

Abstract
The N-monophenylcarbamate analogues of neostigmine methyl sulfate (6) and pyridostigmine bromide (8) together with their precursors (5), (7), and the N(1)-methylammonium analogues of (−)-phenserine (12), (−)-tolserine (14), (−)-cymserine (16) and (−)-phenethylcymserine (18) were synthesized to produce long-acting peripheral inhibitors of acetylcholinesterase or butyrylcholinesterase. Evaluation of their cholinesterase inhibition against human enzyme ex vivo demonstrated that, whereas compounds 5–8 possessed only marginal activity, 12, 14, 16 and 18 proved to be potent anticholinesterases. An extended duration of cholinesterase inhibition was determined in rodent, making them of potential interest as long-acting agents for myasthenia gravis.
Keywords: Myasthenia gravis, pyridostigmine, neostigmine, N-phenylcarbamate analogues of (−)-physostigmine, (−)-phenserine, (−)-cymserine, congenital myasthenic syndromes
1. Introduction
The compounds 3-[(dimethylamino)carboxyl]oxy]-N,N,N-trimethylammonium methyl sulfate, better known as neostigmine methyl sulfate (3),1 and 3-[(dimethylcarbamoyl)oxy]-1-methylpyridinium bromide, pyridostigmine bromide (4)2 (Figure 1) are well known peripheral cholinesterase inhibitors that have been extensively used over the last five decades in the treatment of neuromuscular junction disorders, epitomized by the autoimmune disorder myasthenia gravis and non-autoimmune congenital myasthenic syndromes, and have additionally been used as prophylactics for organophosphate poisoning.3–5 These quaternary compounds are highly water-soluble, and consequently minimally cross the brain-blood barrier. Restricted from entering the brain, they exert potent systemic anticholinesterase activity at skeletal muscles, in the absence of centrally mediated actions or side effects. A short duration of pharmacodynamic action, usually 30 to 120 min,3 allied with rapid metabolism has made the routine clinical use of these compounds troublesome.3–5 This has resulted in necessary multiple dosing (10 doses over 24 hr) as well as the development of slow-release formulations to achieve efficacy.
Figure 1.

Recent studies have demonstrated that specific aromatic carbamate analogues of (−)-physostigmine (9), which include the Alzheimer’s disease experimental drug (−)-phenserine (11), whose history, chemistry and pharmacology have been reviewed,6–9 as well as (−)-tolserine (13), (−)-cymserine (15) and (−)-phenethylcymserine (17), possess far longer durations of in vivo cholinesterase inhibitory action compared to not only their methyl carbamate analogue, (−)-physostigmine (9),7–9 but also to neostigmine and pyridostigmine.3–5,10–12 Phenyl carbamate analogues of neostigmine methyl sulfate (6) and pyridostigmine bromide (8) together with their corresponding precursors (5) and (7), as well as methyl quaternary analogues of (−)-phenserine (12), (−)-tolserine (14), (−)-cymserine (16) and (−)-phenethylcymserine (18) were therefore synthesized to assess their potential pharmacological utility. The assessment of their anticholinesterase activities is reported here as a first step in a search for better agents for treatment of myasthenia gravis that are unencumbered by a short duration of action.
2. Results and Discussion
2.1. Chemistry
As reported previously,1,2 m-dimethylaminophenol (1) and 3-hydroxypyridine (2) can be reacted with different N-alkyl or aryl carbamoyl chlorides to give different N,N-disubstituted carbamates. Following the procedure for the synthesis of (−)-phenserine (11),13 m-dimethylaminophenol (1) and 3-hydroxypyridine (2) were reacted with phenylisocyanate to generate N-monophenyl-substituted carbamates, 5 and 7, in yields of 86% and 70%, respectively. Compounds 5 and 7 then were reacted with methylsulfate and methylbromide, separately, to provide the corresponding quaternary salts, 6 and 8, in yields of 80% and 70%. (−)-Phenserine (11), (−)-tolserine (13), (−)-cymserine (15) and (−)-phenethylcymserine (17) were obtained according to a known procedure13 and then, together with (−)-physostigmine (9), were transformed to quaternary salts 10, 12, 14, 16 and 18 by reaction with methyl bromide, in a manner similar to but different from our prior synthesis of quaternary iodide salts (−)-phenserine (11).14 In this manner our synthesis of a range of quaternary (−)-physostigmine phenylcarbamate bromide salts could be directly compared to pyridostigmine bromide (4)
2.2. Cholinesterase inhibition
The activity of compounds 3 to 18 to inhibit human acetyl- (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8) was assessed by a modification of the Ellman technique,15,16 in which the concentration required to inhibit 50% enzymatic action (IC50 value) was determined in half-log steps over a 5 log concentration range (0.3 nM to 30 uM), and is shown in Table 1. A lower IC50 value is associated with greater anticholinesterase potency.
Table 1.
