

Summary
MAPK and AKT pathways are frequently co-activated in melanoma through overexpression of receptor tyrosine kinases, mutations in their signaling surrogates, such as RAS and BRAF, or loss of negative regulators such as PTEN. Since RAS can be a positive upstream regulator of PI3-K, it has been proposed that the loss of PTEN and the activation of RAS are redundant events in melanoma pathogenesis (Tsao et al., 2000). Here, in genetically engineered mouse models of cutaneous melanomas, we sought to better understand the genetic interactions between HRAS activation and PTEN inactivation in melanoma genesis and progression in vivo. We showed that HRAS activation cooperates with Pten+/- and Ink4a/Arf-/- to increase melanoma penetrance and promote metastasis. Correspondingly, gain- and loss-of-function studies established that Pten loss increases invasion and migration of melanoma cells and non-transformed melanocytes, and that such biological activity correlates with a shift to phosphorylation of AKT2 isoform and E-cadherin down-regulation. Thus, Pten inactivation can drive the genesis and promote the metastatic progression of RAS activated Ink4a/Arf deficient melanomas.
Keywords: melanoma, PTEN, RAS, E-cadherin, AKT2, mouse model
Introduction
RAS activation is a common and potent oncogenic event in human solid tumors, including melanoma. The relevance of RAS-RAF-MAPK pathway in melanoma pathogenesis is evidenced by high frequency BRAF mutations (Davies et al., 2002) and less common yet reciprocal NRAS mutations (Tsao et al., 2004). Activating mutations of NRAS are found in as many as 56% of congenital nevi, 33% of primary and 26% of metastatic melanoma samples (Chin et al., 2006). Although, NRAS is the most commonly mutated RAS family member, mutations on KRAS (2%) and HRAS (1%) are also observed in human melanomas (Forbes et al., 2008). In particular, HRAS mutation was reported to be found in 7.7% of the nodular melanoma subtype (Jafari et al., 1995).
Genetic evidence of a pathogenetic role for the RAS-RAF-MAPK pathway in melanoma derives from a melanoma-prone condition in mice transgenic for melanocyte-directed HRASV12G or NRASQ61K alleles and null for p16INK4a and/or p19ARF tumor suppressors (Ackermann et al., 2005; Chin et al., 1997; Chin et al., 1999; Sharpless et al., 2003). Additionally, BRAF, a key effector of RAS signaling, is the most frequently mutated gene in human melanocytic neoplasms with frequencies of 82% of benign nevi and 66% in melanomas (Davies et al., 2002; Pollock et al., 2003). Melanocyte-directed expression of BRAFV600E can induce nevoid hyperpigmentation phenotype in mice (Dankort et al., 2009; Dhomen et al., 2009; Goel et al., 2009) with rare progression to melanoma (Dhomen et al., 2009; Goel et al., 2009).
While activation of the RAS-RAF signaling cascade has been recognized as an obligate event in melanocyte transformation, Khavari and colleagues have demonstrated that activation of BRAF – MAPK requires concomitant AKT activation to effect melanoma development (Chudnovsky et al., 2005). Consistent with importance of the PI3K-AKT pathway, over 60% of human melanomas exhibit activated AKT (Dhawan et al., 2002), and mutational inactivation and/or deletion of the PI3K negative regulator, PTEN, occurs in 5-15% of uncultured melanoma specimens and metastasis, 17% of melanoma short-term cultures, and 30-40% of established melanoma cell lines (Birck et al., 2000; Guldberg et al., 1997; Lin et al., 2008; Tsao et al., 2004). Mouse modeling has also demonstrated that Pten+/-Ink4a/Arf+/- mice do succumb to melanoma at low frequency (You et al., 2002) and robust melanoma formation and metastases can occur upon combination with activated BRAF (Dankort et al., 2009).
The PI3K-AKT signaling pathway can be activated by receptor tyrosine kinases. Active PI3K phosphorylates and converts the lipid phosphatidylinositol (4,5) bisphosphate (PIP2) into PIP3, which in turn activates AKT through PDK1 mediated phosphorylation. PTEN negatively regulates PI3K signaling by dephosphorylating PIP3, converting it back to PIP2. Therefore, deletion or inactivation of PTEN results in constitutive AKT activation (Cully et al., 2006; Salmena et al., 2008). On the other hand, RAS proteins can positively regulate the PI3K–AKT pathway by direct binding of RAS to the p110 catalytic subunit (Gupta et al., 2007) or by activating an autocrine signaling pathway involving EGFR family ligands (Bardeesy et al., 2005). Since PTEN inactivation and RAS activating mutation can both target the PI3K pathway to drive constitutive AKT activation, it has been proposed that RAS mutation and PTEN inactivation are redundant events in tumorigenesis, as suggested by a reciprocal trend of their mutations in human melanoma and endometrial carcinoma (Ikeda et al., 2000; Tsao et al., 2000). However, constitutive AKT1 activation in mouse has resulted in a milder cancer phenotype than complete PTEN inactivation (Ma et al., 2005; Majumder et al., 2003; Stambolic et al., 2000; Trotman et al., 2003; Wang et al., 2003), pointing to AKT independent activities of PTEN in tumorigenesis (Blanco-Aparicio et al., 2007). Similarly, while expression of oncogenic K-RAS or conditional PTEN deletion in the ovarian epithelium gives rise to preneoplastic ovarian lesions, the combined effect of these two mutations in the ovary leads to invasive and widely metastatic endometrioid ovarian adenocarcinomas with complete penetrance and a disease latency of only 7 weeks (Dinulescu et al., 2005). In this study, we addressed the potential collaborative interactions between RAS activation and PTEN inactivation on the genetic level.
Results
Pten inactivation cooperates with HRASV12G in melanoma genesis in vivo
We had previously shown that Tyr-HRASV12GInk4a/Arf-/- compound mutant mice (hereafter “RAS-Ink4a/Arf”) developed non-metastatic cutaneous melanomas with short latency and high penetrance (Chin et al., 1997; Chin et al., 1999; Kim et al., 2006). The occurrence of PTEN loss in advanced melanoma prompted us to determine whether Pten inactivation can cooperate with HRAS activation to drive melanoma progression. Given that Pten nullizygosity is embryonic lethal (Di Cristofano et al., 1998; Suzuki et al., 1998), we studied the melanoma phenotype of the RAS-Ink4a/Arf model on Pten+/+ and Pten+/- background.
