

Abstract
The hereditary breast and ovarian cancer predisposition genes, BRCA1 and BRCA2, participate in the repair of DNA double strand breaks by homologous recombination. Circumstantial evidence implicates these genes in recombinational responses to DNA polymerase stalling during the S phase of the cell cycle. These responses play a key role in preventing genomic instability and cancer. Here, we review the current literature implicating the BRCA pathway in HR at stalled replication forks and explore the hypothesis that BRCA1 and BRCA2 participate in the recombinational resolution of single stranded DNA lesions termed “daughter strand gaps”, generated during replication across a damaged DNA template.
Keywords: BRCA1, BRCA2, Rad51, Sister chromatid recombination, Daughter strand gap repair
1. Introduction
Cancer-promoting mutations frequently entail the activation of oncogenes and inactivation of tumor suppressor genes [1,2]. These genetic alterations are often mediated by gross chromosomal rearrangements, causing translocations between heterologous chromosomes (creating novel oncogene fusions), large-scale chromosomal deletions (causing loss of heterozygosity at tumor suppressor gene loci) and gene amplifications (activating oncogenes). A key intermediate in this type of genomic instability is thought to be the double strand break (DSB), and a large body of evidence now implicates DSB repair defects in cancer susceptibility. In the absence of extrinsic DNA damage, most chromosomal DSBs in cycling cells, and the associated chromosomal rearrangements, are thought to arise during the S phase of the cell cycle as a result of replication across a damaged DNA template [3-6].
During DNA replication, the parental strands of DNA become dissociated into single-stranded DNA (ssDNA) templates for the synthesis of two identical sister chromatids. A lesion in one of the parental strands may cause the DNA polymerase complex to stall, potentially stalling or collapsing the replication fork, or else uncoupling leading and lagging strand synthesis. Arrested forks may themselves be processed to form a DSB. In some instances, replication may be restarted downstream of the unrepaired parental strand lesion, leaving a “daughter strand gap” (DSG)—a ssDNA lesion that cannot be filled by a conventional DNA polymerase, due to the presence of the DNA polymerase blocking lesion. The DSG has received less attention than its illustrious cousin, the DSB, but recent work in model organisms has reawakened interest in the DSG as a potential intermediate in genomic instability and cancer [7]. DSBs and DSGs can be repaired in an error-free manner by sister chromatid recombination (SCR), a mechanism whereby the damaged chromatid uses the intact sister as a template for repair by homologous recombination (HR) [5,8,9]. SCR competes with alternative error-prone pathways for repair of DSBs and DSGs and is therefore an important bulwark against the threats of genomic instability and cancer.
Several important human tumor suppressor genes function in HR. These include the two major breast/ovarian cancer susceptibility genes, BRCA1 and BRCA2 [10,11], the Bloom’s syndrome gene (BLM) and possibly other genes encoding RecQ-like helicases [12], the Nijmegen Breakage syndrome gene (NBS1) [13], MRE11 [14], the Fanconi Anemia genes (FA) [15], and genes encoding the DSB-activated signaling kinases ATM, ATR and CHK2 [16-18]. Consistent with a primary role in HR and SCR, BRCA1 and BRCA2 are highly expressed during the S phase of the cell cycle and their products form complexes with each other as well as with Rad51, the eukaryotic RecA ortholog [10,11,19]. BRCA1 and BRCA2 orthologs appear to be present in all vertebrate genomes. A BRCA2 homolog has been identified in the fungus Ustilago maydis [20] and homologs of BRCA1 exist in worms and in some plant species [21-24], but both genes appear to be absent from the genomes of Saccharomyces cerevisiae and Schizosaccharomyces pombe. The reason for this patchy representation of BRCA genes across evolution is not well understood. In species that lack BRCA genes, some functions of BRCA1 or 2 may be performed by structurally unrelated proteins. Indeed, recent evidence suggests that the BRCA2-Rad51 interaction has functional or structural similarities to the interactions of two Escherichia coli RecA loading complexes, RecFOR and RecBCD, with RecA [25,26]. HR pathways in E. coli and yeast often show functional redundancy, and the same may be true in multicellular organisms.
2. Relationship between DNA polymerase stalling and HR
The common lesions causing DNA polymerase stalling in somatic cells are thought to be unrepaired oxidative base damage caused by the products of normal aerobic metabolism. Other DNA polymerase stalling lesions include UV pyrimidine dimers, bulky DNA adducts and some other forms of DNA base damage. Intermediates of some DNA repair processes, such as base excision repair, nucleotide excision repair and mismatch repair, entail nicking of the sugar-phosphate backbone. Clearly, if a replication fork were to collide with such a nicked repair intermediate, the ssDNA nick would be converted to a DSB (Fig. 1). Thus, both DNA damage and DNA repair itself may contribute to the load on the HR/SCR pathway. Certain well-characterized DNA adducting carcinogens, such as benzo-[a]-pyrene metabolites, interrupt replication fork progression and are also excised inefficiently from the genome. Carcinogens of this type may promote cancer by causing DNA polymerase stalling and, hence, by increasing demand on the HR/SCR pathway [27,28] or on alternative error-prone pathways such as translesional DNA synthesis (TLS).
Fig. 1.
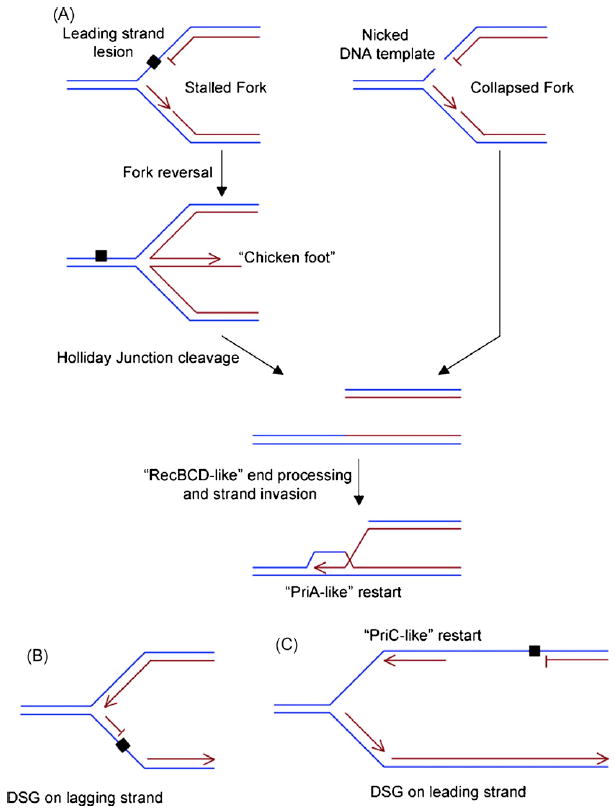
Models of fork restart in eukaryotic cells. (A) Generation of DSBs at stalled forks. Leading strand lesion stalls the fork, which can undergo fork reversal to form a “chicken foot” (Holliday Junction) structure. The HJ is then processed to form a DSB. RecBCD-like functions then load Rad51, catalyzing invasion of the neighboring intact sister chromatid to form a D-loop structure. A eukaryotic PriA-like activity might prime DNA synthesis on the free 3′ end of the D-loop to restart replication in an origin independent manner. (B) Formation of a lagging strand DSG: lagging strand DNA polymerase stalling need not interrupt fork progression. (C) Formation of a leading strand DSG: the lagging strand polymerase may become uncoupled from the stalled leading strand polymerase, leaving a ssDNA gap on the leading strand. A PriC-like activity might reinitiate leading strand synthesis, leaving a DSG on the leading strand. Parental strands are labeled blue. Daughter strands are labeled red. Free 3′ DNA ends are marked with an arrowhead. DNA polymerase stalling lesion is indicated by a black square.