50% Inhibitory Concentration of Compounds Versus Freshly Prepared Human Erythrocyte AChE and Plasma BChE, and computed Log D value
| No. | Compound | AChE | BChE | a Selectivity A/B or B/A | d Log D |
|---|---|---|---|---|---|
| (IC50, nM, ± SEM) | |||||
| 3 | Neostigmine methyl sulfate | 18.8±3.0 | 60±2 | 3.2 AChE | −2.09 |
| 4 | Pyridostigmine bromide | 360±30 | 900±100 | 2.5 AChE | −2.84 |
| 5 | N-Phenylcarbamate of 3-dimethylaminophenol | 2500±130 | 18,000±3000 | 7.2 AChE | 3.06 |
| 6 | N-Phenylcarbamate of 3-phenol-1-trimethyl ammonium methylsulfate | 1875±110 | 18,000±2200 | 9.6 AChE | −1.4 |
| 7 | N-Phenylcarbamate of 3-hydroxypyridine | >30,000b | >30,000b | None | 1.78 |
| 8 | N-Phenylcarbamate of 3-hydroxy-1-methyl pyridinium bromide | >30,000b | 550±200 | BChE | −2.21 |
| 9 | (−)-Physostigmine | 27.9±2.4 | 16.0±2.9 | 1.7 BChE | 0.42 |
| 10 | (−)-N(1)-Methylammonium bromide of physostigmine | 26.1± 6.2 | 130±15 | 5 AChE | −1.04 |
| 11 | (−)-Phenserine | 24.0±6.0 | 1560±60.0 | 65 AChE | 2.22 |
| 12 | (−)-N(1)-Methylammonium bromide of phenserine | 25.4±2.8 | 210±90 | 8.3 AChE | −0.5 |
| 13 | (−)-Tolserine | 10.3±1.6 | 1950±245 | 189 AChE | 2.66 |
| 14 | (−)-N(1)-Methylammonium bromide of tolserine | 14.6±1.2 | 140±60 | 9.6 AChE | −0.17 |
| 15 | (−)-Cymserine | 760±21 | 51±1.0 | 14.9 BChE | 3.51 |
| 16 | (−)-N(1)-Methylammonium bromide of cymserine | 145±14 | 43±23 | 3.4 BChE | 1.0 |
| 17 | (−)-Phenethylcymserine | >30,000b | 6.0±1.0 | >5000 | 5.72 |
| 18 | (−)-N(1)-Methylammonium bromide of phenethylcymserine | 300±22 | 51±2.6 | 5.9 BChE | 2.43 |
A/B or B/A: selectivity for AChE or BChE from IC50 values.
None: Insufficient activity in the range of 0.3 nM to 30 μM to calculate an IC50 value, and thus considered inactive.
The IC50 data of 9, 11, 13, 15 from ref. (Brzostowska et al., 1992)12, and 17 from ref. (Yu et al., 1999)21.
ACD/PhysChem Suite version 12.01 and PrologD (CompuDrug, Pallas).
The introduction of a phenylcarbamoyl side chain into the primary pharmacophore of either neostigmine (3) or pyridostigmine (4), to provide analogues 6 and 8, resulted in a substantial loss of anticholinesterase activity. The IC50 value of neostigmine (3) declined by 100- and 300-fold for AChE and BChE (from 18.8 to 1875 nM, and from 60 to 18,000 nM) for 6, respectively. This same modification in pyridostigmine (4) resulted in entire loss of AChE activity for 8 but, interestingly and unlike 6, the preservation of BChE inhibitory action.
In contrast, the quaternization of (−)-physostigmine (9) and related phenylcarbamates to provide 10, 12, 14, 16 and 18 with charge characteristics akin to neostigmine (3) and pyridostigmine (4), retained or improved AChE inhibitory action. Specifically, for the N(1)-methylamonium bromides (10, 12, 14) of (−)-physostigmine (9), (−)-phenserine (11) and (−)-tolserine (13), the high AChE potency of the parent compounds was maintained, but the differential selectivity of 9 and 11 for AChE was lost consequent to improved BChE inhibition versus the parent compounds. In the case of the BChE-selective inhibitors, (−)-cymserine (15) and (−)-phenethylcymserine (17), quaternization resulted in significantly improved AChE inhibitory potency for 16 and 18 that, together with a loss 10-fold loss of BChE activity for 18, resulted in the loss of BChE selectivity of these quaternary compounds. In each case, however, the resulting AChE IC50 values of these quaternary (−)-physostigmine phenylcarbamates (10, 12, 14, 16 and 18) compared favorably to neostigmine (3) and were more potent than pyridostigmine (4). In addition the AChE and BChE IC50 values compared favorably to those of prior synthesized quaternary (−)-physostigmine phenylcarbamate iodide salts.14
Computation of Log D values of compounds 3, 4, 6, 8, 10, 12, 14, 16 and 18 was undertaken utilizing Advanced Chemistry Development, Inc. (ACD/Labs, Toronto, Ontario, Canada) (ACD/PhysChem Suite version 12.01) and PrologD (CompuDrug, Pallas, Sedona, AZ) software and is shown in Table 1. This log D value is a measure of the aqueous versus lipid solubility of a compound, which relates to its permeability at biological membranes, as determined from the octanol-water distribution coefficient.
The time-dependent cholinesterase inhibition of compound 16, as a representative of a quaternary (−)-physostigmine phenylcarbamate, was assessed in the anesthetized rat following administration by the intrapertioneal (i.p.) route at a dose of 10 mg/kg. As illustrated in Figure 2, onset of plasma cholinesterase inhibition occurred rapidly, inducing peak inhibition of 66% at 60 min that gradually declined to 33% by 24 hr. No central (tremor) or peripheral (lacrimation and salivation) cholinergically-mediated adverse effects were evident.
Figure 2.