Inactivation of one copy of Pten in RAS-Ink4a/Arf mice led to an earlier onset of melanoma and decreased overall melanoma-free survival (Figure 1A and Table 1; p=0.0002). Although median survival was not notably different (median survival 21.6 vs. 18.9 wks, Supplemental Figure 1), melanoma-free survival was significantly decreased by Pten heterozygosity (29.6 vs. 18.9 wks comparing RAS-Ink/Arf mice with Pten +/- vs. Pten+/+ genotypes). In the absence of melanocyte-specific expression of activated RAS, the Pten+/-Ink/Arf-/- mice showed a median survival of 19.4 weeks, and did not develop melanomas (Supplemental Figure 1). In the period prior to the appearance of non-melanoma tumors, 75% of the Pten+/- RAS-Ink4a/Arf mice developed melanoma compared with 35.7% of mice with WT Pten (Table 1). Histopathologically, these primarily spindle cell tumors are similar to those observed in the melanomas from the RAS-Ink4a/Arf model (Chin et al., 1997; Chin et al., 1999). Melanocytic lineage of Pten+/-RAS-Ink4a/Arf cutaneous tumors was confirmed by expression of melanocytic markers on protein and RNA level (Figure 1B and 1C). These findings clearly establish that activated RAS and loss of Pten cooperate to derive the genesis of melanoma on the Ink4a/Arf null background.
Figure 1. Collaboration of PTEN loss and HRAS activation for the pathogenesis of cutaneous melanoma.
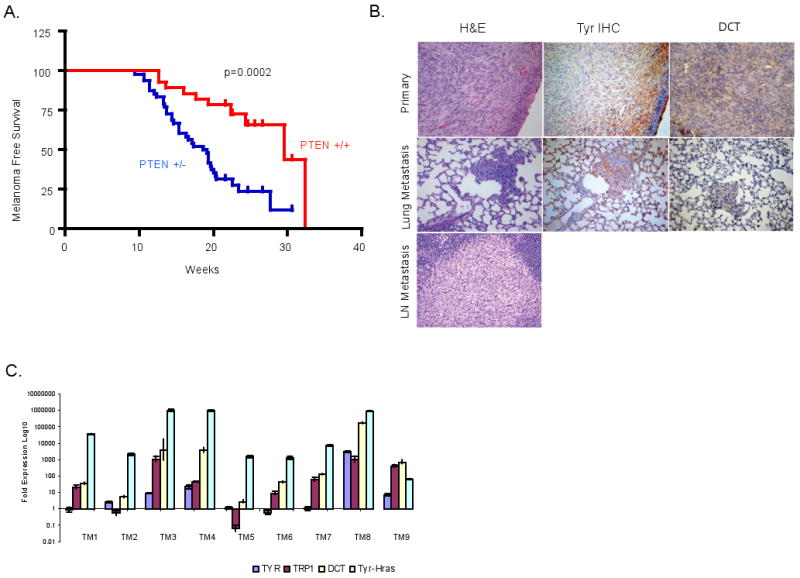
(A) Kaplan-Meier melanoma free survival analysis of Ras-Ink4a/Arf mice with wild type (n=28) or heterozygous (n=48) Pten allele. Non-melanoma tumors were censored from this analysis. Statistically significant differences were detected between the two cohorts (p=0.0002) (B) H&E (left), Tyrosinase (middle) and Dct (right) staining of primary melanoma (top panels) and lung metastasis (middle panels). Bottom panel shows H&E of a probable melanoma metastasis on a lymph node, based on cell morphology. (C) Real time reverse transcription PCR analysis for mRNA level of Tyr-Hras transgene and Tyr, Trp1 and Dct melanocytic markers in cutaneous melanomas from Ras-Ink4a/Arf animals that were wild type (TM1-4) or heterozygous for Pten allele (TM5-9) relative to the ones from non-transformed INK4a-/- melanocytes.
Table 1.
Incidence of melanoma and median survival of RAS-Ink4a/Arf animals on Pten wild type and heterozygous background
| RAS Ink4a/Arf | Pten +/+ | Pten +/- |
|---|---|---|
| Melanoma | 10 | 36 |
| Non-Melanoma | 18 | 12 |
| Median survival (weeks) | 30 | 19 |
Extensive experience with the RAS-Ink4a/Arf model has established that metastasis does not occur in this model (Bardeesy et al., 2001; Bardeesy et al., 2005; Chin et al., 1997; Chin et al., 1999). Among the 21 tumor-bearing RAS-Ink4a/Arf mice heterozygous for Pten, full histological surveys uncovered one case of melanoma metastasis to the lung and one case with metastsis in a draining lymph node, although tissue availability only enabled melanocyte marker confirmation in the former (Tyrosinase and Dct/TRP2 positive; Figure 1B). Although sample size is small, these observations prompted an examination for a potential role of Pten in suppression of melanoma metastasis.