The fundamental relationship between DNA polymerase stalling and HR was deduced four decades ago, in a series of classic experiments on the E. coli RecA gene and the nucleotide excision repair (NER) genes, UvrABC [29]. uvrA mutants are absolutely unable to excise UV lesions. Single mutants of recA or uvrA are mildly sensitive to UV light, but the recA uvrA double mutant is absolutely intolerant of UV damage; RecA is essential for survival in the presence of UV lesions that cannot be excised. Clues regarding the role of RecA in this process came from physical analysis of newly synthesized daughter strands in UV exposed uvrA mutants [30]. UV light caused a reduction in the average size of newly synthesized daughter strands, but during recovery from UV damage, daughter strands reverted to normal length. This suggested that replication across a UV lesion generates gaps in the newly synthesized daughter strands, and that RecA facilitates resolution of these daughter strand gaps. The RecA-mediated process was termed “daughter strand gap repair”—although it is strictly a DNA damage tolerance process, since it does not remove the original DNA polymerase stalling lesion. DSG repair in E. coli requires the RecFOR complex, which initiates RecA loading at the free 5′ DNA end of the dsDNA-ssDNA junction. The relationship between DNA polymerase stalling and recombination in E. coli has been reviewed elsewhere (for example, [31] and this issue) and is discussed only briefly here.
The 4.6Mb circular chromosome of E. coli is replicated from a single bidirectional origin of replication. For the cell to successfully duplicate the genome, each fork must traverse >2Mb without interruption. Given the density of DNA lesions caused by the products of normal aerobic metabolism, it is thought that most replication forks in E. coli will encounter a DNA polymerase stalling lesion. Interruption of lagging strand synthesis need not arrest the fork, but might leave a DSG on the lagging strand, corresponding to incomplete Okazaki fragment synthesis (Fig. 1). Arrest of the leading strand poses other problems [31]. Fork reversal with resolution of the resulting Holliday Junction (HJ) or “chicken foot” structure plays a part in processing of the stalled fork in some circumstances. If the HJ is cleaved, the resulting DSB is processed by RecBCD and loaded with RecA, permitting invasion of the unbroken sister chromatid [32]. Replication is reinitiated at the resulting free 3′ end of the recombination joint (“D-loop”) by the PriA-dependent φX174 primasome, a process termed recombination-dependent replication (RDR) [31]. PriA can also prime replication on free 3′ ends generated by RecA-independent processes.
Leading strand polymerase stalling need not necessarily result in lagging strand arrest, which may continue uncoupled from the stalled leading strand DNA polymerase, with the formation of an extensive tract of ssDNA on the leading strand [33]. Recently, Heller and Marians discovered that PriC can prime DNA synthesis directly on such gapped leading strand templates, leaving a DSG on the leading strand [34] (Fig. 1). This indicates that the replication machinery of E. coli tolerates DSGs in both the leading and lagging strands and can reinitiate DNA replication downstream of the fork on either strand by recombination dependent or recombination independent mechanisms (reviewed elsewhere in this issue).
In lower eukaryotes, the relationship between DNA polymerase stalling and HR is increasingly well documented. A genetic screen using a S. cerevisiae mutant of PCNA, pol30, revealed synthetic lethality with rad52 epistasis group mutants [35]. pol30 mutants were found to produce daughter strands of lower than normal molecular weight, suggesting that the pol30 mutation causes spontaneous gaps in the daughter strands. This is consistent with the idea that a defect in DNA polymerase processivity – presumably the basic problem in pol30 mutants – generates more frequent DSGs and puts an extra burden on the HR pathway. Specific evidence of a relationship between DNA polymerase stalling and HR came from studies in S. pombe in which a replication fork barrier was introduced into an ectopic chromosomal locus and was found to promote recombination and chromosomal rearrangements at that locus [27].
Direct visualization of stalled replication forks in yeast by electron microscopy has provided evidence of abnormal DNA structure at stalled forks, including fork reversal in checkpoint-defective rad53 mutants of S. cerevisiae [36]. Presumably, as in E. coli, processing of the “chicken foot” structure of the reversed fork by endonucleases such as Mus81 may generate a DSB [37]; alternatively, the stalled fork might be attacked directly. A recent study by Lopes et al. in UV-irradiated NER-deficient rad14 mutants of S. cerevisiae revealed the existence of DSGs in both leading and lagging strands downstream of the replication fork [38]. This suggests that S. cerevisiae may possess a recombination-independent mechanism of leading strand replication restart, since recombination-mediated restart need not generate leading strand DSGs [34]. There are no clear orthologs of PriC in eukaryotes; the mediators of this putative restart mechanism are unknown.
If replication restart on both strands can be accomplished without recombination, the problem of DSG repair might be addressed subsequent to the passage of the replication complex, allowing scheduled DNA synthesis to progress independently of the slower DSG repair processes [38]. Lopes et al. examined the effect of inactivating TLS or HR functions on the distribution of DSGs in UV irradiated rad14 mutants, and found that gaps nearer the fork accumulate in TLS mutants, whereas those distant from the fork accumulate more prominently in HR mutants. HR-mediated repair correlated with the presence of X-shaped DNA molecules (HJs) in the replicon—DNA structures that are predicted to arise during recombination-mediated DSG repair (Fig. 2).
Fig. 2.
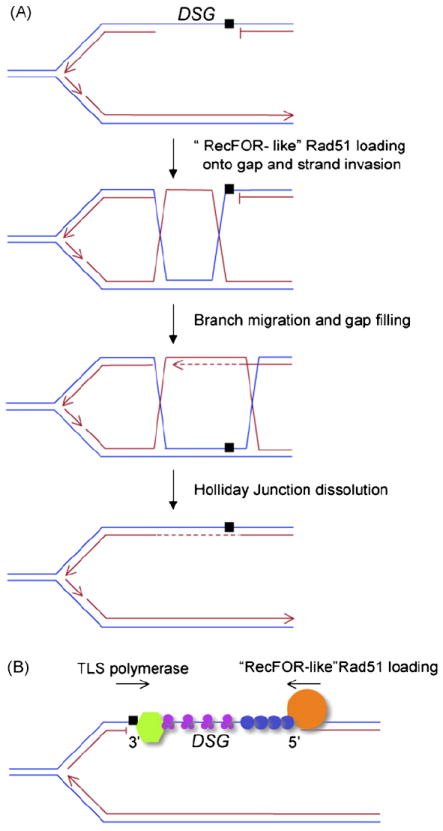
HR mediated daughter strand gap repair. (A) Figure shows a leading strand DSG. RecFOR-like loading of Rad51 facilitates strand exchange with the sister chromatid without a DSB intermediate. Branch migration switches the polymerase stalling lesion to the donor sister, allowing continued synthesis of the previously blocked nascent strand. Double Holliday Junction dissolution by the Bloom’s syndrome helicase may preferentially generate non-crossover products [159]. (B) Model depicting possible competition between TLS polymerase (green hexagon) and RecFOR-like HR functions (orange circle) for DSG repair. Figure shows a lagging strand DSG. TLS and Rad51 loading are initiated at opposite ends of the DSG. Purple circles, RPA trimer; blue circles, Rad51 oligomerizing on ssDNA. Black arrows depict direction of polymerase elongation by TLS or of Rad51 nucleation on ssDNA. DNA polymerase stalling lesion is indicated by a black square. FA proteins may also regulate this choice [160].