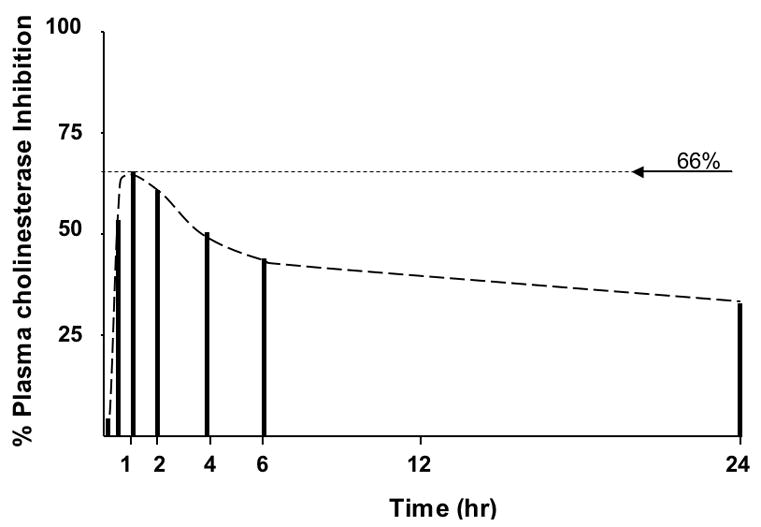
Time-dependent plasma cholinesterase inhibition following a single intraperitoneal dose of compound 16 in rat.
2.3. Discussion
Myasthenia gravis, the most common disorder affecting the postsynaptic element of the neuromuscular junction, is an autoimmune disease. It afflicts up to 2 per 100,000 population, primarily 20 to 30 year old women and older (70 to 80 year) men.17 The majority cases (85%) are associated with an IgG antibody raised against the postsynaptic nicotinic acetylcholine receptor, reducing the number of effective receptors to a third of normal levels.3–5 Impaired neuromuscular transmission together with diminished generation of postsynaptic action potentials results in decreased muscle contraction, leading to the classical clinical features of fluctuating muscle weakness and fatigability.18 Albeit an autoimmune disorder and routinely treated by a wide array of immunosuppressive strategies,4 cholinesterase inhibitors remain crucial to symptomatic treatment, particularly for congenital myasthenic syndromes that are a group of non-autoimmune disorders caused by mutations in several proteins that compose the neuromuscular junction.5 Unfortunately, neostigmine methyl sulfate (3) and pyridostigmine bromide (4), are plagued by the need for frequent dosing, side effects in the elderly and erratic absorption after oral administration.19
The inhibitory interaction between carbamate-based cholinesterase inhibitors, as epitomized by 3, 4 and 9, and the cholinesterase enzymes, AChE and BChE, occurs within a narrow gorge of some 20 Å depth that contains several domains that bind and cleave ACh,20–22 and provide a rationale for the described modifications to generate improved inhibitors.23–26 The three-dimensional structure of the gorge is roughly equivalent to two hemispheres that sandwich a catalytic center between two loops (the acyl- and omega-loops) that form the sidewalls of the active site gorge, with key amino acid differences between AChE and BChE in the gorge region differentiating their respective shapes and catalytic properties.20–22 Four different domains within the gorges of both AChE and BChE have been defined and mapped. These include: (i) a peripheral anionic site (PAS) at the mouth of the gorge that is the first binding domain encountered by a positively charged substrate or inhibitor as it enters, (ii) a slightly deeper cationic-π site, where the quaternary ammonium of choline interacts, (iii) a deeper still acylation site that is the active center of the enzyme and involves a catalytic triad and, (iv), an acyl-binding pocket at the gorge base.20,23
The acylation site defines the active center involved in the catalysis of ACh and is centered around an active serine residue that, in human AChE, comprises a catalytic triad of Ser203, His447 and Glu334 (in human BChE: Ser198, His438, Glu325). The latter two amino acids act as proton acceptors/donors to provide a ‘charge relay’ system and the subsequent activation of the hydroxyl group of Ser203. Substrates, exemplified by ACh, then bind covalently to the active site serine consequent to the attack at the carbonyl group of the substrate by the hydroxyl anion of the serine that leads to a tetrahedral alkoxy anion enzyme-substrate intermediate. This tetrahedral intermediate then collapses, the choline moiety of ACh leaves and the serine becomes acylated, yielding a transient ‘acyl-enzyme’ complex. The presence of a water molecule allows nucleophilic attack on the acyl-enzyme intermediate that, via a tetrahedral intermediate, rapidly collapses (in the order of milliseconds) to restore a free catalytic site and carboxylate product.27–31 By contrast, in the presence of a carbamate-based cholinesterase inhibitor, attack of the carbonyl function of the carbamate similarly through Ser203 generates a temporary (in the order of min to hr) ‘carbamylated drug-enzyme complex’ that is incapable of further substrate catalysis.16,20,21,24
This complex, however, is considerably more stable than the acetyl-enzyme one described for ACh, and its time-dependent hydrolysis, or rate of decarbamylation, is in large part contingent on the structure of the carbamate moiety.16,24 Whereas simple short-chain alkyl carbamates (e.g., H and CH3) are subject to relatively rapid decarbamylation32 to provide short-acting anticholinesterases (e.g., physostigmine: human pharmacodynamic half-life (T1/2) 30 min,33 long-chain alkyl carbamates (e.g., (CH2)n where n = 7 or 8)34,35 and simple aryl carbamates (e.g., phenyl)7,8,36,37 provide more stable carbamylated drug-enzyme complexes that undergo slower decarbamylation and, thereby, induce a longer duration of cholinesterase inhibition. Consequently, the alkyl heptyl and phenyl carbamate anticholinesterases, (−)-eptylstigmine and (−)-phenserine (11), possess inhibition T1/2s of 10 to 11 hr.38,39
One of several factors that may further contribute to the interaction between the phenylcarbamate moiety of a carbamate-based anticholinesterase and the acyl binding site of AChE is the stability provided by both hydrophobic and π electron interactions due to π-π stacking of the phenylcarbamate between the flanking phenyl moieties of two phenylalanines (Phe295 and Phe297) that are present on the acyl-loop and form the base of the gorge of human AChE.16,20,21,24 These key amino acids are not present within BChE, where the gorge base is formed by the smaller aliphatic ones Leu286 and Val288.20,28 This allows the accommodation within BChE of larger substrates and inhibitors, such as (−)-cymserine (15) and (−)-phenethylcymserine (17) that bear a 4’ isopropyl moiety on their phenylcarbamate.23