Loss of PTEN enhances invasion of primary and transformed melanocytes
We next assessed the impact of Pten status on the migration and invasion activity of RAS-Ink4a/Arf melanoma cells in vitro (Figure 2). First, we asked whether Pten protein levels in early-passage melanoma cultures derived from spontaneously arising tumors track with migratory and invasive activity. We selected three tumors with high, intermediate or absent Pten protein levels (AL4, CN41 and CN44, respectively) (Figure 2A); early passage CN44 cells show absence of both Pten mRNA and protein with retention of the Pten gene consistent with epigenetic silencing (Figure 2B). Using the modified Boyden chamber assay, the level of Pten expression negatively correlated with invasion through the Matrigel (Figure 2C). Second, we observed an overall lower level of invasive activity across 4 independent RAS-Ink4a/Arf Pten+/+ melanoma cell lines when compared with 5 independent RAS-Ink4a/Arf Pten+/- melanoma cell lines (Figure 2D). Third, these correlations in tumors aligned well with Pten knockdown and reconstitution experiments in vitro and in vivo. Specifically, RNAi-mediated knockdown of Pten in a Pten+/+ RAS-Ink4a/Arf melanocyte and melanoma cell lines using two independent shRNAs (sh4 and sh11 with documented efficient knockdown of Pten protein levels) showed increased invasion in non-transformed melanocyte culture (C140) (Figure 3A) as well as in melanoma cells CN116 (Figure 3B), both were derived from RAS-Ink4a/Arf mice with intact Pten. Similar results were obtained with AL4 RAS-Ink4a/Arf Pten+/+ melanoma cell line (Supplemental Figure 2), demonstrating increased invasion in Boyden chamber in vitro compared to the control which was transduced with empty vector (EV). While we noted an increase in proliferation by Pten loss (Figure 3C), the level of invasion was still markedly increased when normalized for proliferation rates (see material and method). In vivo, Pten knockdown resulted in reduced melanoma-free survival upon subcutaneous implantation of these melanoma cells in severe combined immunodeficient (SCID) mice (Figure 3D). Finally, Adenoviral-mediated Pten reconstitution in CN44 melanoma cells decreased invasion relative to GFP-expressing adenovirus control cells (Figure 3E, p=0.02). Together, these data show that Pten levels can influence the invasive potential of primary and transformed melanocytes.
Figure 2. Migration and invasion capability of Ras-Ink4a/Arf melanoma cells inversely correlates to the PTEN protein level.
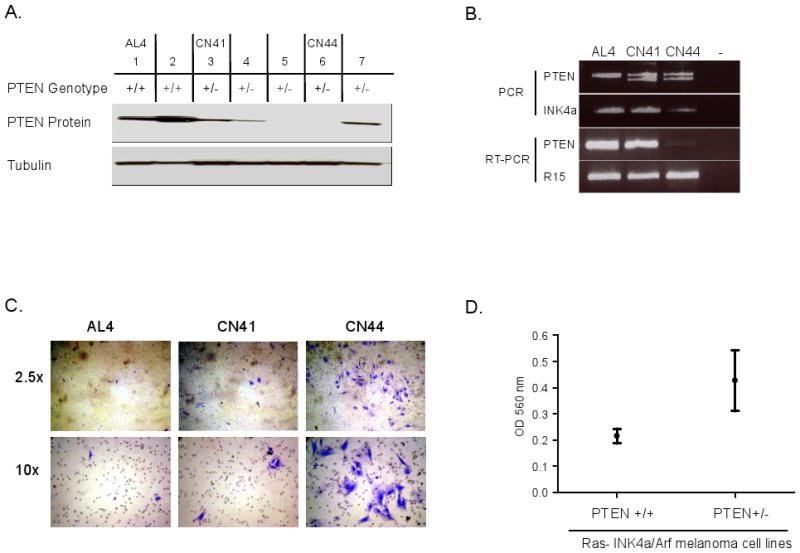
(A) Immunoblot analyses of PTEN in cell lysates from early-passage Ras-Ink4a/Arf melanoma. The corresponding Pten genotype is shown on top of the blot. Note the different level of Pten in AL4 (high), CN41 (intermediate), and CN44 (loss), which were used in B and C. (B) (Top) PCR analysis on genomic DNAs of early passage AL4, CN41, and CN44 melanoma cells of Pten (exon 5) and Ink4a allele. (Bottom) Semi-quantitative reverse transcription PCR analysis for Pten mRNA expression (20 cycles). Ribosomal protein R15 mRNA expression serves as an internal control (19 cycles). (C) Boyden Chamber assay. Photographs of a representative field of the stained membrane show highest level of invasion through the Matrigel by CN44 melanoma cells. (D) Optical quantitation of Boyden Chamber assay on Ras-Ink4a/Arf-Pten+/+ (n=4) and Ras-Ink4a/Arf-Pten+/- (n=5) early passage melanoma cell lines. Pten wt cells showed overall lower invasive capacity (OD560 mean= 0.2152 ± 0.02781, N=4) when compared to Pten heterozygous cells (OD560 mean= 0.4268 ± 0.1160, N=5).
Figure 3. RNAi mediated Pten inactivation accelerates tumorigenesis and promotes invasion of Ras-Ink4a/Arf melanoma and melanocyte cell lines.
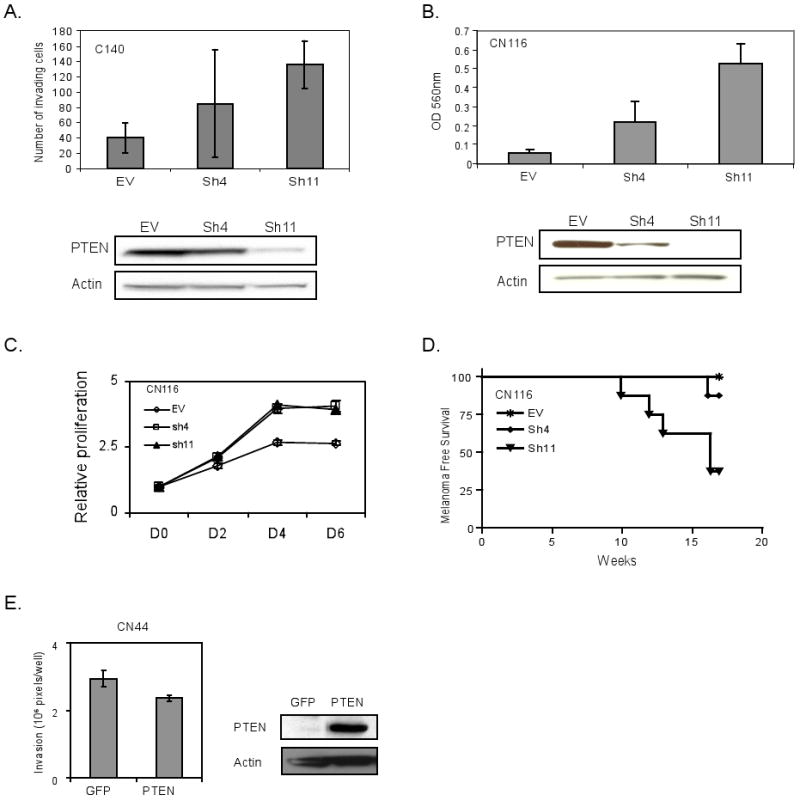
C140 (Ras-Ink4a/Arf-Pten+/+) non-transformed melanocytes (A) and CN116 (Ras-Ink4a/Arf-Pten+/+) melanoma cell line (B-D) were infected with pSuper.Retro empty vector (EV) or two independent shRNAs targeting Pten- Sh4 and Sh11. CN44 (Pten+/-Ras-Ink4a/Arf-) melanoma cell line with Pten protein loss (E) was transduced with adenovirus expression GFP or PTEN. Western blots in A, B, and E show Pten level. Actin serves as a loading control).