Rad6/Rad18-mediated ubiquitylation of PCNA serves as a scaffold for loadingTLS polymerases [39,40], but little is known about the mutual regulation of TLS and HR in DSG repair. TLS and HR could, in principle, attack opposite ends of the same DSG simultaneously; TLS polymerases might attempt to bypass the lesion at the blocked free 3′ end of the interrupted daughter strand, while RecFOR-like functions catalyze loading of Rad51 onto ssDNA, a process initiated at the free 5′ end of the dsDNA/ssDNA junction at the other side of the gap, some hundreds of base pairs away (Fig. 2). Simple competition could determine which process completes DSG repair. Presumably, TLS would be engaged provided that it can bypass the lesion, since HR is a slower process than replication. This is consistent with the findings of Lopes et al., in which TLS polymerases accounted for an early “wave” of DSG repair in the wake of the fork, and HR for DSG repair occurring later. Despite this, HR seems to play a quantitatively greater role than TLS in DSG repair, suggesting that TLS often fails to bypass the lesion successfully. Interestingly, resolution of DSGs closer to the fork is also impaired in rad53 checkpoint mutants, raising the possibility that DNA damage checkpoint signaling regulates engagement of TLS polymerases [38].
Studies in mammalian cells of replicating UV-irradiated plasmids have revealed DNA polymerase stalling on both the leading and lagging strands and showed the existence of ssDNA gaps on the lagging strand, presumably caused by interruption of Okazaki fragment synthesis [41-44]. This suggests that DSGs also arise during mammalian DNA replication, although it is not yet clear whether they can form on the leading strand.
3. Recombination-dependent replication restart in eukaryotes: an incomplete picture
Despite the generic similarities noted above between prokaryotes and eukaryotes, there are important differences that could affect the relationship between replication arrest and HR. The eukaryotic chromosome is packaged into chromatin, which adds additional complexity to the control of all chromosomal processes, including recombination [45-48]. Unlike E. coli, eukaryotic chromosomes possess numerous potential origins of replication, of which only a fraction are used to initiate DNA synthesis in each cell cycle. Indeed, the spacing between origins in eukaryotes is typically ~30–300 kb and is as low as 8–12 kb during early Xenopus development [49]. Thus, eukaryotic forks typically traverse much shorter distances than the >2MB needed for successful replication in E. coli. Replication fork collapse on a eukaryotic chromosome could in theory be compensated for by the arrival of a neighboring fork, possibly accompanied by the firing of a previously unused nearby origin of replication. This complicates the study of replication restart in eukaryotes. Although recent data, noted above, does suggest the existence of replication restart mechanisms in yeast, it remains to be determined whether HR has a role in this process. There are no clear eukaryotic homologs of E. coli PriA or PriC, but perhaps structurally unrelated eukaryotic proteins perform equivalent functions.
If a Rad51-dependent replication restart mechanism does operate in eukaryotes, there must be a means to convert the Rad51-mediated recombination joint (“D-loop”) into a replication fork. In yeast, repair synthesis during gene conversion does not normally entail formation of a replication fork, since gene conversion can be accomplished without the use of lagging strand synthesis [50] (Fig. 3). The gene conversion mechanism, termed “synthesis-dependent strand annealing” (SDSA), typically terminates after copying only a few hundred base pairs from the donor template—sufficient to patch the information lost at the site of chromosome breakage [51-53]. Under some circumstances, an alternative, highly processive copying mechanism, termed “break-induced replication” (BIR) can be engaged [54] (Fig. 3). BIR was first described as a RAD51-independent error-prone DSBR pathway and is known to mediate several types of chromosomal instability in yeast [55-58]. BIR is as processive as conventional chromosomal replication and it therefore probably entails formation of a replication fork at the recombination joint [59] (Fig. 3). The recent description of a RAD51-dependent form of BIR in S. cerevisiae suggests that BIR may not merely be an error-prone system of DSBR [59,60]. RAD51-dependent BIR might be a true analog of (RecA-dependent) RDR in E. coli.
Fig. 3.
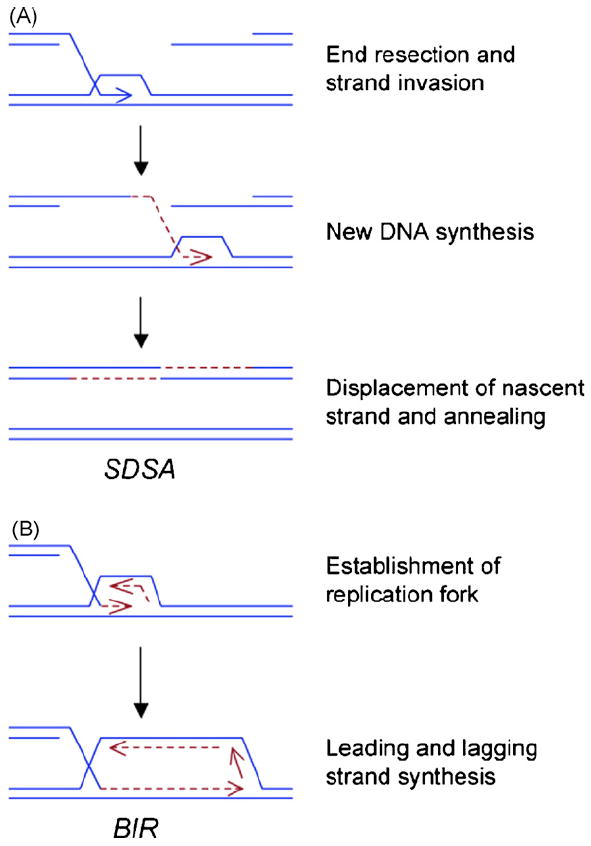
Mechanism of gene conversion by synthesis dependent strand annealing (SDSA) and break-induced replication (BIR). (A) In the SDSA model, following end processing, the 3′ end invades the sister chromatid and initiates repair synthesis without necessarily involving lagging strand synthesis. Repair synthesis copies a small stretch of information, after which the nascent strand is displaced and pairs with ssDNA of complementary sequence on the second end of the DSB. B. In the BIR model, a replication fork is established at the recombination joint, initiating both leading and lagging strand synthesis. Free 3′ DNA ends are marked with an arrowhead. Newly synthesized DNA is depicted by a dotted red line.
In mammalian cells, the existence of a BIR pathway (whether Rad51-dependent or not) has not been rigorously established. Gene conversion tracts are longer in cells lacking certain Rad51 paralogs; however, the longest gene conversion tracts measured in mammalian cells to date are only ~6 kb in length—much less than the hundreds of kilobases copied during BIR in yeast [61-63]. Interestingly, in Drosophila melanogaster, conventional gene conversion mediated by SDSA appears to be capable of copying tens of kilobases from the donor template [64]. Dysfunction in the fly ortholog of Rad51 leads to an unexpected decrease in average gene conversion tract length—the opposite of what one would expect were BIR engaged when Rad51 is dysfunctional [65]. These observations underscore the need to seek more direct evidence of a BIR pathway in higher eukaryotes. It is possible that BIR is such a potent engine of genomic instability that it cannot be tolerated by certain higher eukaryotes, where high levels of chromosomal instability will translate into failed embryonal development and high cancer incidence.
In summary, although some connections between DNA polymerase stalling and recombination are likely conserved across evolution, it is important to distinguish between DSB repair at broken forks, DSG repair in the wake of the fork and replication restart itself. Each of these could, in principle, be accomplished by Rad51-dependent or Rad51-independent mechanisms. Some differences between species are to be expected, shaped by evolutionary “facts on the ground”—the structure of the chromosome, the distribution of replication origins, the nature of the chromatin response to DSBs, the distribution of repetitive elements within the genome and the needs of multicellular organisms to develop organized tissues and avoid cancer.