Based on these factors, two approaches were taken to generate longer-acting anticholinesterases for myasthenia gravis: (i) replacing the dimethyl carbamoyl side chain of the known drugs neostigmine methyl sulfate (3) and pyridostigmine bromide (4) with a phenyl carbamoyl side chain to form new quaternary salts (6) and (8) and, (ii) modifying the known long-acting anticholinesterases, (−)-phenserine (11) and (−)-tolserine (13), by reacting them with iodomethane to form their corresponding quaternary salts 12 and 14. In addition, following our development of reversible, carbamate-based selective BChE inhibitors, (−)-cymserine (15) and (−)-phenethylcymserine (17), the same approach was applied to generate their corresponding quaternary salts (16) and (18). Whereas the former approach resulted in a dramatic loss of anticholinesterase activity, indicating that phenylcarbamates of neostigmine and pyridostigmine (6, 8) cannot be readily accommodated within the binding domains of AChE, the latter approach proved effective in generating potent charged inhibitors (10, 12, 14, 16 and 18) that, albeit less cholinesterase subtype selective than their parent compounds, were of similar or greater potency to neostigmine (3) and pyridostigmine (4). As would be predicted and shown in Table 1, the carbamoyl side-chain replacement as in 6 and 8 resulted in moderate decline in aqueous solubility as reflected in Log D values, compared to 3 and 4. By contrast and also expected, the quaternary salts 12, 14, 16 and 18 were less lipophilic than their respective parent compounds. Although these quaternary salts were not as water-soluble as either neostigmine (3) or pyridostigmine (4) and thus may not have as restricted a blood-brain barrier permeability, they would be not expected to enter brain as freely as 11, 13, 15 and 17, whose brain partition are high.7,8 As cognitive impairment has been reported to occur in myasethenia gravis,40 but remains controversial,41 some brain entry of drug could be of value. As illustrated in Figure 2, the preliminary time-dependent cholinesterase inhibition of 16 in rat is relatively long-acting, and in line with aryl carbamates of (−)-physostigmine in both animal models and humans.7,8,39
In summary, in the quest for longer acting peripheral anticholinesterases modifying the structures of neostigmine methyl sulfate (3) and pyridostigmine bromide (4) led new quaternary salts 6 and 8 that lacked cholinesterase inhibitory action. In the case of phenylcarbamate analogues of physostigmine, the quaternary salts retained activity, compared to parent compounds 11, 13, 15 and 17, and appear long-acting.
3. Conclusion
To overcome the short duration of action of neostigmine methyl sulfate (3) and pyridostigmine bromide (4) in the symptomatic treatment of myasthenia gravis, the N-monophenylcarbamate analogues of neostigmine (6) and pyridostigmine (8) were synthesized. Evaluation of their cholinesterase inhibition demonstrated that 6 and 8 possessed only marginal activity. As an alternative approach, the longer-acting cholinesterase inhibitory phenylcarbamate analogues of (−)-physostigmine 11, 13, 15 and 17 were transferred into their corresponding N(1)-methylammonium bromide analogues (12, 14, 16 and 18) and retained significant potency that, for AChE, was equimolar to or great than their parent compounds. These new quaternary salts (12, 14, 16 and 18) proved more water-soluble than their parent compounds, and 16, as a representative of the class, proved to be long acting in rodent. Together, these results suggest that such agents are of interest as drug candidates for myasthenia gravis and congenital myasthenic syndromes.
4. Experimental
4.1. Chemistry
Melting points (uncorrected) were measured with a Fisher-Johns apparatus; 1H NMR was recorded on a Bruker (Bellevica, MA) AC-300 spectrometer; MS (m/z) were measured on an Agilent 5973 GC-MS (CI); elemental analyses were performed by Atlantic Microlab, Inc. All reactions involving non-aqueous solutions were performed under an inert atmosphere.
N-Phenylcarbamate of 3-dimethylaminophenol (5)
3-Dimethylaminophenol (1) (500mg, 3.65 mmol) was dissolved in anhydrous Et2O (50 ml) and a small piece of Na (about 1 mg) was added. After stirring for 2 min under nitrogen at r. t., phenylisocyanate (434 mg, 3.65 mmol) was added drop wise; thereafter, the solvent was immediately evaporated under vacuum. The residue was crystallized from CH2CL2 to provide crystalline product 5 (650 mg, 70%): mp 150–152 °C; 1HNMR (CDCL3): δ 9.00 - 7.70 (m, 9H, Ar-H), 4.20 (s, 6H, N-(CH3)2), 2.87 (br., H, NH); CI-MS, m/e 257 (MH+). Anal. for (C15H16N2O2), C, H, N.
N-Phenylcarbamate of 3-phenol-1-trimethylammonium methylsulfate (6)
Compound 5 (128 mg, 0.5 mmol) was dissolved in acetone (2 ml), and (CH3)2SO4 (0.1 ml) was added. The mixture was kept in a refrigerator overnight. A crystallized product was filtered and recrystallized from EtOH to give compound 6 as white crystals (153 mg, 80%): mp 125–127 °C; 1HNMR (CD3OD): δ 7.90 – 7.20 (m, 9H, Ar-H), 3.62 (m, 12H, 4CH3). Anal. (C17H22N2O6S), C, H, N.