(A, B &E) Boyden Chamber assay. Cell invasion through a Matrigel was quantified by counting cristal violet stained cells under a microscope (5 fields per well) (A), by measuring absorbance at O.D. 560nm after dye extraction (B), or by pixel quantification (E). Results reflect normalization for differences in proliferation (see Material and Methods). (C) Cell proliferation curve. The effect of Pten loss on growth of CN116 melanoma cells in 1% serum containing media was measured over 6 days by quantification of crystal violet staining. (D) Kaplan-Meier melanoma free survival analysis. SCID mice were subcutaneously injected into the flanks with half million cells of CN116 Ras-Ink4a/Arf melanoma cells infected with pSuper.Retro Sh4 (N=8), Sh11 (N=8) or empty vector (N=8). Inactivation of Pten by Sh11 resulted in earlier melanoma onset (P value=0.0081).
PTEN inactivation correlates with downregulation of E-cadherin
Many PTEN biological functions have been linked to its lipid phosphatase activity through which PIP3 is converted to PIP2 resulting in inactivation of AKT. At the same time, it is clear that PTEN exerts functions that are apparently mediated via AKT-independent pathways. Of relevance to the current study, PTEN has been shown to modulate actin cytoskeletal organization, focal contacts, and directional cell motility via focal adhesion kinase (FAK) (Tamura et al., 1999) and can physically interact with E-cadherin at junctional complexes to stabilize cell-cell contacts, thereby suppressing invasiveness (Kotelevets et al., 2005; Vogelmann et al., 2005). In our model system, we sought to better understand the molecular action of Pten by assessment of known targets of Pten pathway (Figure 4).
Figure 4. E-cadherin protein levels are correlated to Pten level.
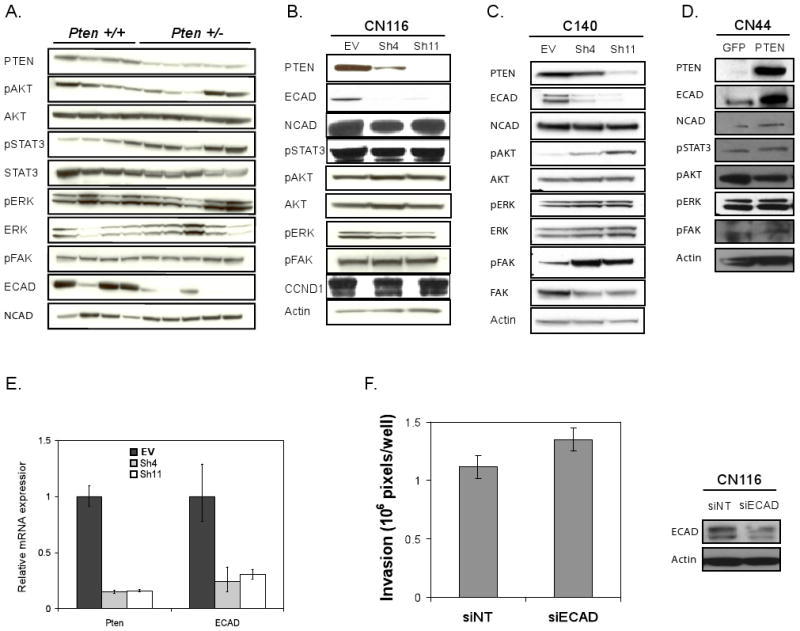
(A) Molecular profile comparison of Ras-Ink4a/Arf Pten+/+ and Pten +/- early passage melanoma cell lysates by immunoblotting with antibodies labeled on left. The corresponding Pten genotype is shown on top of the blot. E-cadherin protein levels are lower on Pten +/- melanoma cells compared to on Pten +/+ cells. (B-D) Immunoblot analysis CN116 (B) and C140 (C) with EV, sh4, or sh11, and CN44 (D) cells with GFP or PTEN expression with antibodies indicated. (E) Real time rt-PCR analysis of Pten and E-Cadherin expression in CN116 cells with EV, Sh4, or Sh11. (F) Boyden chamber assay (left) of CN116 cells following siRNA-mediated E-Cadherin downregulation (siECAD) compared to non-targeting control (siNT) and western blot analysis (right).