4. Role of BRCA1 and BRCA2 in HR regulation
BRCA1 and BRCA2 null mice die early in development, around the time of gastrulation. The BRCA1−/− phenotype (death ~E7.5) is slightly more severe than the BRCA2−/− phenotype (death ~E8.5). BRCA1−/− BRCA2−/− double mutant embryos die ~E7.5, hinting at an epistatic relationship between the two gene mutations [19,66,67]. In the days prior to embryonic death, BRCA mutant embryos reveal evidence of a stress response, with p53 activation, p21 induction and a reduced rate of cell proliferation in comparison to wild-type littermates [66,68]. An interpretation of this phenotype was suggested by the finding that BRCA1 interacts with the mammalian RecA ortholog, Rad51, in discrete nuclear foci during the S and G2 phases of the cell cycle [69]. BRCA2 interacts stoichiometrically with Rad51 [19,70,71] and forms a complex with BRCA1, suggesting that BRCA1 and BRCA2 function on a common pathway regulating HR [11,72]. The activation of p53 in BRCA null embryos is likely part of a DNA damage checkpoint response arising as a consequence of defective recombination in these embryos. Consistent with this, deletion of p53 prolongs the survival of BRCA null embryos by a few days [67,68,73]. This is a significant effect, given the rapid developmental transitions that normally occur during gastrulation. Rad51 null embryos die very early in development (~E6.5), and their survival is also slightly prolonged on a p53 null background [74,75].
Primary cells explanted from BRCA2 and BRCA1 homozygous mutant embryos develop spontaneous chromosome breakage and gross chromosomal rearrangements at early passage [76-79]. BRCA1 and BRCA2, together with many other recombination proteins, also decorate the axial element of the developing synaptonemal complex during meiotic prophase I, a likely site of DSB formation and processing [69,72]. This suggests a possible role for BRCA1 and BRCA2 in meiotic recombination, for which some functional evidence now exists [80]. (A role for BRCA1 in transcriptional silencing during meiosis is also likely [81,82].) Attempts to cultivate BRCA1 or BRCA2 null primary mammalian cells in vitro have been unsuccessful, perhaps reflecting the severity of the recombination defect in these cells. As a result, most work on the recombination functions of BRCA genes has made use of cells expressing hypomorphic BRCA alleles. Direct evidence of a role for BRCA1 and BRCA2 in HR came from quantifying recombinational repair of a site-specific DSB in BRCA mutant cell lines. Either BRCA1 or BRCA2 mutation reduces the efficiency of HR induced by a site-specific DSB [83-87]. Error-prone repair of the DSB by single strand annealing (SSA) is increased in BRCA2 mutants [87,88]—a pattern reminiscent of HR mutants in yeast and in other vertebrate cells. In contrast, SSA is partly dependent on BRCA1, suggesting that BRCA1 also participates in an earlier step in HR, perhaps working with the MRN complex to control processing of the DSB end [88,89]. These studies do not assay HR at stalled forks, since the induced DSB has no relation to replication arrest. It is not clear whether DSG repair entails any kind of processing of the dsDNA–ssDNA iunction. In the limited studies that have been performed so far on different BRCA alleles, there appears to be a correlation between defective tumor suppressor function and loss of HR/DSBR function [90-92], suggesting that the HR functions of BRCA1 and BRCA2 contribute to tumor suppression.
Clues regarding the role of BRCA1 and BRCA2 in HR control in somatic cells and hence, potentially, in tumor suppression came initially from the finding that BRCA1 relocalizes rapidly from S/G2 phase nuclear foci to sites of replication arrest in S phase cells treated with hydroxyurea (HU) [93]. HU inhibits ribonucleotide reductase and thereby allows depletion of cell nucleotide pools in S phase, leading to DNA synthesis arrest. BRCA1 undergoes rapid S-phase-specific HU-induced phosphorylation with similar kinetics to DNA synthesis inhibition [93]. This phosphorylation event is regulated by the ATR signaling kinase, placing BRCA1 at an interesting intersection between the DNA damage checkpoint response and HR control [94]. BRCA1 is recruited to sites of stalled replication together with BRCA2, Rad51 and other recombination proteins, suggesting that this is part of a generic response of HR proteins to replication arrest. The idea that BRCA gene products have a significant HR function during the S phase was corroborated by the finding that chromosomes from BRCA mutant embryos develop “chromatid-type” structural aberrations [95,96]. This type of chromosomal rearrangement reflects asymmetric damage to sister chromatids and has traditionally been considered to arise from replication across a damaged DNA template and from recombination errors in S phase.
Hydroxyurea induces fork arrest but generates few DSBs in normal cells. Indeed, direct visualization of HU-arrested forks in yeast revealed the predicted formation of tracts of ssDNA on the lagging strand, reflecting incomplete synthesis of Okazaki fragments [36]. Structurally, HU-induced lagging strand gaps may resemble DSGs, and the recruitment of BRCA1, BRCA2, Rad51 and their associated proteins to sites of replication arrest in HU-treated cells suggests a response to ssDNA, even in the absence of DSB formation. This led to the proposal that BRCA1 and BRCA2 function in daughter strand gap repair [11,28].
Efficient Rad51 focus formation at DSBs induced by laser scissors or ionizing radiation (IR) is dependent upon BRCA1 and BRCA2, and Rad51 retention on chromatin following HU treatment is impaired in the absence of wild-type BRCA2, suggesting a possible role for BRCA1 and 2 in loading Rad51 onto ssDNA [97-100]. Lomonosov et al. visualized replication structures in rDNA of BRCA2 mutant mouse embryonic fibroblasts (MEF) during HU treatment. After prolonged treatment in HU, BRCA2 mutant MEFs revealed increased breakage within the rDNA locus [101]. This suggests that prolonged delay in fork progression leads to fork instability in BRCA2 mutant cells—for example, this might reflect the conversion of DSGs near the arrested fork to DSBs. The immediate structural consequences of fork arrest in BRCA mutant cells remain unknown.
A second set of data that speaks to the role of BRCA1 and BRCA2 at stalled forks came from treatment of BRCA mutant cells with poly(ADP-ribose)polymerase (PARP) inhibitors [102-104]. PARP is a co-factor in base excision repair and its inhibition should delay re-ligation of nicked intermediates of this repair process. Replication across the nicked template would generate DSBs either on the leading strand (with fork collapse) or on the lagging strand (without necessarily arresting the fork). BRCA1 and BRCA2 mutant primary embryonic stem cells were each found to be highly sensitive to PARP inhibitors [102,103]. The same sensitivity is seen in a BRCA2 mutant tumor cell line, and in cells defective in other HR genes [104,105]. These findings are consistent with a defect in DSB repair at stalled/broken forks in BRCA mutant cells. PARP inhibitors may have exciting prospects for therapy of BRCA-linked cancers, as may other drugs that target the “Achilles’ heel” of cells that are defective for HR.
A role for BRCA1, BRCA2 and Rad51 in HR repair of both DSBs and DSGs arising at stalled forks seems likely, each entailing recombination with the neighboring sister chromatid (Figs. 1 and 2). Whether the BRCA proteins have any role in replication restart at stalled forks is unclear. It should be noted that the resumption of bulk DNA synthesis following release of cells from an HU block is not a proven example of replication restart, since replication in such experimental conditions might reflect the firing of previously unused origins of replication. The HU-arrested fork may also differ from the D-loop formed at a broken fork reconstituted by HR, since the former might be primed to continue conventional DNA synthesis, whereas the latter would require de novo priming by a PriA-like activity (Fig. 1).