N-Phenylcarbamate of 3-hydroxypyridine (7)
3-Hydroxypyridine (500 mg, 5.26 mmol) was dissolved in anhydrous Et2O (50 ml) and a small piece of Na (about 1 mg) was added. After stirring for 2 min under nitrogen at r. t., phenylisocyanate (626 mg, 5.26 mmol) was added dropwise. The solvent was then immediately evaporated under vacuum to provide a crystalline residue, which was recrystallized from CH2CL2 to give product 7 as white crystals (964 mg, 85%): mp 112–113 °C; 1HNMR (CDCL3): δ 8.70 - 7.00 (m, 9H, Ar-H), 1.70 (br., 1H, NH). CI-MS, m/e: 215 (MH+). Anal. for (C12H10N2O2), C, H, N.
N-Phenylcarbamate of 3-hydroxy-1-methylpyridinium bromide (8)
Compound 7 (107 mg, 0.5 mmol) was dissolved in anhydrous Et2O (2 ml), and 1 ml of bromomethane (2M in Et2O) was then added into the ether solution. The mixture was maintained at r.t. for 2 days to give crystalline product. Recrystallization of this product from EtOH provided crystalline compound 8 (108 mg, 70%): mp. 148–149 °C; 1HNMR (CD3OD): δ 8.30 - 7.50 (m, 9H, Ar-H), 4.20 (s, 3H, N-CH3). Anal. (C13H13BrN2O2. HBr.H2O), C, H, N.
N(1)-Methylammonium bromide of (−)-physostigmine (10)
(−)-Physostigmine 9 (78.8 mg, 0.286 mmol) was dissolved in anhydrous CH3CN (1.5 ml), and 0.572 ml of bromomethane (2M in Et2O) was added. The solution was stirred at r.t. overnight. Thereafter, evaporation of solvent and reagent left a residue that was crystallized from H2O to provide crystalline product 10 (84.7 mg, 80%): mp. 134 – 136 °C; [α]D23 −124.6 °(c = 0.2 EtOH); 1HNMR (DMSO-d6), δ7.65 (s, 1H, N-H), 7.13 (s, 1H, C4-H), 6.90 (d, 1H, C6-H), 6.65 (d, 1H, C7-H), 5.10 (s, 1H, C8-H), 3.60 (m, 1H, C2a-H), 3.15 (s, 6H, N1+-(CH3)2), 3..00 (m, 1H, C2b-H), 2.85 (s, 3H, N8-CH3), 2.72 (d, 3H, N-CH3), 2.50-2.30 (m, 2H, C3-H2), 1.55 (s, 3H, C3a-CH3). Anal. (C16H24BrN3O2.H2O) C, H, N.
N(1)-Methylammonium bromide of (−)-phenserine(12)
(−)-Phenserine 11 (168 mg, 0.5 mmol) was dissolved in anhydrous CH3CN (2 ml), and 1 ml of bromomethane (2M in Et2O) was added. The solution was stirred at r.t. overnight. Evaporation of solvent and reagent provided a residue, which was crystallized from H2O to give crystalline product 12 (184 mg, 85%): mp. 185 - 188 °C; [α]D23 −116.6 ° (c = 0.1 EtOH); 1HNMR (DMSO-d6), δ10.18 (s, 1H, N-H), 7.70 (d, 1H, C4’-H), 7.40 (m, 2H, C3’-H and C5’-H), 7.30 (d, 2H, C2’-H and C6’-H), 7.18 (m, 2H, C4-H and C6-H), 6.90 (d, 1H, C7-H), 5.18 (s, 1H, C8-H), 3.70 (m, 1H, C2a-H), 3.35 (s, 6H, N1+-(CH3)2), 3..15 (m, 1H, C2b-H), 2.95 (s, 3H, N8-CH3), 2.60-2.40 (m, 2H, C3-H2), 1.65 (s, 3H, C3a-CH3). Anal. (C16H24BrN3O2.1/2H2O) C, H; N: Calcd. 9.52; Found 8.95.
N(1)-Methylammonium bromide of (−)-tolserine (14)
(−)-Tolserine 13 (370 mg, 1.05 mmol) was dissolved in anhydrous CH3CN (4.2 ml), 2.1 ml of bromomethane (2M in Et2O) was added, and the solution stirred at r.t. overnight. Thereafter, evaporation of solvent and reagent gave a residue that was crystallized from H2O to provide crystalline product 14 (374 mg, 80%): mp. 136–140 °C; [α]D23 −82.6 ° (c = 0.2 EtOH); 1HNMR (DMSO-d6), δ10.01 (s, 1H, N-H), 7.40 (d, 2H, C3’-H and C5’-H), 7.20 (d, 2H, C2’-H and C6’-H), 7.18 (s, 1H, C4-H), 7.00 (s, 1H, C6-H), 6.70 (d, 1H, C7-H), 5.18 (s, 1H, C8-H), 3.55 (m, 1H, C2a-H), 3.15 (s, 6H, N1+-(CH3)2), 2.95 (m, 1H, C2b-H), 2.85 (s, 3H, N8-CH3), 2.50-2.30 (m, 2H, C3-H2), 1.50 (s, 3H, C3a-CH3). Anal. (C24H32N3O2Br.H2O) C, H, N.