Early passage RAS-Ink4a/Arf Pten+/- melanoma cells showed consistent E-cadherin downregulation relative to RAS-Ink4a/Arf Pten+/+ controls; in contrast, the levels of p-AKT, p-FAK, and p-ERK did not track consistently with Pten genotype (Figure 4A). While increased p-STAT3 was observed in RAS-Ink4a/Arf Pten+/- cell lines relative to RAS-Ink4a/Arf Pten+/+ controls, Pten knockdown in CN116 and AL4 RAS-Ink/Arf Pten+/+ melanoma cells failed to show any enhancement in p-STAT3 as well as p-AKT and p-FAK (Figure 4B; Supplemental figure 2); whereas, Pten knockdown in primary RAS-Ink4a/Arf-/- melanocytes resulted in increased phosphorylation of these molecules (Figure 4C). In contrast, Pten knockdown results in consistent down-regulation of E-cadherin expression in both melanocyte and melanoma cell lines. Conversely, Pten reconstitution was associated with no change in p-STAT3, p-FAK, and p-ERK, a modest decrease in p-AKT, and robust increase in E-Cadherin (Figure 4D). In both CN116 and AL4 cells, Pten knockdown correlated well with loss of E-cadherin protein and mRNA expression (Figure 4B&E and Supplemental Figure 2B&C). Since loss of E-Cadherin has been linked to epithelial to mesenchymal transition and progression, our data raised the possibility that PTEN-mediated modulation of E-cadherin expression may be integral to the impact of PTEN on the invasive phenotype of melanoma. Indeed, RNAi-mediated knockdown of E-Cadherin resulted in increased invasion of CN116 cells (Figure 4F, p=0.04).
PTEN inactivation enhances invasion of human melanoma cells
To establish the human relevance of our findings, we next assessed the effect of PTEN loss on invasion as well as on p-AKT in human melanoma cell lines. In line with our murine studies, siRNA-mediate knockdown of PTEN in WM1366, human melanoma cells harboring NRAS Q61I mutation, resulted in increased invasion without a significant increase of AKT phosphorylation compared to siNT (non-targeting) control (1.34 fold, p=0.00008) (Figure 5A). In addition, we investigated the effect of PTEN reconstitution in human melanoma cells WM793A and 1205Lu harboring BRAF V600E mutation and PTEN loss. Previously, Stahl et al have shown that reconstitution of PTEN expression in PTEN null melanoma cell lines leads to significant decrease in AKT phosphorylation and apoptosis (Stahl et al., 2003). Here, to assess the impact of PTEN on invasion, PTEN level was titrated to avoid activation of cell death associated with high level expression (Figure 5B). At this level of ectopic PTEN expression in WM793A and 1205Lu cells, PTEN reduced invasion compared to control without significant decrease of AKT phosphorylation (WM793A: 49%; p=0.003 and 1205Lu: 24%; p=0.048) (Figure 5B and data not shown). This is consistent with the study reported by Dankort et al showing the collaboration between Braf and Pten in melanoma progression (Dankort et al., 2009). Together these studies support the view that PTEN inactivation enhances invasion of mouse and human melanoma cells harboring RAS/RAF/MAPK pathway activation without significant changes in total AKT phosphorylation.
Figure 5. PTEN inhibits invasion of human melanoma cells with RAS/RAF/MAPK activation.

(A) Western blot analysis with indicated antibodies (top) and invasion chamber analysis (bottom) of NRAS mutated human melanoma cell WM1346 with siNT (non-targeting) control or with siRNA targeting PTEN (siPTEN).
(B) WM793A human melanoma cell lines harboring BRAF mutation and loss of PTEN expression were transduced with adenovirus encoding control GFP or with PTEN. Immunoblot (top left), Annexin V/PI apoptosis (bottom left), and invasion chamber analysis (right) are shown.
PTEN inactivation preferentially activates AKT2 among AKT isoforms
Intriguingly, treatment with PI3K inhibitor, LY294002, in RAS-Ink4a/Arf melanoma cells led to a decrease in total p-AKT and abrogation of the increased invasion brought about by Pten knockdown (Figure 6A), suggesting that PI3K-AKT signaling does contribute to the invasion phenotype we observed. Although modulation of PTEN does not track well with total p-AKT levels in RAS-Ink/Arf melanoma cells (Figures 3 and 4), the fact that all three isoforms of AKT (AKT1, 2, and 3) are phosphorylated in RAS-Ink/Arf melanoma cells (data not shown and Supplemental figure 3) raised the possibility that Pten may regulate invasion through switching among activated AKT isoforms (AKT1, 2, and 3) without substantially affecting the total amount of active AKT.
Figure 6. PTEN loss has more impact on pAKT2 among AKT isoforms.
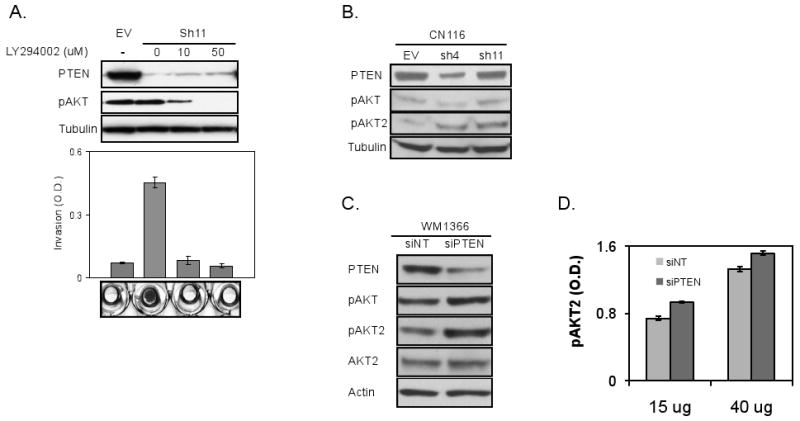
(A) LY294002 treatment decreased p-AKT level of CN116 cells with Pten loss (sh11) (top, immunoblots with indicated antibodies), which correlates with invasive capability (bottom, Boyden chamber assay). Invasion was quantitated by measuring absorbance of crystal violet dye. Representative images of invasion chamber are shown.
(B) Immunoblot analysis of CN116 RAS Ink/Arf melanoma cells with EV, Sh4, or Sh11. Note much higher increase of pAKT2 compared to total pAKT by loss of Pten protein expression.
(C&D) PTEN expression was knocked down with siRNA targeting PTEN (siPTEN) in WM1366 and its effect on AKT phosphorylation (total and AKT2) was measured by immunoblotting (C). p-AKT2 level was analyzed by ELISA (D).
