

Abstract
Structural characterization, identification and quantification of xenobiotics and their metabolic products commonly require the use of at least two different techniques. This has been the case in the analysis of metabolic products of doxorubicin, a widely-used fluorescent anthracycline for the treatment of tumors and leukemia. In this work, we combine HPLC with a tandem laser-induced fluorescence (LIF) and mass spectrometry (MS) detection scheme for the characterization of doxorubicin and its metabolites produced in the post-mitochondrial fraction prepared from Fisher 344 rat liver. LIF detection allowed quantification of the metabolic compounds while MS detection aided in the identification of the metabolites. Using this HPLC-LIF-MS methodology, the disapperance of doxorubicin and the appearance of 7-deoxydoxorubicinone and 7-deoxydoxorubicinolone were monitored over the course of 40 minutes. This application demonstrates the potential of the tandem LIF-MS detection scheme in quantification and characterization of biotransformations of fluorescent xenobiotics of biomedical and environmental relevance. Furthermore, this detection scheme would be particularly relevant in the analysis of fluorescent analytes in complex samples and in validation of methods for the analysis of such samples that typically rely only on LIF detectors.
Keywords: In vitro metabolism, doxorubicin, high-performance liquid chromatography, mass spectrometry, laser-induced fluorescence
Introduction
The combination of optical and mass detectors in tandem is a powerful resource to improve upon the characterization and analysis of complex samples and samples containing unknown compounds. Optical detection (e.g. UV-Vis absorption or fluorescence) allows for the quantitative data to be collected without the bias of ionization conditions. Mass spectrometry detection (MS) provides structural information of analytes that cannot be observed by spectroscopic methods. Surprisingly, very few reports describe combining UV absorption or laser-induced fluorescence (LIF) detectors with MS detectors. Crow, et al., used HPLC-UV-MS to characterize components of synthetic, toxic oil associated with toxic oil syndrome.1 Akbay, et al., reported the use of micellar electrokinetic chromatography with a tandem UV and MS detector to analyze a complex mixture of chiral beta-blockers.2 Capillary electrophoresis (CE) with LIF-MS was used to obtain quantitative and structural data from an extract of dried leaves of Psychotria viridis.3 CE-LIF-MS has also been used for the direct characterization of N-linked glycans in therapeutic antibodies.4 Here we describe an HPLC-LIF-MS system and its use in the analysis of metabolites resulting from the biotransformation of doxorubicin in rat liver extracts.
Defining the pharmacological and toxicological profiles of xenobiotics requires both identifying and quantifying the products of their biotransformations. Mass spectrometry is an ideal method to obtain molecular structure data that assists in the identification of compounds, but more cumbersome when used for quantification purposes.5 Using mass spectrometry for quantification would require the use of isotopically labeled standards or compounds with analogous molecular structures, which are not always available. In contrast, other detection schemes, such as fluorescence detection, are suitable for quantification of fluorescent xenobiotics (e.g. anthracyclines),6–8 but inadequate for molecular characterization of unknown biotransformation products.
Doxorubicin is an anticancer anthracycline used to treat many cancer types, from solid tumors to leukemia. The metabolism of doxorubicin has been monitored in blood plasma, urine and cell cultures.8–14 These analyses were performed with a separation technique such as thin-layer chromatography,9 CE12 or HPLC8, 10, 11, 13, 14 combined with varying detection schemes. For quantitation purposes, fluorescence detection is typically preferred8, 9, 12–14 due to its selectivity and sensitivity. Unfortunately, the molecular structure of the detected metabolites cannot be determined with such detection scheme and their identity could only be speculated based on comigration with a few standards of known metabolites. Furthermore, the CE separation conditions are incompatible with mass spectrometry. Mass spectrometry is commonly used by monitoring characteristic mass peaks of doxorubicin and other anthracyclines through multiple reaction monitoring experiments.10, 11 However, for quantification using mass spectrometry, an internal standard or isotopically labeled standard is typically used.7, 10, 15 A tandem LIF-MS detector would eliminate the need of adding external standards to accomplish the desired quantification. Furthermore, the LIF sensitivity would help detect low abundance metabolites, even if they are not readily detected by MS.