N(1)-Methylammonium bromide of (−)-cymserine (16)
(−)-Cymserine 15 (379 mg, 1.0 mmol) was dissolved in anhydrous CH3CN (4.5 ml) and 2 ml of bromomethane (2M in Et2O) was added. The solution was stirred at r.t. overnight, following which evaporation of solvent and reagent provided a residue that was crystallized from H2O to give crystalline product 14 (315 mg, 80%): mp. 160–162 °C; [α]D23 −92.2 ° (c = 0.2 EtOH); 1HNMR (DMSO-d6), δ10.02 (s, 1H, N-H), 7.55-7.30 (m, 5H, Ar-H and C4-H), 7.20 (d, 1H, C5-H), 6.90 (d, 1H, C7-H), 5.30 (s, 1H, C8-H), 3.70 (m, 1H, C2a-H), 3.40 (s, 6H, N1+-(CH3)2), 3..20 (m, 1H, C2b-H), 3.05 (s, 3H, N8-CH3), 2.60-2.50 (m, 2H, C3-H2), 2.45 (s, 3H, Ar-CH3), 1.75 (s, 3H, C3a-CH3). Anal. (C24H32BrN3O2.H2O) C, H, N.
N(1)-Methylammonium bromide of (−)-phenethylcymserine(18)
(−)-Phenethylcymserine 17 (469 mg, 1.0 mmol) was dissolved in anhydrous CH3CN (5 ml), 2 ml of bromomethane (2M in Et2O) was added, and the solution was stirred at r.t. overnight. Evaporation of solvent and reagent provided a residue that was cystalized from H2O to give crystalline product 18 (462 mg, 82%): mp. 150–151 °C; [α]D23 −142.8 ° (c = 0.2 EtOH); 1HNMR (DMSO-d6), δ10.02 (s, 1H, N-H), 7.45-6.70 (m, 12H, Ar-H) 5.22 (s, 1/2H,1/2 C8-H), 5.18 (s, 1/2H,1/2 C8-H), 3.70-2.80 (m, 10H, C2-H2, N-CH2CH2. (Me)2CH- and N8-CH3), 3.40 (s, 6H, N1+-(CH3)2), 3..20 (m, 1H, C2b-H), 3.05 (s, 3H, N8-CH3), 2.50-2.30 (m, 2H, C3-H2), 1.55 (s, 1.5H, 1.5C3a-CH3). 1.45 (s, 1.5H, 1.5C3a-CH3) Anal. (C31H38BrN3O2.1/2H2O) C, H, N.
4.2 Quantitation of anticholinesterase activity
The anticholinesterase activity of compounds 3 – 18 was assessed by quantifying their capacity to inhibit the ability of freshly prepared human AChE and BChE to enzymatically degrade their respective specific substrates, acetyl-(β-methyl)thiocholine and s-butyrylthiocholine (0.5 mmol/L) (Sigma Chemical Co., St. Lois, MO).15,42 Samples of AChE and BChE were derived from freshly collected human whole red blood cells and plasma, respectively. Compounds were dissolved in Tween 80/EtOH 3:1 (v:v; <150 μL total volume) and were diluted in 0.1 M Na3P04 buffer (pH 8.0) in half-log concentrations to provide a final concentration range that spanned 0.3 nM to 30 uM. Tween 80/EtOH was diluted to in excess of 1 in 5000. No inhibitory action of either AChE or BChE was detected in separate experiments in which the anticholinesterase activity of the known compound, 9, was quantified in excess and without Tween 80/EtOH.
For the preparation of BChE, freshly collected blood was centrifuged (10,000 g, 10 min, 4° C) and plasma was removed and diluted 1:125 with 0.1 M Na3P04 buffer (pH 7.4). Plasma was carefully checked to insure an absence of haemolysis. For AChE preparation, erythrocytes were washed five times in isotonic saline, lysed in 9 volumes of 0.1 M Na3P04 buffer (pH 7.4) containing 0.5% Triton-X (Sigma) and then were diluted with an additional 19 volumes of buffer to a final dilution of 1:200.
Analysis of anticholinesterase activity was undertaken by utilizing a 25 μL sample of each enzyme preparation, and was undertaken at their optimal working pH, 8.0,15 in 0.1 M Na3P04 buffer (0.75 mL total volume). Compounds were preincubated with enzymes (30 min, r.t.) and then were incubated with their respective substrates and with 5,5′-dithiobis-2-nitrobenzoic acid (25 min, 37° C). The substrate/enzyme interaction was immediately halted by the addition of excess enzyme inhibitor ((−)-physostigmine (9) 1 × 10−5 M) and production of a yellow thionitrobenzoate anion was then measured by spectrophotometer at 412 nm λ. To correct for nonspecific substrate hydrolysis, aliquots were co-incubated under conditions of absolute enzyme inhibition (by the addition of 1 ×10−5 M (−)-physostigmine (9), and the associated alteration in absorbance was subtracted from that observed through the concentration range of each test compound. Each agent was analyzed on four separate occasions and assayed along-side (−)-physostigmine (9), as a control and external standard whose activity we have previously reported.42 The mean enzyme activity at each concentration of test compound was then expressed as a percent of the activity in the absence of compound. This was transformed into a logit format (where logit = ln(%activity/100 minus %activity)) and then was plotted as a function of its log concentration. Inhibitory activity was calculated as an IC50, defined as the concentration of compound (nM) required to inhibit 50% of enzymatic activity, which was determined from a correlation between log concentration and logit activity. Only results obtained from correlation coefficients of r2≥−0.98 were considered acceptable. Studies that did not obtain this threshold were repeated.
A preliminary pharmacodynamic study to determine time-dependent cholinesterase inhibition was undertaken in the anesthetized (isofuorane, Abbott, Chicago) male Fischer-344 rat (Charles River, Wilmington, MA) (approximately 250 g) with a femoral artery catheter to remove blood samples. A predrug blood sample was taken to determine resting (no inhibition) cholinesterase activity. Compound 16 was administered a 10 mg/kg i.p. and blood samples were taken at predefined times from 5 min to 24 hr. These were centrifuged (8000xg, 30 s) and the plasma was collected and frozen (−80° C) for determination of cholinesterase inhibition. In rat plasma, unlike that in humans, both AChE and BChE are present. Therefore a specific inhibitor of BChE, Iso-OMPA (1×10−4 M), was used during the quantitative determination of AChE inhibition. Throughout this procedure, the animal was assessed for any acute adverse effects, particularly associated with cholinergic overdrive (e.g., tremor, salivation).