To explore further the thesis that specific AKT isoform confers differential activity in melanoma cells, we first examined AKT3 activation since AKT3 is reported to be the predominant AKT isoform in human melanoma (Stahl et al., 2004). Here, we ectopically expressed a constitutively active form of AKT3 (myristoylated AKT3) in CN116A cells. While it dramatically elevated AKT3 phosphorylation at Ser 473 without affecting p-FAK and p-STAT3, myr-AKT3 expression actually inhibited invasion (Supplemental figure 4A). Of note, this result is consistent with the anti-invasive role of myristoylated AKT1 in breast cancer cells reported by Bissell and colleagues (Liu et al., 2006). On the other hand, a dominant negative form of AKT2 expression (Yuan et al., 2003)in invasive CN44 melanoma cells led to inhibition of invasion (Supplemental figure 4B), supporting a pro-invasive role for AKT2. Furthermore, in line with the thesis that PTEN loss results in switching of AKT isoform, siRNA-mediated knockdown of PTEN in the human melanoma cell line WM1366 enhanced level of p-AKT2 without significantly impacting on total pan-isoform p-AKT when compared to siNT control (Figure 6C). This can be further corroborated by ELISA measurement, where p-AKT2 levels were found to be significantly higher in PTEN knockdown cells compared to non-targeting siRNA controls (Figure 6D, 15ug; 1.26 fold (p=0.024) and 40ug; 1.15 fold (p=0.023)). Accordingly, in the mouse, loss of Pten in CN 116 and AL4 melanoma cells was associated with increased p-AKT2 without significant change on total p-AKT (Figure 6B and Supplemental Figure 4). Together, these data point to PTEN inactivation in RAS activated melanomas promotes invasion through preferential activation of AKT2.
Discussion
Utilizing genetically engineered models, we investigated the interactions of three melanoma prone genetic elements, namely RAS activation, Pten loss, and Ink/Arf deficiency. Previously, RAS activation in mouse model has been shown to cooperate with Ink/Arf inactivation to drive non-metastatic melanomas (Chin et al., 1997) and loss of Pten when combined with loss of Ink/Arf function contributes to melanoma tumorigenesis at low frequency (You et al., 2002). However, there still exists considerable uncertainty surrounding the genetic interactions of these lesions. Here, using genetically engineered mouse models in vivo and mouse and human cell systems in vitro, we demonstrated synergistic interaction of these three components in melanoma genesis and progression, manifested as accelerated melanoma development and enhanced migration and invasion of melanoma cells from Pten+/-RAS-Ink/Arf mice compared to Pten+/+Ras-Ink/Arf mice. In addition, we have further elucidated the key molecular events acting as points of convergence and distinction of their downstream signaling pathways. Firstly, we observed E-Cadherin downregulation associated with PTEN loss. Secondly, we have shown differential regulation of AKT isoforms by PTEN loss in RAS mutated melanomas. In particular, we observed preferential increase of phospho-AKT2, much less appreciated isoform in melanoma tumorigenesis.
Previous work has proposed that RAS activation and PTEN extinction are functionally and genetically redundant based on the relative reciprocity of NRAS and PTEN alterations observed in human melanomas and cell lines (Tsao et al., 2000). However, in human melanoma, we found that 14% (2/14) of the NRAS mutated tumor samples harbor Pten loss or mutation (data not shown). Conversely, 17% (2/12) of the melanoma samples with PTEN loss or mutation also contain NRAS mutation (L. Chin, unpublished data). Thus, our observations in the genetically engineered mouse models are relevant for the human disease, as further supported by our work in human cell lines.
PTEN is a major tumor suppressor located on chromosome 10q23-24, the frequently deleted region in multiple tumor types (Li et al., 1997; Steck et al., 1997). In human melanoma, PTEN is deregulated via genetic mechanisms (mutation and allelic loss) (Guldberg et al., 1997; Tsao et al., 1998)) as well as via epigenetic silencing such as methylation (Mirmohammadsadegh et al., 2006) or altered subcellular localization (Trotman et al., 2007). PTEN methylation status serves as a prognostic marker for poor survival (Lahtz et al.). In our study, murine melanomas derived from from Pten+/-RAS-Ink4a/Arf mice commonly exhibit loss of Pten protein expression, consistent with the observation in human.
AKT kinase family consists of 3 highly homologous isoforms: AKT1/PKB-alpha, AKT2/PKB-beta, and AKT3/PKB-gamma. Although structurally similar, each isoforms of AKT have non-overlapping functions in cancer (Gonzalez and McGraw, 2009). Phenotypic analyses of polyoma middle T (PyMT) and ErbB2/Neu-driven mammary adenocarcinoma mouse models suggest that Akt1 may inhibit and Akt2 may enhance the invasion (Maroulakou et al., 2007). This is consistent to previous reports that AKT2 expression in breast and ovarian cancer cells increased adhesion, invasion, and metastasis, associated with β1 integrin upregulation (Arboleda et al., 2003). Several mechanisms have been proposed to achieve isoform specific signaling such as distinct tissue distribution, differential activation by extracellular stimuli, distinct intrinsic catalytic activity and substrage specificity, and differential subcellular localization of the AKT isoforms (Gonzalez and McGraw, 2009). Although the role of PTEN loss in AKT activation is well established, the effect of PTEN loss on the activity of each AKT isoform has not been addressed in detail.
Among the 3 isoforms, AKT3 is considered to be the predominant isoform activated in melanomas by amplification and somatic activating mutations (Davies et al., 2008; Stahl et al., 2004). Here, in our mouse melanomas with activated RAS signaling, Pten loss is associated with enhanced invasion accompanied by a shift to increase phosphorylation of AKT2 isoform, which has been linked to invasiveness, without significant change in total amount of AKT activation (Figure 6). Interestingly, Tsichils and colleagues recently reported that it is the balance between AKT1 and AKT2 rather than the amount of total AKT activity that differentially regulates microRNA expression, whereby AKT2 induces miR-200 microRNA family, which in turn decreases E-Cadherin expression (Iliopoulos et al., 2009). In this study, we report an increase on pAKT2 and decrease of E-Cadherin protein levels upon Pten inactivation. It will be of interest to asses if mir-200 is induced in our model and if it is responsible for the observed downregulation of E-Cadherin. In this regard, it is of interest to note that, although real time quantitative PCR analysis revealed that E-cadherin was downregulated at the transcriptional level following downregulation of Pten expression in RAS Ink/Arf melanoma cells, upregulation of its known transcriptional regulators including Slug, Snail, and Twist was not observed (data not shown). Zeb1 showed small nonsignificant upregulation by Pten loss (1.23 fold for sh4, 1.38 fold for sh11 vs. EV, data not shown).
