could affect salt bridges and the secondary structure of this segment.
Functional analysis of mutated plexins
Based on phylogenetic conservation and prediction of potential functional relevance of affected residues, we focused our attention on mutations R538H in the extracellular domain of PLXNB3, and E1154K and H1736Y in the extracellular and intracellular domains of PLXNA4. To establish whether these amino acid changes affect plexin function, we introduced them by site-directed mutagenesis in the corresponding normal cDNA subcloned in mammalian expression constructs. The mutated proteins were then ectopically expressed in COS cells and their functional competence was experimentally tested, using well established assays. The mutated extracellular domains of PlexinB3 and PlexinA4 were challenged in binding assays, by using the specific cognate ligands (Sema5A and Sema6A, respectively) fused to alkaline-phosphatase (AP) for detection, as previously shown (Artigiani et al., 2004). Importantly, although the expression of the mutated receptor was comparable to the normal counterpart, the binding of Sema5A to the extracellular domain of PLXNB3 carrying p.R538H mutation was strikingly impaired (Figure 4A). In addition we found that the ectodomain of PLXNA4 carrying the p.E1154K mutation interacted with its ligand Sema6A with reduced efficiency as compared to the wild type receptor (Supp. Figure S6).
Figure 4. Functional impact of Plexin mutations found in cancers.
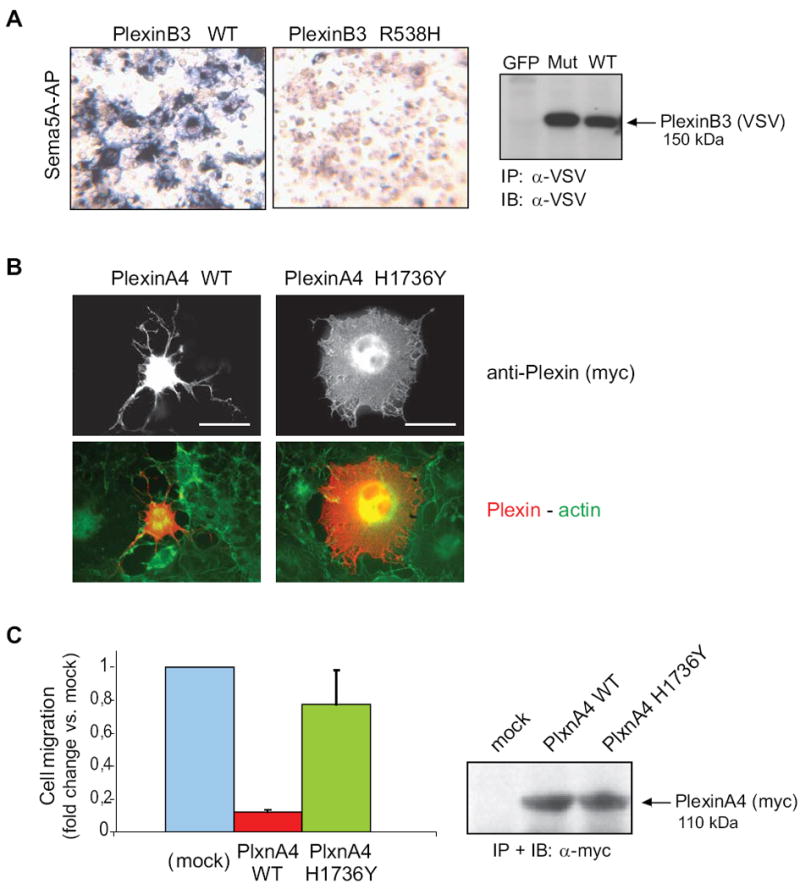
A, the functional impact of mutation p.R538H in the extracellular domain of PLXNB3 was assessed in COS cells (see Supplementary Methods for details) by challenging the receptor with a soluble form of the specific ligand Sema5A fused to alkaline phosphatase (Sema5A-AP), as previously shown (Artigiani et al., 2004). The comparable expression of the receptor proteins in the cells used for binding assays was confirmed by Western blotting (on the right). The results shown are representative of three independent experiments. The mutated extracellular domain of PLXNB3 has lost the ability to interact with Sema5A. B, the collapsing activity of either wild type or p.H1736Y mutated PLXNA4 was tested in COS cells, by expressing a constitutively active form of the receptor (see Methods), followed by immunofluorescence analysis with anti-Myc-tag antibodies (revealing the plexin) and phalloidin-FITC cell counterstaining (see merged images, at the bottom). Scale bar = 30 μm. The comparable expression of the two proteins was confirmed by Western blotting (on the right). Over 60% of the cells expressing wild-type active PlexinA4 displayed a collapsed phenotype (as shown in representative field), while the mutated plexin had lost this ability (yielding less then 10% collapsed cells). The results shown are representative of three independent experiments. C, wild type and p.H1736 mutated PLXNA4 (as in B) were further expressed in MDA-MB435 human melanoma cells, to investigate the impact of the mutation on the ability of plexin signals to inhibit tumor cell migration (see Methods for experimental details). Spontaneous cell migration was significantly inhibited in cells expressing the wild type activated form of PLXNA4, as expected, whereas cells expressing the mutated receptor were almost unaffected, indicating a functional loss. Protein expression levels were comparable (shown on the right).