Plasma samples were thawed, and a 10 ul sample was then added immediately to 1390 ul 0.1 mol/L sodium phosphate buffer, pH 7.4, with and without iso-OMPA. Three minutes later, 100 ul 5,5′-dithiobis-2-nitrobenzoic acid (0.5 mmol/L; Sigma Chemical Co.) plus S-butyrylthiocholine in 0.1 mol/L sodium phosphate buffer, pH 7.4, was added. The production of a yellow 5-thio-2-nitro-benzoate anion (produced by the reaction of 5,5′-di-thiobis-2-nitrobenzoic acid with thiocholine released by enzymatic hydrolysis of butyrylthiocholine) was measured precisely 20 min later by spectrophotometer (412 nm λ). All reagents were maintained at r.t. and plasma samples were assayed together in duplicate. Cholinesterase inhibition was calculated by subtracting the absorption value of samples with iso-OMPA from those without and expressing the difference (related to substrate hydrolysis by cholinesterase alone) as a percent of the predrug level. The total assay time was 23 min, which minimized drug decomposition and rejuvenation of enzyme in the plasma sample.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, and by National Institutes of health grants (AG18379 and AG18884) to DKL. The authors are grateful for the aid and input of Scott MacDonald, Advanced Chemistry Development, Inc., Toronto, ON, Canada, for determination of Log D values of charged compounds. Animal studies were undertaken on an approved protocol in accord with the Animal Care and Use Committees of the Intramural Research Program, National Institute on Aging, and followed National Institutes of Health guidelines.
Footnotes
The authors declare no conflicts of interest relating to the described research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Qian-sheng Yu, Drug Design & Development Section, Laboratory of Neurosciences, Biomedical Research Center, National Institute on Aging, National Institutes of Health, 251 Bayview Blvd. Baltimore, MD 21224, USA.
Harold W. Holloway, Drug Design & Development Section, Laboratory of Neurosciences, Biomedical Research Center, National Institute on Aging, National Institutes of Health, 251 Bayview Blvd. Baltimore, MD 21224, USA
Weiming Luo, MedStar Research Institute, Hyattsville, MD 20782, USA.
Debomoy K. Lahiri, Department of Psychiatry, Institute of Psychiatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA
Arnold Brossi, School of Pharmacy, University of North Carolina at Chapel Hill, NC 27599, USA.
Nigel H. Greig, Drug Design & Development Section, Laboratory of Neurosciences, Biomedical Research Center, National Institute on Aging, National Institutes of Health, 251 Bayview Blvd. Baltimore, MD 21224, USA.
References
- 1.Aeschliman JA. 1905990. US Patent. 1933
- 2.Urban R. 2572579. US Patent. 1951
- 3.Howland RD, Mycek MJ, Harvey RA, Champe PC, Mycek MJ. Lippincott’s Illustrated Reviews. 3. 2008. Pharmacology; p. 51. [Google Scholar]
- 4.Gold R, Schneider-Gold C. Neurotherapeutics. 2008;5:535–41. doi: 10.1016/j.nurt.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argov Z. Curr Opin Neurol. 2009;22:493–7. doi: 10.1097/WCO.0b013e32832f15fa. [DOI] [PubMed] [Google Scholar]
- 6.Polonovski M. Bl Soc Chim Fr. 1916;19:47–50. summarized in Beilstein E 1954, II, 23:333. [Google Scholar]
- 7.Greig NH, Pei X-F, Soncrant TT, Ingram DK, Brossi A. Med Res Reviews. 1995;15:3–31. doi: 10.1002/med.2610150103. [DOI] [PubMed] [Google Scholar]
- 8.Greig NH, Sambamurti K, Yu QS, Brossi A, Bruinsma GB, Lahiri DK. Curr Alzheimer Res. 2005;2:281–90. doi: 10.2174/1567205054367829. [DOI] [PubMed] [Google Scholar]
- 9.Klein J. Expert Opin Investig Drugs. 2007;16:1087–97. doi: 10.1517/13543784.16.7.1087. [DOI] [PubMed] [Google Scholar]
- 10.Varin F, Couture J, Gao H. J Chromatogr B Biomed Sci Appl. 1999;723:319–23. doi: 10.1016/s0378-4347(98)00535-0. [DOI] [PubMed] [Google Scholar]
- 11.Broggini M, Benvenuti C, Botta V, Fossati A, Valenti M. Methods Find Exp Clin Pharmacol. 1991;13:193–8. [PubMed] [Google Scholar]
- 12.Aquilonius SM, Hartvig P. Clin Pharmacokinet. 1986;11:236–49. doi: 10.2165/00003088-198611030-00005. [DOI] [PubMed] [Google Scholar]
- 13.Brzostowska M, He X-S, Greig NH, Brossi A. Phenylcarbamates of (−)-eseroline, (−)-N1-noreseroline and (−)-physovenol: selective inhibitors of acetyl and, or butyrylcholinesterase. Med Chem Res. 1992;2:238–246. [Google Scholar]