In conclusion, we provide genetic evidence in vivo that RAS activation and Pten loss cooperate to drive melanoma genesis and progression in the mouse. Our data encourages more systematic and comprehensive characterization for the activity of each AKT isoforms at distinct stages of melanoma tumorigenesis and in clinical trials when addressing the effect of AKT inhibitors.
Material and Methods
Mouse tumor cohorts
Tyrosinase enhancer-promoter-driven H-RASV12G transgenic mice in FVB (N10) (Chin et al., 1997) were crossed onto the Pten mutant background in mixed FVB/C57Bl/6 strain (Podsypanina et al., 1999) and the Ink4a/Arf-/- background in FVB (N10) (Serrano et al., 1996) to generate the compound mice Tyr-H-RASV12GInk4a/Arf-/-Pten+/+ and Tyr-H-RASV12G; INK4a/Arf-/-Pten+/-. Mice were observed biweekly for development of tumors or appearance of ill health. Premoribund animals or animals with significant tumor burdens were sacrificed, and detailed autopsies were performed. Tumor specimens were fixed in 10% formalin and embedded in paraffin for histological analysis as previously described (Chin et al., 1997). In cases in which sufficient specimens were available, primary tumors were adapted to culture to establish derivative cell lines and/or flash-frozen for subsequent analyses.
Melanocyte and melanoma cell culture
Melanoma cell lines were derived from mouse tumors by digestion with collagenase and Hyaluronidase (2 mg/ml each; Sigma-Aldrich, St Louis,MO, USA) for 2 hours followed by cultivation with RPMI 1640 media (Gibco BRL, Gaithersburg, MD, USA) containing 10% FBS and 1% penicillin/streptomycin. Melanocyte cultures were generated from newborn mouse epidermis as described (Sviderskaya et al., 1997) and maintained in RPMI 1640 containing 5% FBS, 1% penicillin/streptomycin, 200 pM cholera toxin, 200 nM 12-Otetradecanoylphorbol- 13-acetate (TPA).
RNA analysis
For analyses of gene expression, total RNA was isolated from primary cutaneous melanomas or from cultured cells using Trizol (Gibco BRL, Gaithersburg, MD, USA) according to manufacturer's protocol. Total RNA was treated with RQ1 DNAse (Promega, Madison, WI, USA) and 1ug total RNA was used for reverse transcription reaction using Superscript II polymerase (Invitrogen, Carlsbad, CA, USA) primed with oligo(dT). Coding regions were amplified by PCR or quantitative real time PCR using SYBR Green (Applied Biosystems, Foster City, CA, USA) on an Mx3000P real-time PCR system (Stratagene, La Jolla, CA, USA). Ribosomal protein R15 was used as an internal expression control. Primer sequences are as follows: Tyr: F-ccagaagccaatgcacctat and R-agcaataacagctcccacca; Trp1: F-attctggcctccagttacca and R-ggcttcattcttggtgcttc; Dct: F-aacaacccttccacagatgc and R-tctccattaagggcgcatag; RAS transgene: F- cagatcaaacgggtgaagg and R- cacttgcagctcatgcag; R15: F-cttccgcaagttcacctacc and R-tacttgagggggatgaatcg; Pten: F- ttgcaatcctcagtttgtgg and R- tggctgagggaactcaaagt; E-Cadherin: F- ctttaagcccagcactcagg and R- cctgcttcctgagaaaatgc
Protein analyses
For immunoblots, lysates were resolved on 4-12% Nu-Page minigels (Invitrogen). Western blots were probed with antibodies against AKT, phospho-AKT (Ser473), PTEN, p44/42 and phospho-p44/42 Map Kinase (Thr202/Tyr204), phospho-STAT3 (Ser727) from Cell Signaling Technology (Danvers, MA). Anti-FAK [pY397] antibody was obtained from Invitrogen Biosource, E-CAD and N-CAD from BD Transduction Laboratories (San Jose, California), and p-AKT2 (Ser474) from Abcam (Cambridge, MA). Beta-Actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), beta-Tubulin (Sigma), and GAPDH (Sigma) protein expression served as loading control. All antibodies were used at 1:1000 dilutions. p-AKT2 level was measured with PathScan p-AKT2 (Ser 474) Sandwitch ELISA Kit (Cell Signaling Technology) following manufacturer's protocol. Immunohistochemical staining of Tyrosinase (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and DCT (TRP2) (provided by Dr. Glenn Merlino (National Cancer Institute) was performed on formalin-fixed paraffin-embedded tissues with heat induced epitope retrieval (0.01M citrate buffer, pH6.0).
siRNAs, Plasmids, and viral infection
In order to knock-down Pten using RNAi, Pten shRNA oligos were designed using iRNAi software (http://www.mekentosj.com/irnai). Pten shRNA oligos were cloned into the pRetroSuper retroviral vector (Brummelkamp et al., 2002). Viral supernatants were produced using standard protocols. Retroviral vectors were transfected into 293T cells using the pCL-Eco helper plasmid. Retroviral supernatants isolated at 36 and 48h after transfection were diluted 1:1 in culture medium and used to infect melanoma cell lines in the presence of 4 μg/ml Polybrene. At 24 h postinfection, the cells were selected for 2 days in growth medium containing 2.5 μg/ml puromycin. Western blot analysis of cell extracts from cells infected with the Pten shRNA containing retroviral vectors showed that two Pten shRNA oligos (Pten sh4: 5′ GGCACAAGAGGCCCTAGAT 3′ and Pten sh11: 5′AGACAAGGCCAACCGATAC 3′) show efficient Pten knock-down. Adenoviruses for PTEN was provided by Dr. Jin Q. Cheng (Moffitt Cancer Center). The siRNAs targeting human PTEN was purchased from Cell Signaling Technology and mouse E-Cadherin from Dharmacon (Lafayette, CO, USA).