In order to test the functional competence of mutated intracellular domain of PLXNA4 (p.H1736Y), we generated a ligand-independent constitutively active form of the human receptor (by analogy to that previously reported for PLXNA1 and PLXNB1, by Takahashi and Strittmatter, 2001; Oinuma et al., 2004). While the expression of constitutively activated PLXNA4 led to the expected cellular collapse phenotype, the p.H1736Y mutated form was equally expressed but functionally incompetent (Figure 4B). Notably, p.H1736Y is very close to the conserved residue in the R-RasGAP domain (Arg1746) that is required for catalytic activity and for the collapsing response (Rohm et al., 2000), which could explain the loss-of-function phenotype. It has been shown previously that, in addition to eliciting cellular collapse, plexin signals may inhibit directional migration (Barberis et al., 2004; Swiercz et al., 2008). To further confirm the impact of the H1736Y mutation in tumor cell migration, we compared the function of either wild type or mutated PLXNA4 in MDA-MB435 melanoma cells. Figure 4C shows that the migration of tumor cells expressing wild type activated PLXNA4 is strongly inhibited, while this is not seen in cells expressing the p.H1736Y variant. In sum, we have demonstrated that three of the novel plexin mutations found in human cancers dramatically impair ligand binding and functional activity of the receptors.
DISCUSSION
Members of plexin gene family encode high-affinity receptors for the semaphorins. Plexin signalling has been shown to regulate integrin function, cell migration and cell survival (Kruger et al., 2005; Casazza et al., 2007). A number of studies suggest that these activities involve the regulation of small GTPases, such as RhoA and R-Ras. Several additional mechanisms of action have been proposed, including the plexin-mediated interaction and activation of oncogenic tyrosine kinase receptors (Franco and Tamagnone, 2008). Semaphorin signals elicit a variety of, sometimes antagonistic, functional outcomes in tumor cells. For example, while certain semaphorins have been shown to inhibit tumor growth and angiogenesis, others seem to promote tumor growth or metastasis (Rizzolio and Tamagnone, 2007). These different functional effects are thought to depend on distinctive plexin-based receptor complexes. Importantly, although a number of functional evidences implicate plexins in cancer progression, their molecular alterations in cancer tissues have been investigated in a few studies, limited to selected family members or tumour types (Wong et al., 2007; Jones et al., 2008). Completion of the genome project has now enabled the systematic analysis of genetic alterations in human tumors, thus leading to the identification of candidate cancer genes that may represent new therapeutic targets (Bardelli and Velculescu, 2005; Sjoblom et al., 2006; Wood et al., 2007). We therefore performed a systematic profiling of the entire plexin gene family (mutations and gene copy number) in two deadly cancer types: melanoma and pancreatic ductal adenocarcinomas.