5. Molecular functions of BRCA1 and BRCA2
BRCA1 and BRCA2 encode large nuclear proteins that have no structural resemblance to each other. BRCA1 contains an N-terminal RING domain and two tandem C-terminal BRCT repeats. The structure of the large central region of BRCA1 – a region absent from certain splice variants of mammalian BRCA1 and entirely missing from the worm and plant homologs of BRCA1 – is unknown. BRCA1 exists as a constitutive heterodimer with BARD1, a smaller, structurally related polypeptide that also contains an N-terminal RING domain and two tandem C-terminal BRCT repeats [106]. The double RING domain of BRCA1/BARD1 has E3 ubiquitin ligase function in vitro [107-110]. Although the preferred linkage for polyubiquitylation in vitro appears to be an unconventional one involving lysine 6 of ubiquitin, BRCA1/BARD1 may also catalyze other linkages [111,112]. Not all of these ubiquitin linkages are associated with protein degradation, and detectable foci of ubiquitylation at DNA damage sites are dependent upon BRCA1 [113,114]. A number of cellular targets of the BRCA1/BARD1 E3 ubiquitin ligase have been proposed, but the list of direct targets of BRCA1/BARD1 is probably incomplete [115,116]. An intact RING domain is necessary for efficient BRCA1-mediated DSB repair [91], and several cancer-predisposing point mutant alleles of BRCA1 affect the RING domain, inactivating its E3 ubiquitin ligase function [108,117]. E3 ubiquitin ligase activity in BRCA1 immunoprecipitates is enhanced following exposure of cells to IR, raising the possibility that this activity is regulated by DNA damage signaling [114].
The BRCT repeats of BRCA1 are implicated in regulated interactions with certain phosphorylated proteins [118,119], including BACH1/FANCJ, a BRCA1-interacting DEAH-type 5′-to-3′ DNA helicase [120,121], and CtIP, a transcriptional repressor with DNA damage checkpoint functions [122,123]. Certain point mutant disease-predisposing alleles of BRCA1 disrupt these interactions, indicating that these interactions are clinically relevant. BRCA1 forms complexes with a large number of other protein interaction partners, including the Mre11/Rad50/NBS1 (MRN) heterotrimer (a complex involved in the early recognition of DSBs), BRCA2/FANCD1, Rad51 and other Fanconi Anemia gene products, as well as with a number of chromatin-modifying factors [124-126]. A recent biochemical purification of BARD1 revealed at least three distinct BRCA1/BARD1 complexes: one containing BACH1, the DNA damage responsive protein, TopBP1 and the Mlh1 mismatch repair protein; one containing MRN and CtIP; one containing BRCA2 and Rad51. Each of these complexes contains other polypeptides, and there may be other complexes of BRCA1/BARD1 yet to be described [100]. The interaction of BRCA1/BARD1 with TopBP1 and with MRN is induced following exposure of cells to IR. Given the numerous biochemical interactions of BRCA1/BARD1, it seems likely that BRCA1/BARD1 will have multiple different ubiquitylation target substrates.
BRCA1 accumulates on both ssDNA near the break and on a tract of chromatin neighboring the break, marked by serine 139-phosphorylation of the variant histone, H2AX [127,128]. Phosphorylation of H2AX (“γ-H2AX”) is a rapid, evolutionarily conserved event in the DSB response of eukaryotes and marks chromatin for hundreds of kilobases flanking the DSB [129]. H2AX contributes to DSBR, including SCR, by mechanisms that may involve cohesin complexes [46-48,130,131]. This “double life” of BRCA1 on ssDNA and on chromatin further underscores the diversity of functions of this protein, which include HR control, transcriptional regulation/gene silencing and DNA damage checkpoint control. Current evidence suggests that BRCA1’s HR function is executed independently of H2AX and it is therefore likely that BRCA1 controls HR at least in part through interactions with proteins such as BACH1, MRN and BRCA2/Rad51 on DNA near the break.
Some insight into the functional relationship between BRCA1 and its various partners has come from study of nuclear foci forming at DSBs induced by IR or in partial nuclear volumes by use of UV laser scissors. The accumulation of BRCA2, Rad51, BACH1 and CtIP at or near such DSBs is impaired in cells lacking wild-type BRCA1 and is restored by re-expression of wild-type BRCA1 [100]. In contrast, the MRN complex is recruited to DSBs in a BRCA1-independent manner. Thus, BRCA1 seems to function as a scaffold for the assembly of certain recombination enzymes at DSBs and may have additional enzymatic roles there, either directly as an E3 ubiquitin ligase or indirectly as a regulator of BACH1 helicase function.
Like many DNA damage response factors, BRCA1 undergoes phosphorylation by the Atm or Atr kinases in response to recombinogenic DNA lesions [94,132]. The activation of Atm by DSBs is mediated by its interaction with the MRN complex, while RPA-coated ssDNA activates Atr and associated checkpoint signaling proteins [133,134]. Hence, the signaling response to DSGs is predicted to be mediated by Atr. The Atr activation pathway in some ways resembles the bacterial SOS response since, in each case, a ssDNA nucleoprotein filament that acts as an intermediate in recombinational repair doubles as a trigger to the DNA damage response. These similarities are limited, however, since the SOS response is mediated by a co-protease activity of the RecA-ssDNA filament for cleavage of the LexA transcriptional repressor, and there is no evidence that Rad51-ssDNA has an equivalent function. Precisely how signaling by Atm and Atr affect BRCA1 function is not well understood. One might speculate that BRCA1/BARD1 has several distinct “civilian” functions (transcription regulation, gene silencing, etc.) on chromatin in the undamaged cell; in times of crisis, characterized by the presence of unresolved recombinogenic DNA lesions, BRCA1, modified by Atm or Atr signaling, might switch to “battle” mode as a DNA damage signaling and HR factor (Fig. 4).
Fig. 4.
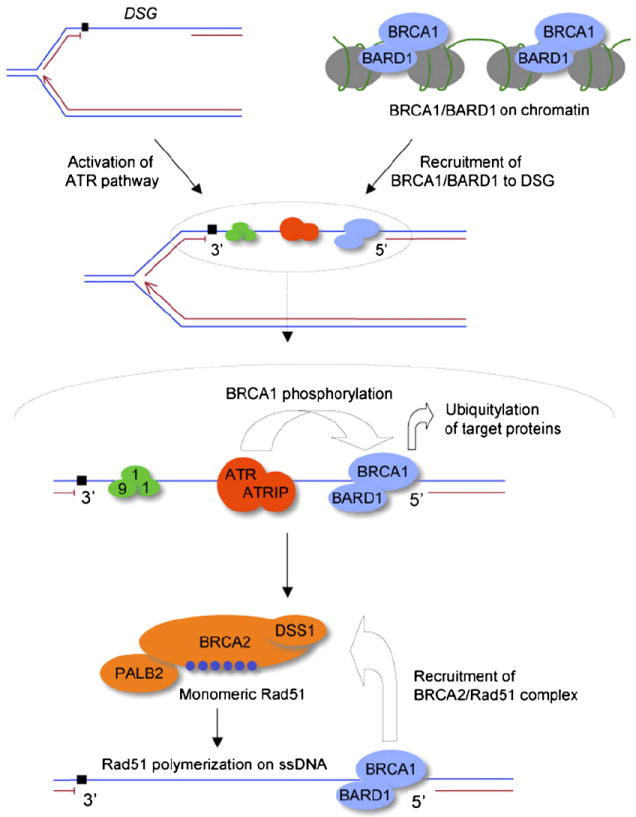
Proposed roles of BRCA1 and BRCA2 in daughter strand gap repair. BRCA1/BARD1 (light blue) performs transcription/silencing functions on chromatin in the undamaged cell. An RPA-coated DSG (shown here on the lagging strand; RPA not shown) recruits Atr/AtrIP (red) and the 9-1-1 complex (green). Recruitment of BRCA1/BARD1 to DSGs may be independent of Atr. Activated Atr phosphorylates BRCA1, promoting interaction of BRCA1/BARD1 with other DNA damage response proteins (not shown), as discussed in the text. The BRCA1/BARD1 heterodimer may ubiquitylate target proteins at the DSG, and facilitates recruitment of the BRCA2/Rad51 complex (orange) for HR-mediated DSG repair. Rad51 is shown in dark blue. DNA polymerase stalling lesion is indicated by a black square. The HR function of BRCA1 depicted here is independent of the H2AX response [48].