Boyden Chamber
Invasion assay was performed in Matrigel Invasion Chambers (24 well plate format from BD BioCoat™ San Jose, CA, USA) or in Chemotaxis Chambers (96 well format from NeuroProbe (Gaithersburg, MD, USA) following coating with Matrigel (BD). Briefly, cells were trypsinized, rinsed twice with PBS, resuspended in serum-free RPMI 1640 media, and were loaded on upper chamber and allowed to invade through the Matrigel towards 10% FBS serum for 20 hours. Non-invasive cells were removed and remaining cells were fixed and stained with Cristal Violet stain. Invasive cells were either counted under a microscope (5 fields per well) or quantitated via Pixel quantification (Richard Rosenman V1.2), or Cristal Violet dye was extracted with 10% Acetic Acid and cells were quantified at OD 560nm. For loading control and to normalize for differences in cell proliferation, cells were seeded in a regular 24 well plate and allowed to grow for the same time as the incubation in the Boyden chambers after which cells were stained with Cristal Violet. Cells were quantified at OD 560nm after dye extraction and this number was used to normalize the invasion value. Statistical analysis was done by t-test.
Supplementary Material
Acknowledgments
C. Nogueira was supported by a fellowship from FCT (PRAXIS / BD / 21794 / 99). M. Kim was supported by the Claudia Adams Barr Program, the Dermatology Foundation, and Melanoma Research Foundation. J. H. Dannenberg was supported by Damon-Runyon Cancer Research Foundation and the Dutch Cancer Society. This work is supported by grants from the NIH (UO1 CA84313; RO1 CA93947) to L. Chin and from the Bankhead Coley Pilot Research Award and American Cancer Society Institutional Research Grant (#93-032-13) to M. Kim. We thank Dr. Jin Q. Cheng (Moffitt Cancer Center) for helpful discussion and adenoviruses for PTEN and DN-AKT2 expression, and Dr. Ronald A. DePinho for critical reading of this manuscript.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005;65:4005–11. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001;21:2144–53. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Kim M, Xu J, Kim RS, Shen Q, Bosenberg MW, et al. Role of epidermal growth factor receptor signaling in RAS-driven melanoma. Mol Cell Biol. 2005;25:4176–88. doi: 10.1128/MCB.25.10.4176-4188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birck A, Ahrenkiel V, Zeuthen J, Hou-Jensen K, Guldberg P. Mutation and allelic loss of the PTEN/MMAC1 gene in primary and metastatic melanoma biopsies. J Invest Dermatol. 2000;114:277–80. doi: 10.1046/j.1523-1747.2000.00877.x. [DOI] [PubMed] [Google Scholar]
- Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–86. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–82. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997;11:2822–34. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005;37:745–9. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–8. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan P, Singh AB, Ellis DL, Richmond A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappaB and tumor progression. Cancer Res. 2002;62:7335–42. [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr Protoc Hum Genet. 2008;Chapter 10(Unit 10):11. doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel VK, Ibrahim N, Jiang G, Singhal M, Fee S, Flotte T, et al. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene. 2009;28:2289–98. doi: 10.1038/onc.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–8. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldberg P, thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57:3660–3. [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Yoshinaga K, Suzuki A, Sakurada A, Ohmori H, Horii A. Anticorresponding mutations of the KRAS and PTEN genes in human endometrial cancer. Oncol Rep. 2000;7:567–70. doi: 10.3892/or.7.3.567. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009;2:ra62. doi: 10.1126/scisignal.2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari M, Papp T, Kirchner S, Diener U, Henschler D, Burg G, et al. Analysis of ras mutations in human melanocytic lesions: activation of the ras gene seems to be associated with the nodular type of human malignant melanoma. J Cancer Res Clin Oncol. 1995;121:23–30. doi: 10.1007/BF01202725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–81. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. Faseb J. 2005;19:115–7. doi: 10.1096/fj.04-1942fje. [DOI] [PubMed] [Google Scholar]
- Lahtz C, Stranzenbach R, Fiedler E, Helmbold P, Dammann RH. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J Invest Dermatol. 130:620–2. doi: 10.1038/jid.2009.226. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Lin WM, Baker AC, Beroukhim R, Winckler W, Feng W, Marmion JM, et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008;68:664–73. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, et al. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc Natl Acad Sci U S A. 2006;103:4134–9. doi: 10.1073/pnas.0511342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Ziel-van der Made AC, Autar B, van der Korput HA, Vermeij M, van Duijn P, et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 2005;65:5730–9. doi: 10.1158/0008-5472.CAN-04-4519. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A. 2003;100:7841–6. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66:6546–52. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L. Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene. 2003;22:5055–9. doi: 10.1038/sj.onc.1206809. [DOI] [PubMed] [Google Scholar]
- Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–90. [PubMed] [Google Scholar]
- Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–10. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000;60:3605–11. [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–78. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Sviderskaya EV, Bennett DC, Ho L, Bailin T, Lee ST, Spritz RA. Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J Invest Dermatol. 1997;108:30–4. doi: 10.1111/1523-1747.ep12285621. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–41. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60:1800–4. [PubMed] [Google Scholar]
- Vogelmann R, Nguyen-Tat MD, Giehl K, Adler G, Wedlich D, Menke A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J Cell Sci. 2005;118:4901–12. doi: 10.1242/jcs.02594. [DOI] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- You MJ, Castrillon DH, Bastian BC, O'Hagan RC, Bosenberg MW, Parsons R, et al. Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. Proc Natl Acad Sci U S A. 2002;99:1455–60. doi: 10.1073/pnas.022632099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZQ, Feldman RI, Sussman GE, Coppola D, Nicosia SV, Cheng JQ. AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: implication of AKT2 in chemoresistance. J Biol Chem. 2003;278:23432–40. doi: 10.1074/jbc.M302674200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.