Copy number analysis of plexin genes identified a distinct pattern of gains and losses. Copy numbers ranged from as low as 0.32 (suggesting a complete loss of the region analysed) to as many as 11.67 (indicating at least 11 copies of the amplicon). In PDACs, loss of plexin gene copies was rather frequent, while no significant increase in copy number was seen. Remarkable increases of PLXNA4 gene copy numbers were observed in multiple melanoma samples, but not in PDACs. Assessment of the region involving PLXNA4 gene amplification showed that the amplicon also included BRAF, an oncogene whose mutation and copy number increase are known to occur in melanoma (Stark and Hayward, 2007), suggesting that this gene may be implicated as the driving force for the occurrence of the amplicon in 7q32.3.
The direct sequencing of plexin genes in PDAC and melanomas (which included more than 4.36 Mb of tumor DNA) led to the identification of somatic mutations in three genes: PLXNA4, PLXNB3 and PLXNC1. It was recently reported that the intracellular domain of PLXNB1 is mutated at very high frequency (89%) in prostate cancers (Wong et al., 2007), indicating that this gene may play a major role in the progression of this cancer lineage. Notably, in our study we failed to detect PLXNB1 mutations in a wide panel of solid tumors, including prostate, glioblastoma multiforme, melanoma, PDACs, lung and breast cancers. Moreover, our data indicate that the direct sequencing of low DNA amounts, especially if derived from paraffin-embedded samples that were utilized in the study of Wong and colleagues, can lead to PCR artifacts. Accordingly, we suggest that the mutation frequency of Plexin-B1 in prostate cancer should be reconsidered.
While this work was under completion, Jones and coworkers have reported a genome-wide analysis of genetic alterations in 24 samples of pancreatic carcinoma (Jones et al., 2008). All nine plexin genes were analyzed, but (likely because genomic maps were incomplete at that stage) only 15 out of 35 exons of PLXNA4 gene were sequenced by Jones et al. In that study, two plexin mutations were found in independent PDAC samples: PLXNA1 p.N443Y and PLXNB1 p.A639V. We have not identified the same mutations in our panel of PDACs, which is consistent with the fact that these events are rare. Jones and colleagues do not provide evidence on the potential functional relevance of these mutations. Notably, both changes affect amino acid residues in the extracellular domain of the receptors that are not conserved in the gene family. Altogether, our present study and that of Jones et al. have identified five different mutations in plexin genes in 36 analyzed PDACs: two in PLXNC1, and one each in PLXNA1, PLXNB1 and PLXNB3. This makes a frequency of 14% PDAC cases with mutations in at least one plexin gene. Moreover, based on our present data, four plexin mutations were found in a total of 24 samples analyzed (i.e. mutated plexinome in 17% of cases).
The missense mutations of PLXNA4, PLXNB3 and PLXNC1 that we have identified in melanomas and PDACs were all confirmed by multiple PCR amplification and sequencing. They have not been previously reported and occur in both the extracellular and the cytoplasmic domains of the plexin receptors (Figure 3). Most of the changes affect highly conserved residues located in known structural domains, enabling us to speculate about the possible effects of these mutations on receptor function. Our in silico evaluation showed that the corresponding amino acid residues are expected to be surface exposed, suggesting that these mutations should not impair basic protein folding, while they could affect the intramolecular interaction between plexin domains and/or their affinity for interacting molecules. We therefore hypothesized that either the ligand binding capabilities or the signalling properties of the mutated plexins could be functionally affected. This was experimentally verified by introducing the corresponding nucleotide changes in the cDNA of PLXNA4 and PlexinB3. Cell based assays demonstrated that two of the mutations (i.e. PLXNA4 p.H1736Y and PLXNB3 p.R538H) strongly impair plexin function. In particular, while PLXNA4 signaling can inhibit tumor cell migration, the mutated p.H1736Y allele has lost this activity. Notably, another mutation in PLXNA4 (p.E1154K), identified in an independent sample, also results in loss of function as shown by reduced ligand binding. Considering that plexins are believed to form receptor complexes at the cell surface, these loss-of-function mutations may affect semaphorin signalling even in presence of one normal residual allele. Moreover, the occurrence of second hit, either genetic (such as gene copy losses as we observed) or epigenetic, could lead to a complete abrogation of the corresponding protein function.