BRCA2 binds Rad51 directly via multiple BRC repeats in the central part of BRCA2 [99,135,136]. The BRCA2 C-terminus forms a tight complex with a conserved protein, DSS1, and can also interact with Rad51 [19,71,137]. The crystal structure of the BRCA2 C-terminus bound to DSS1 revealed similarity with the heterotrimeric ssDNA-binding complex, RPA, implying a direct interaction of BRCA2/DSS1with ssDNA [138]. The crystal structure of a BRCA2 BRC4-Rad51 fusion protein indicates that the BRC domain interacts with the core of Rad51 and, by a process of molecular mimicry, prevents the homo-oligomerization of Rad51 [136]. Consistent with this, isolated BRC fragments of BRCA2 inhibit Rad51-mediated strand exchange in vitro and interfere with HR in vivo [135,139]. In contrast, certain combined domains of BRCA2 promote Rad51-mediated strand exchange in vitro, suggesting that there may be intramolecular collaboration between distinct domains of BRCA2 [140,161]. The U. maydis BRCA2 homolog, Brh2, possesses only one BRC domain and has been purified with DSS1 in vitro [20,25,141]. Brh2/DSS1 efficiently catalyzes the loading of Rad51 onto the free 5′ end of a dsDNA-ssDNA junction, provided that the ssDNA is pre-coated with RPA (as it would be in vivo). Rad51 loading was observed on a gapped, circular plasmid, suggesting that a free DSB end is not required for Brh2-mediated loading of Rad51 [25]. This function resembles that of E. coli RecFOR, a complex that loads RecA onto ssDNA gaps for DSG repair in E. coli [142]. A recent crystal structure of E. coli RecB bound to RecA revealed an interaction of RecB with the core of RecA, similar to that of the BRCA2 BRC domain interaction with Rad51 [26]. Thus, certain Rad51 loading functions of BRCA2 are reflected in two distinct RecA loading complexes in E. coli. It is not clear whether BRCA2 is the only Rad51 loading complex in mammalian cells. By analogy with E. coli, one might expect eukaryotic cells to possess more than one mechanism for loading Rad51 onto ssDNA.
Although purified Brh2/DSS1 can facilitate loading of Rad51 in vitro, BRCA2/DSS1/Rad51 does not act alone in vivo. BRCA2 interacts stoichiometrically with a newly identified protein, PALB2, which is required for accumulation of BRCA2/Rad51 at DSBs induced in partial nuclear volumes in vivo [92]. Consistent with this, certain point mutations in BRCA2 that disrupt its interaction with PALB2 are also implicated in breast/ovarian cancer predisposition. BRCA2 and, very recently, PALB2 have been shown to undergo biallelic mutation in some cases of Fanconi Anemia [143-146]. Rad51 focus formation is defective in cells from some FA complementation groups, including those harboring mutations in BRCA2 or PALB2. This raises the possibility that other FA gene products function with BRCA1, PALB2 and BRCA2 to regulate Rad51 loading under some circumstances. It will be interesting to determine whether PALB2 interacts with other FA gene products and with BRCA1/BARD1.
The loading of Rad51 onto ssDNA is clearly a critical control step in cell physiology: either defective or excessive Rad51 loading might cause genomic instability and cancer. Our current knowledge of the Rad51 loading mechanism in multicellular organisms suggests that it is mediated by a complex set of protein–protein and protein–DNA interactions, orchestrated by Atm and Atr signaling and perhaps by more diverse cell signaling pathways [147]. By analogy with yeast, there may also be negative regulators, operating to inhibit Rad51 loading or disassemble Rad51 nucleoprotein filaments and suppress inappropriate recombination events [148]. The appearance in multicellular organisms of novel HR regulatory genes (BRCA1/BARD1, Fanconi Anemia genes) may in part represent an evolution-driven titration of Rad51 loading activity to a “set point” that is appropriate to the demands of multicellularity.
6. When the levee breaks: DSGs, genomic instability and cancer
The current focus on DSB repair functions of BRCA1 and BRCA2 may reflect an experimental bias in the field—there are tractable assays for HR-mediated DSB repair in mammalian cells but not for HR-mediated DSG repair. If mammalian cells generate predominantly DSGs in response to DNA polymerase blocking lesions, the HR functions of BRCA1 and BRCA2 at DSGs may be quantitatively as important for tumor suppression as their functions at DSBs.
An unresolved DSG may be a key intermediate in genomic instability. Perhaps the innate fragility of persistent ssDNA carries a risk of degradation to a DSB. Alternatively, an unresolved DSG might be able to interact with free DSB ends generated elsewhere in the genome. Indeed, although the repair of two experimentally induced DSBs on heterologous chromosomes can cause chromosome translocations [149], the DNA intermediates of spontaneous chromosome translocations are unknown. Conceivably, interactions between a DSB and a DSG, or between two DSGs, might occasionally resolve as a chromosome rearrangement.
This review has not touched upon the contributions of BRCA1 and BRCA2 to checkpoint functions [150-152], transcriptional regulation [153,154], gene silencing [81,82,155], telomere function [96] or (for BARD1) mRNA processing [156,157]. Both HR and non-HR functions of BRCA genes might account for the tissue specificity of cancer risk in BRCA gene mutation carriers [158] and the latter may be important modulators of the clinical phenotypes associated with BRCA gene inactivation.