- 14.Pei X, Yu QS, Holloway HW, Brossi A, Greig NH. Med Chem Res. 1999;9:50–60. [Google Scholar]
- 15.Ellman GL, Courtney KD, Andres V, Jr, Feather-stone RM. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 16.Yu Q, Holloway HW, Flippen-Anderson JL, Hoffman B, Brossi A, Greig NH. J Med Chem. 2001;44:4062–71. doi: 10.1021/jm010080x. [DOI] [PubMed] [Google Scholar]
- 17.McGrogan A, Sneddon S, de Vries CS. Neuroepidemiology. 2010;34:171–183. doi: 10.1159/000279334. [DOI] [PubMed] [Google Scholar]
- 18.Conti-Fine BM, Mikani M, Kaminski HJ. J Clin Invest. 2006;116:2843–2854. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer T. In: The Pharmacological Basis of Therapeutics. 9. Goodman SL, editor. Mcgraw-Hill; Tx: 1996. p. 173. [Google Scholar]
- 20.Darvesh S, Hopkins D, Geula C. Nat Rev Neurosci. 2003;4:131–8. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 21.Soreq H, Zakut H. Human cholinesterases and anticholinesterases. Academic Press; New York, NY: 1993. [Google Scholar]
- 22.Soreq H, Seidman S. Nat Rev Neurosci. 2001;2:294–302. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- 23.Yu Q, Holloway HW, Utsuki T, Brossi A, Greig NH. J Med Chem. 1999;42:1855–1861. doi: 10.1021/jm980459s. [DOI] [PubMed] [Google Scholar]
- 24.Greig NH, Yu QS, Brossi A, Matin E, Lahiri DK, Darvesh S. In: Medicinal Chemistry of Alzheimer’s Disease. Martinez A, editor. Transworld Research Network, Research Signpost; Kerala, India: 2008. pp. 79–109. [Google Scholar]
- 25.Savini L, Gaeta A, Fattorusso C, Catalanotti B, Campiani G, Chiasserini L, Pellerano C, Novellino E, McKissic D, Saxena A. J Med Chem. 2003;46:1–4. doi: 10.1021/jm0255668. [DOI] [PubMed] [Google Scholar]
- 26.Darvesh S, Darvesh KV, McDonald RS, Mataija D, Walsh R, Mothana S, Lockridge O, Martin E. J Med Chem. 2008;51:4200–12. doi: 10.1021/jm8002075. [DOI] [PubMed] [Google Scholar]
- 27.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 28.Harel M, Sussman JL, Krejci E, Bon S, Chanal P, Massoulie J, Silman I. Proc Natl Acad Sci USA. 1992;89:10827–10831. doi: 10.1073/pnas.89.22.10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zacharias N, Dougherty DA. Trends Pharmacol Sci. 2002;23:281–287. doi: 10.1016/s0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]
- 30.Gao D, Zhan CG. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2005;109:23070–23076. doi: 10.1021/jp053736x. [DOI] [PubMed] [Google Scholar]
- 31.Saxena A, Redman AM, Jiang X, Lockridge O, Doctor BP. Biochemistry. 1997;36:14642–14651. doi: 10.1021/bi971425+. [DOI] [PubMed] [Google Scholar]
- 32.Yu QS, Greig NH, Holloway HW, Flippen-Anderson F, Brossi A. Med Chem Res. 2000;10:186–199. [Google Scholar]
- 33.Asthana S, Greig NH, Hegedus L, Holloway HH, Raffaele KC, Schapiro MB, Soncrant TT. Clin Pharmacol Ther. 1995;58:299–309. doi: 10.1016/0009-9236(95)90246-5. [DOI] [PubMed] [Google Scholar]
- 34.Marta M, Gatta F, Pomponi M. Biochim Biophys Acta. 1992;1120:262–6. doi: 10.1016/0167-4838(92)90246-a. [DOI] [PubMed] [Google Scholar]
- 35.Brufani M, Castellano C, Marta M. Pharmacol Biochem Behav. 1987;26:625–629. doi: 10.1016/0091-3057(87)90176-6. [DOI] [PubMed] [Google Scholar]
- 36.Kamal MA, Greig NH, Alhomida AS, Al-Jafari AA. Biochem Pharmacol. 2000;60:561–70. doi: 10.1016/s0006-2952(00)00330-0. [DOI] [PubMed] [Google Scholar]
- 37.Kamal MA, Klein P, Yu QS, Tweedie D, Li Y, Holloway HW, Greig NH. J Alzheimers Dis. 2006;10:43–51. doi: 10.3233/jad-2006-10108. [DOI] [PubMed] [Google Scholar]
- 38.Mant T, Troetel WM, Imbimbo BP. J Clin Pharmacol. 1998;38:610–7. doi: 10.1002/j.1552-4604.1998.tb04467.x. [DOI] [PubMed] [Google Scholar]
- 39.Greig NH, Ruckle J, Comer P, Brownell L, Holloway HW, Flanagan DR, Jr, Canfield CJ, Burford RG. Curr Alzheimer Res. 2005;2:483–92. doi: 10.2174/156720505774330564. [DOI] [PubMed] [Google Scholar]
- 40.Paul RH, Cohen RA, Gilchrist JM, Aloia MS, Goldstein JM. J Neurol Sci. 2000;179(S 1–2):59–64. doi: 10.1016/s0022-510x(00)00367-1. [DOI] [PubMed] [Google Scholar]
- 41.Sitek EJ, Biliska MM, Wieczorek D, Nyka WM. Neurol Sci. 2009;30:9–14. doi: 10.1007/s10072-008-0001-y. [DOI] [PubMed] [Google Scholar]
- 42.Luo W, Yu QS, Zhan M, Parrish D, Deschamps JR, Kulkarrni SS, Holloway HW, Alley GM, Lahiri DK, Brossi A, Greig NH. J Med Chem. 2005;48:986–994. doi: 10.1021/jm049309+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.