A recent systematic analysis of 18,191 well annotated coding sequences in breast and colorectal cancers revealed that the landscape of cancer genomes comprises a handful of commonly mutated gene ‘mountains’ but is dominated by a much larger number of infrequently mutated gene ‘hills’ (Wood et al., 2007). Whether the few mutations that typically occur in ‘hill-type’ cancer genes behave as passengers or drivers with respect to tumor progression is presently the matter of intensive and controversial research. Our data suggest that plexins are mutated at low frequency in melanoma and pancreatic cancers thus behaving like ‘hill type’ cancer genes at least in these tumor types. Notably, we found that at least two of the newly identified mutations in plexins have a functional impact on the corresponding proteins thus suggesting that they act as drivers in cancer progression.
In conclusion, we have completed the first systematic genetic profiling of the plexinome in human tumors leading to the identification of somatic mutations in three plexin genes: PLXNA4, PLXNB3 and PLXNC1. The evidence that the mutated alleles PLXNA4 p.H1736Y and PLXNB3 p.R538H impair the biological function of the corresponding proteins suggests that mutated plexins could play a functional role in cancer progression.
Supplementary Material
Acknowledgments
The authors wish to thank Lorena Capparuccia and all the members of the Laboratory of Molecular Genetics at IRCC, for help and useful suggestions. Drs. Christina Kiel and Luis Serrano (CRG-EMBL Systems Biology Unit, Barcelona) and Mehdi Bagheri Hamaneh (Case Western Res. Univ.) helped with homology modeling of plexin RasGAP domains. We thank Dr S. Leenstra, Dr. T Hulsebos and Professor Troost (Departments of Neurosurgery, Neuropathology and Neurogenetics, Academic Medical Center) for making glioblastoma tumour samples available. Thanks to A. Puschel and H. Fujisawa for semaphorin and plexin cDNA expression constructs. This research was supported by grants of the Italian Association for Cancer Research (AIRC; to A.Bardelli, P.M.C. and L.T.) and of the Regione Piemonte (to A.Bardelli and L.T.). Moreover, A.Bardelli was supported by grants of the Association for International Cancer Research (AICR-UK), EU-FP6 MCSCs contract 037297, EU-FP7 CancerGene contract 218071 and CRT Progetto Alfieri. M.B. is recipient of NIH grant GM73071.
Footnotes
As a reference for previously reported changes and SNPs, we consulted the following public databases: http://www.ensembl.org; http://genome.ucsc.edu/cgi-bin/hgGateway; http://www.sanger.ac.uk/genetics/CGP/cosmic/
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Akbari M, Hansen MD, Halgunset J, Skorpen F, Krokan HE. Low copy number DNA template can render polymerase chain reaction error prone in a sequence-dependent manner. J Mol Diagn. 2005;7:36–39. doi: 10.1016/s1525-1578(10)60006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artigiani S, Conrotto P, Fazzari P, Gilestro GF, Barberis D, Giordano S, Comoglio PM, Tamagnone L. Plexin-B3 is a functional receptor for semaphorin 5A. EMBO Rep. 2004;5:710–714. doi: 10.1038/sj.embor.7400189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67:3545–3550. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, Urist M, McMasters KM, Ross MI, Kirkwood JM, Atkins MB, Thompson JA, Coit DG, Byrd D, Desmond R, Zhang Y, Liu PY, Lyman GH, Morabito A. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol. 2001;19:3622–3634. doi: 10.1200/JCO.2001.19.16.3622. [DOI] [PubMed] [Google Scholar]