Acknowledgments
We thank members of the Scully lab for their contributions and for critical reading of the manuscript. We also thank Dr. Jim Haber and Dr. Lorraine Symington for stimulating discussions. This work was supported by NIH grants CA95175 and GM073894, ACS grant RSG 107471, a Leukemia and Lymphoma Society Research Scholar Award and by the Pew Scholars Program in the Biomedical Sciences.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 3.Kowalczykowski SC. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem Sci. 2000;25:156–165. doi: 10.1016/s0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- 4.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 5.Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proc Natl Acad Sci USA. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann AR, Fuchs RP. Gaps and forks in DNA replication: rediscovering old models. DNA Repair (Amst) 2006;5:1495–1498. doi: 10.1016/j.dnarep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 11.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 13.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, III, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 14.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 15.D’Andrea AD, Grompe M. The, Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 16.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 17.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 18.Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 19.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking BRCA2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. see comments. [DOI] [PubMed] [Google Scholar]
- 20.Kojic M, Kostrub CF, Buchman AR, Holloman WK. BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2002;10:683–691. doi: 10.1016/s1097-2765(02)00632-9. [DOI] [PubMed] [Google Scholar]
- 21.Boulton SJ, Martin JS, Polanowska J, Hill DE, Gartner A, Vidal M. BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr Biol. 2004;14:33–39. doi: 10.1016/j.cub.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Joukov V, Chen J, Fox EA, Green JB, Livingston DM. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc Natl Acad Sci USA. 2001;98:12078–12083. doi: 10.1073/pnas.211427098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lafarge S, Montane MH. Characterization of Arabidopsis thaliana ortholog of the human breast cancer susceptibility gene 1: AtBRCA1, strongly induced by gamma rays. Nucl Acids Res. 2003;31:1148–1155. doi: 10.1093/nar/gkg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reidt W, Wurz R, Wanieck K, Chu HH, Puchta H. A homologue of the breast cancer-associated gene BARD1 is involved in DNA repair in plants. EMBO J. 2006;25:4326–4337. doi: 10.1038/sj.emboj.7601313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA–ssDNA junction. Nature. 2005;433:653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 26.Spies M, Kowalczykowski SC. The RecA binding locus of RecBCD is a general domain for recruitment of DNA strand exchange proteins. Mol Cell. 2006;21:573–580. doi: 10.1016/j.molcel.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 28.Scully R, Puget N, Vlasakova K. DNA polymerase stalling, sister chromatid recombination and the BRCA genes. Oncogene. 2000;19:6176–6183. doi: 10.1038/sj.onc.1203971. [DOI] [PubMed] [Google Scholar]
- 29.Howard-Flanders P, Boyce RP. DNA repair and genetic recombination: studies on mutants of Escherichia coli defective in these processes. Radiat Res. 1966;6:156–184. [PubMed] [Google Scholar]
- 30.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 31.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 32.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 33.Pages V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 34.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439:557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 35.Merrill BJ, Holm C. The RAD52 recombinational repair pathway is essential in pol30 (PCNA) mutants that accumulate small single-stranded DNA fragments during DNA synthesis. Genetics. 1998;148:611–624. doi: 10.1093/genetics/148.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 37.Whitby MC. Junctions on the road to cancer. Nat Struct Mol Biol. 2004;11:693–695. doi: 10.1038/nsmb0804-693. [DOI] [PubMed] [Google Scholar]
- 38.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 39.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 40.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 41.Mezzina M, Menck CF, Courtin P, Sarasin A. Replication of simian virus 40 DNA after UV irradiation: evidence of growing fork blockage and single-stranded gaps in daughter strands. J Virol. 1988;62:4249–258. doi: 10.1128/jvi.62.11.4249-4258.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svoboda DL, Vos JM. Differential replication of a single, UV-induced lesion in the leading or lagging strand by a human cell extract: fork uncoupling or gap formation. Proc Natl Acad Sci USA. 1995;92:11975–11979. doi: 10.1073/pnas.92.26.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cordeiro-Stone M, Zaritskaya LS, Price LK, Kaufmann WK. Replication fork bypass of a pyrimidine dimer blocking leading strand DNA synthesis. J Biol Chem. 1997;272:13945–13954. doi: 10.1074/jbc.272.21.13945. [DOI] [PubMed] [Google Scholar]
- 44.Cordeiro-Stone M, Makhov AM, Zaritskaya LS, Griffith JD. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J Mol Biol. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 45.Fritsch O, Benvenuto G, Bowler C, Molinier J, Hohn B. The INO80 protein controls homologous recombination in Arabidopsis thaliana. Mol Cell. 2004;16:479–485. doi: 10.1016/j.molcel.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 46.Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 47.Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 48.Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R. Control of sister chromatid recombination by histone H2AX. Mol Cell. 2004;16:1017–1025. doi: 10.1016/j.molcel.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilbert DM. Making sense of eukaryotic DNA replication origins. Science. 2001;294:96–100. doi: 10.1126/science.1061724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Ira G, Tercero JA, Holmes AM, Diffley JF, Haber JE. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:6891–6899. doi: 10.1128/MCB.24.16.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson HH, Sweetser DB, Nickoloff JA. Effects of terminal nonhomology and homeology on double-strand-break-induced gene conversion tract directionality. Mol Cell Biol. 1996;16:2951–2957. doi: 10.1128/mcb.16.6.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sweetser DB, Hough H, Whelden JF, Arbuckle M, Nickoloff JA. Fine-resolution mapping of spontaneous and double-strand break-induced gene conversion tracts in Saccharomyces cerevisiae reveals reversible mitotic conversion polarity. Mol Cell Biol. 1994;14:3863–3875. doi: 10.1128/mcb.14.6.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malkova A, Ivanov EL, Haber JE. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kraus E, Leung WY, Haber JE. Break-induced replication: a review and an example in budding yeast. Proc Natl Acad Sci USA. 2001;98:8255–8262. doi: 10.1073/pnas.151008198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bosco G, Haber JE. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 1998;150:1037–1047. doi: 10.1093/genetics/150.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301:1908–1911. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- 58.Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–1399. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol. 2005;25:933–944. doi: 10.1128/MCB.25.3.933-944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol Cell Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagaraju G, Odate S, Xie A, Scully R. Differential regulation of short- and long-tract gene conversion between sister chromatids by Rad51C. Mol Cell Biol. 2006;26:8075–8086. doi: 10.1128/MCB.01235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Mol Cell. 2002;10:387–395. doi: 10.1016/s1097-2765(02)00595-6. [DOI] [PubMed] [Google Scholar]
- 63.Stark JM, Jasin M. Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol. 2003;23:733–743. doi: 10.1128/MCB.23.2.733-743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–267. doi: 10.1126/science.1077198. [DOI] [PubMed] [Google Scholar]
- 65.McVey M, Adams M, Staeva-Vieira E, Sekelsky JJ. Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics. 2004;167:699–705. doi: 10.1534/genetics.103.025411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, Firpo E, Hui CC, Roberts J, Rossant J, Mak TW. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 67.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 68.Suzuki A, de la Pompa JL, Hakem R, Elia A, Yoshida R, Mo R, Nishina H, Chuang T, Wakeham A, Itie A, Koo W, Billia P, Ho A, Fukumoto M, Hui CC, Mak TW. BRCA2 is required for embryonic cellular proliferation in the mouse. Genes Dev. 1997;11:1242–1252. doi: 10.1101/gad.11.10.1242. [DOI] [PubMed] [Google Scholar]
- 69.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 70.Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene Brca2. J Biol Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- 71.Mizuta R, LaSalle JM, Cheng HL, Shinohara A, Ogawa H, Copeland N, Jenkins NA, Lalande M, Alt FW. RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc Natl Acad Sci USA. 1997;94:6927–6932. doi: 10.1073/pnas.94.13.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- 73.Hakem R, de la Pompa JL, Elia A, Potter J, Mak TW. Partial rescue of Brca1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat Genet. 1997;16:298–302. doi: 10.1038/ng0797-298. [DOI] [PubMed] [Google Scholar]
- 74.Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 77.Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, Guan XY, Ried T, Deng CX. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17:3115–3124. doi: 10.1038/sj.onc.1202243. [DOI] [PubMed] [Google Scholar]
- 78.Tutt A, Gabriel A, Bertwistle D, Connor F, Paterson H, Peacock J, Ross G, Ashworth A. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol. 1999;9:1107–1110. doi: 10.1016/s0960-9822(99)80479-5. [DOI] [PubMed] [Google Scholar]