- Barberis D, Artigiani S, Casazza A, Corso S, Giordano S, Love CA, Jones EY, Comoglio PM, Tamagnone L. Plexin signaling hampers integrin-based adhesion, leading to Rho-kinase independent cell rounding, and inhibiting lamellipodia extension and cell motility. Faseb J. 2004;18:592–594. doi: 10.1096/fj.03-0957fje. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, Markowitz S, Willson JK, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Velculescu VE. Mutational analysis of gene families in human cancer. Curr Opin Genet Dev. 2005;15:5–12. doi: 10.1016/j.gde.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Basile JR, Castilho RM, Williams VP, Gutkind JS. Semaphorin 4D provides a link between axon guidance processes and tumor-induced angiogenesis. Proc Natl Acad Sci U S A. 2006;103:9017–9022. doi: 10.1073/pnas.0508825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti S, Frattini M, Arena S, Zanon C, Cappelletti V, Coradini D, Daidone MG, Pilotti S, Pierotti MA, Bardelli A. PIK3CA cancer mutations display gender and tissue specificity patterns. Hum Mutat. 2008;29:284–288. doi: 10.1002/humu.20648. [DOI] [PubMed] [Google Scholar]
- Bielenberg DR, Hida Y, Shimizu A, Kaipainen A, Kreuter M, Kim CC, Klagsbrun M. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest. 2004;114:1260–1271. doi: 10.1172/JCI21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Fazzari P, Tamagnone L. Semaphorin signals in cell adhesion and cell migration: functional role and molecular mechanisms. Adv Exp Med Biol. 2007;600:90–108. doi: 10.1007/978-0-387-70956-7_8. [DOI] [PubMed] [Google Scholar]
- Catalano A, Caprari P, Moretti S, Faronato M, Tamagnone L, Procopio A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood. 2006;107:3321–3329. doi: 10.1182/blood-2005-06-2445. [DOI] [PubMed] [Google Scholar]
- Christensen C, Ambartsumian N, Gilestro G, Thomsen B, Comoglio P, Tamagnone L, Guldberg P, Lukanidin E. Proteolytic processing converts the repelling signal Sema3E into an inducer of invasive growth and lung metastasis. Cancer Res. 2005;65:6167–6177. doi: 10.1158/0008-5472.CAN-04-4309. [DOI] [PubMed] [Google Scholar]
- Conrotto P, Corso S, Gamberini S, Comoglio PM, Giordano S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene. 2004;23:5131–5137. doi: 10.1038/sj.onc.1207650. [DOI] [PubMed] [Google Scholar]
- Franco M, Tamagnone L. Tyrosine phosphorylation in semaphorin signalling: shifting into overdrive. EMBO Rep. 2008 doi: 10.1038/embor.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D, Tamagnone L, Comoglio PM. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat Cell Biol. 2002;4:720–724. doi: 10.1038/ncb843. [DOI] [PubMed] [Google Scholar]
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- Hofreiter M, Jaenicke V, Serre D, Haeseler Av A, Pääbo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001;29:4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nat Rev Mol Cell Biol. 2005;6:789–800. doi: 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Marchetti A, Felicioni L, Buttitta F. Assessing EGFR mutations. N Engl J Med. 2006;354:526–528. doi: 10.1056/NEJMc052564. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld G, Kessler O. The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat Rev Cancer. 2008;8:632–645. doi: 10.1038/nrc2404. [DOI] [PubMed] [Google Scholar]
- Oinuma I, Katoh H, Negishi M. Molecular dissection of the semaphorin 4D receptor plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. J Neurosci. 2004;24:11473–11480. doi: 10.1523/JNEUROSCI.3257-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger J, Wick W, Weller M. Human malignant glioma cells express semaphorins and their receptors, neuropilins and plexins. Glia. 2003;42:379–389. doi: 10.1002/glia.10210. [DOI] [PubMed] [Google Scholar]
- Riva A, Kohane IS. A SNP-centric database for the investigation of the human genome. BMC Bioinformatics. 2004;5:33. doi: 10.1186/1471-2105-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolio S, Tamagnone L. Semaphorin signals on the road to cancer invasion and metastasis. Cell Adh Migr (Landes) 2007;1:62–68. doi: 10.4161/cam.1.2.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rody A, Holtrich U, Gaetje R, Gehrmann M, Engels K, von Minckwitz G, Loibl S, Diallo-Danebrock R, Ruckhaberle E, Metzler D, Ahr A, Solbach C, Karn T, Kaufmann M. Poor outcome in estrogen receptor-positive breast cancers predicted by loss of plexin B1. Clin Cancer Res. 2007;13:1115–1122. doi: 10.1158/1078-0432.CCR-06-2433. [DOI] [PubMed] [Google Scholar]
- Rohm B, Rahim B, Kleiber B, Hovatta I, Puschel AW. The semaphorin 3A receptor may directly regulate the activity of small GTPases. FEBS Lett. 2000;486:68–72. doi: 10.1016/s0014-5793(00)02240-7. [DOI] [PubMed] [Google Scholar]
- Roodink I, Raats J, van der Zwaag B, Verrijp K, Kusters B, van Bokhoven H, Linkels M, de Waal RM, Leenders WP. Plexin D1 expression is induced on tumor vasculature and tumor cells: a novel target for diagnosis and therapy? Cancer Res. 2005;65:8317–8323. doi: 10.1158/0008-5472.CAN-04-4366. [DOI] [PubMed] [Google Scholar]
- Shi M, Sundramurthy K, Liu B, Tan SM, Law SK, Lescar J. The crystal structure of the plexin-semaphorin-integrin domain/hybrid domain/I-EGF1 segment from the human integrin beta2 subunit at 1.8-A resolution. J Biol Chem. 2005;280:30586–30593. doi: 10.1074/jbc.M502525200. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Stark M, Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67:2632–2642. doi: 10.1158/0008-5472.CAN-06-4152. [DOI] [PubMed] [Google Scholar]
- Suto F, Ito K, Uemura M, Shimizu M, Shinkawa Y, Sanbo M, Shinoda T, Tsuboi M, Takashima S, Yagi T, Fujisawa H. Plexin-A4 mediates axon-repulsive activities of both secreted and transmembrane semaphorins and plays roles in nerve fiber guidance. J Neurosci. 2005;25:3628–37. doi: 10.1523/JNEUROSCI.4480-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz JM, Worzfeld T, Offermanns S. ErbB-2 and met reciprocally regulate cellular signaling via plexin-B1. J Biol Chem. 2008;283:1893–1901. doi: 10.1074/jbc.M706822200. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Strittmatter SM. PlexinA1 autoinhibition by the plexin sema domain. Neuron. 2001;29:429–439. doi: 10.1016/s0896-6273(01)00216-1. [DOI] [PubMed] [Google Scholar]
- Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Shibata T, Sakiyama T, Yoshida T, Tamura T. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:6829–6837. doi: 10.1200/JCO.2005.01.0793. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol. 2000;10:377–383. doi: 10.1016/s0962-8924(00)01816-x. [DOI] [PubMed] [Google Scholar]
- Tomizawa Y, Sekido Y, Kondo M, Gao B, Yokota J, Roche J, Drabkin H, Lerman MI, Gazdar AF, Minna JD. Inhibition of lung cancer cell growth and induction of apoptosis after reexpression of 3p21.3 candidate tumor suppressor gene SEMA3B. Proc Natl Acad Sci U S A. 2001;98:13954–13959. doi: 10.1073/pnas.231490898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Zhang H, Kumanogoh A, Takegahara N, Suto F, Kamei J, Aoki K, Yabuki M, Hori M, Fujisawa H, Kikutani H. Dual roles of Sema6D in cardiac morphogenesis through region-specific association of its receptor, Plexin-A1, with off-track and vascular endothelial growth factor receptor type 2. Genes Dev. 2004;18:435–447. doi: 10.1101/gad.1167304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C, Pontén F, Moberg C, Söderkvist P, Uhlén M, et al. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol. 1999;155:1467–1471. doi: 10.1016/S0002-9440(10)65461-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong OG, Nitkunan T, Oinuma I, Zhou C, Blanc V, Brown RS, Bott SR, Nariculam J, Box G, Munson P, Constantinou J, Feneley MR, Klocker H, Eccles SA, Negishi M, Freeman A, Masters JR, Williamson M. Plexin-B1 mutations in prostate cancer. Proc Natl Acad Sci U S A. 2007;104:19040–19045. doi: 10.1073/pnas.0702544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gunput RA, Pasterkamp RJ. Semaphorin signaling: progress made and promises ahead. Trends Biochem Sci. 2008;33:161–170. doi: 10.1016/j.tibs.2008.01.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.