- 79.Connor F, Bertwistle D, Mee PJ, Ross GM, Swift S, Grigorieva E, Tybulewicz VL, Ashworth A. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat Genet. 1997;17:423–430. doi: 10.1038/ng1297-423. [DOI] [PubMed] [Google Scholar]
- 80.Xu X, Aprelikova O, Moens P, Deng CX, Furth PA. Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development. 2003;130:2001–2012. doi: 10.1242/dev.00410. [DOI] [PubMed] [Google Scholar]
- 81.Ganesan S, Silver DP, Greenberg RA, Avni D, Drapkin R, Miron A, Mok SC, Randrianarison V, Brodie S, Salstrom J, Rasmussen TP, Klimke A, Marrese C, Marahrens Y, Deng CX, Feunteun J, Livingston DM. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell. 2002;111:393–405. doi: 10.1016/s0092-8674(02)01052-8. [DOI] [PubMed] [Google Scholar]
- 82.Turner JM, Aprelikova O, Xu X, Wang R, Kim S, Chandramouli GV, Barrett JC, Burgoyne PS, Deng CX. BRCA1, histone H2AX phosphorylation, and male meiotic sex chromosome inactivation. Curr Biol. 2004;14:2135–2142. doi: 10.1016/j.cub.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 83.Moynahan ME, Chiu JW, Koller BH, Jasin M. BRCA1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 84.Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed] [Google Scholar]
- 85.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. see comment. [DOI] [PubMed] [Google Scholar]
- 86.Xia F, Taghian DG, DeFrank JS, Zeng ZC, Willers H, Iliakis G, Powell SN. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci USA. 2001;98:8644–8649. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang YG, Saidi A, Frappart PO, Min W, Barrucand C, Dumon-Jones V, Michel on J, Herceg Z, Wang ZQ. Conditional deletion of Nbs1 in murine cells reveals its role in branching repair pathways of DNA double-strand breaks. EMBO J. 2006;25:5527–5538. doi: 10.1038/sj.emboj.7601411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22:5784–5791. doi: 10.1038/sj.onc.1206678. [DOI] [PubMed] [Google Scholar]
- 91.Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- 92.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 93.Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. D ynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 94.Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston DM, Elledge SJ, Abraham RT. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. doi: 10.1101/gad.851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel KJ, Yu VPCC, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement of BRCA2 in DNA repair. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 96.McPherson JP, Hande MP, Poonepalli A, Lemmers B, Zablocki E, Migon E, Shehabeldin A, Porras A, Karaskova J, Vukovic B, Squire J, Hakem R. A role for Brca1 in chromosome end maintenance. Hum Mol Genet. 2006;15:831–838. doi: 10.1093/hmg/ddl002. [DOI] [PubMed] [Google Scholar]
- 97.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 98.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59:3547–3551. [PubMed] [Google Scholar]
- 99.Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, Venkitaraman AR. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol Cell. 2003;12:1029–1041. doi: 10.1016/s1097-2765(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 100.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose)polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 103.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 104.McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of poly(ADP-ribose)polymerase: an issue of potency. Cancer Biol Ther. 2005;4:934–936. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- 105.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor J, Tutt MAN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose)polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 106.Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 107.Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev. 2002;12:86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 108.Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 109.Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci USA. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 111.Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem. 2003;278:34743–34746. doi: 10.1074/jbc.C300249200. [DOI] [PubMed] [Google Scholar]
- 112.Nishikawa H, Ooka S, Sato K, Arima K, Okamoto J, Klevit RE, Fukuda M, Ohta T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279:3916–3924. doi: 10.1074/jbc.M308540200. [DOI] [PubMed] [Google Scholar]
- 113.Morris JR, Solomon E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet. 2004;13:807–817. doi: 10.1093/hmg/ddh095. [DOI] [PubMed] [Google Scholar]
- 114.Polanowska J, Martin JS, Garcia-Muse T, Petalcorin MI, Boulton SJ. A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J. 2006;25:2178–2188. doi: 10.1038/sj.emboj.7601102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 116.Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brzovic PS, Meza JE, King MC, Klevit RE. BRCA1 RING domain cancer-predisposing mutations: structural consequences and effects on protein–protein interactions. J Biol Chem. 2001;276:41399–l1406. doi: 10.1074/jbc.M106551200. [DOI] [PubMed] [Google Scholar]
- 118.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 119.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 120.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. BACH1 a novel helicase-like 39 protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 121.Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 123.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 125.Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 126.Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, Shiekhattar R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol Cell. 2003;12:1087–1099. doi: 10.1016/s1097-2765(03)00424-6. [DOI] [PubMed] [Google Scholar]
- 127.Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol. 2003;81:123–129. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- 128.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, Lou Z, Bassing CH, Manis JP, Chen J, Carpenter PB, Alt FW. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 132.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of BRCA1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 133.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 134.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 135.Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–282. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- 136.Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 137.Marston NJ, Richards WJ, Hughes D, Bertwistle D, Marshall CJ, Ashworth A. Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals. Mol Cell Biol. 1999;19:4633–4642. doi: 10.1128/mcb.19.7.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 139.Stark JM, Hu P, Pierce AJ, Moynahan ME, Ellis N, Jasin M. ATP hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J Biol Chem. 2002;277:20185–20194. doi: 10.1074/jbc.M112132200. [DOI] [PubMed] [Google Scholar]
- 140.Shivji MK, Davies OR, Savill JM, Bates DL, Pellegrini L, Venkitaraman AR. A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucl Acids Res. 2006;34:4000–4011. doi: 10.1093/nar/gkl505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kojic M, Yang H, Kostrub CF, Pavletich NP, Holloman WK. The BRCA2-interacting protein DSS1 is vital for DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2003;12:1043–1049. doi: 10.1016/s1097-2765(03)00367-8. [DOI] [PubMed] [Google Scholar]
- 142.Morimatsu K, Kowalczykowski SC. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell. 2003;11:1337–1347. doi: 10.1016/s1097-2765(03)00188-6. [DOI] [PubMed] [Google Scholar]
- 143.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. see comment. [DOI] [PubMed] [Google Scholar]
- 144.Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 145.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 147.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 148.Symington LS, Heyer WD. Some disassembly required: role of DNA translocases in the disruption of recombination intermediates and dead-end complexes. Genes Dev. 2006;20:2479–2486. doi: 10.1101/gad.1477106. [DOI] [PubMed] [Google Scholar]
- 149.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–8894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 150.Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 151.Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, Essers J, van Duijn-Goedhart A, Wiggers I, Swaminathan S, van Buul PP, Errami A, Tan RT, Jaspers NG, Sharan SK, Kanaar R, Zdzienicka MZ. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol. 2002;22:669–679. doi: 10.1128/MCB.22.2.669-679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–879. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- 153.Starita LM, Parvin JD. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol. 2003;15:345–350. doi: 10.1016/s0955-0674(03)00042-5. [DOI] [PubMed] [Google Scholar]
- 154.Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, Milner J, Brown LA, Hsu F, Gilks B, Nielsen T, Schulzer M, Chia S, Ragaz J, Cahn A, Linger L, Ozdag H, Cattaneo E, Jordanova ES, Schuuring E, Yu DS, Venkitaraman A, Ponder B, Doherty A, Aparicio S, Bentley D, Theillet C, Ponting CP, Caldas C, Kouzarides T. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115:523–535. doi: 10.1016/s0092-8674(03)00930-9. see comment. [DOI] [PubMed] [Google Scholar]
- 155.Turner JM, Mahadevaiah SK, Fernandez-Capetillo O, Nussenzweig A, Xu X, Deng CX, Burgoyne PS. Silencing of unsynapsed meiotic chromosomes in the mouse. Nat Genet. 2005;37:41–47. doi: 10.1038/ng1484. [DOI] [PubMed] [Google Scholar]
- 156.Kleiman FE, Manley JL. Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science. 1999;285:1576–1579. doi: 10.1126/science.285.5433.1576. [DOI] [PubMed] [Google Scholar]
- 157.Kleiman FE, Manley JL. The BARD1-CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell. 2001;104:743–753. doi: 10.1016/s0092-8674(01)00270-7. [DOI] [PubMed] [Google Scholar]
- 158.Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res. 2000;2:324–330. doi: 10.1186/bcr76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 160.Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 161.San Filippo J, Chi P, Sehorn MG, Etchin J, Krejci L, Sung P. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J Biol Chem. 2006;281:11649–11657. doi: 10.1074/jbc.M601249200. [DOI] [PMC free article] [PubMed] [Google Scholar]