In this paper, we report on the development and use of a tandem LIF-MS detection scheme coupled with an HPLC system. Using this setup, we demonstrated the simultaneous separation (by HPLC), quantification (by LIF detection) and structural characterization (by MS) of doxorubicin and two of its metabolites, 7-deoxydoxorubicinolone and 7-deoxydoxorubicinone. MS data was not suitable for quantification, with relative standard deviations as high as 78% for peak area measurements in standard solutions. On the other hand, fluorescence measurements showed a relative standard deviation between repeat analyses of 7.5%. The HPLC-LIF-MS method described here could be easily extended to characterize and quantify other fluorescent compounds in increasingly complex samples of biomedical and environmental relevance occurring in vivo, in vitro, and in environmental systems. Furthermore, the tandem detection scheme would aid in validating HPLC separations of complex samples prior to the implementation of such separations in instruments with only LIF or MS detection.
Experimental
Materials
All materials were used as received. Water from a Millipore filter system (18 M3) was used for HPLC and buffer preparation. Acetonitrile, glucose-6-phosphate dehydrogenase type I and D-glucose-6-phosphate dipotassium salt hydrate were purchased from Sigma-Aldrich (St. Louis, MO). Formic acid, tris(hydroxymethyl)aminomethane (TRIS) and magnesium chloride were purchased from Fisher Scientific (Pittsburgh, PA). Sucrose was purchased from MP Biomedicals, Inc. (Illkirch, France). Monobasic potassium phosphate, perchloric acid, sodium hydroxide, hydrochloric acid, citric acid and potassium chloride were purchased from Mallinckrodt (Phillipsburgh, NJ). NADP was purchased from Roche Diagnostics, GmBH (Mannheim, Germany). Doxorubicin was a generous gift from Meiji Seika Kaisha, LTD (Tokyo, Japan). Standards of doxorubicinol, doxorubicinone and 7-deoxydoxorubicinone were obtained from Qvantas, Inc. (Newark, DE).
Solutions
Tissue storage buffer consisted of sucrose (0.2 M), potassium phosphate (0.05 M) and potassium chloride (0.15 M) in water and adjusted to pH 7.4 with 0.1 M NaOH. Incubation buffer consisted of TRIS (0.05 M) and potassium chloride (0.15 M) in water. Glucose-6-phosphate dehydrogenase was reconstituted in citrate buffer (5 mM citric acid adjusted to pH 7.5 with 1 M sodium hydroxide) per the instructions accompanying the enzyme (Sigma-Aldrich G4134). The aqueous component of the HPLC mobile phase was Millipore filtered water with 0.1% Formic acid. The mobile phase was filtered with a 0.2 micron nylon filter (Gelman Sciences).
Tissue Procurement
10-month old Fisher 344 rats were purchased from the National Institute on Aging and housed in a research animal facility at the University of Minnesota. They were fed ad libitum standard laboratory rat chow and kept on a 12 hour light/dark cycle.
On the day of tissue harvesting, the rats were anesthetized with sodium pentobarbital (50 mg/kg body weight). Tissue was immediately removed and placed in the storage buffer on ice until homogenized. The rats were sacrificed after tissue harvesting. The animal protocol was approved by the University of Minnesota Institutional Animal Care and Use Committee.
In Vitro Metabolism
In vitro metabolism procedures were adapted from the literature.15, 16 1.6 g of rat liver tissue was homogenized in 3 mL of incubation buffer using 15 strokes of a Dounce homogenizer. The homogenizer was rinsed with 2 mL of incubation buffer and this solution was then added to the homogenate. Centrifugations were performed at 4° C. The tissue homogenate was centrifuged at 600×g for 10 minutes to remove tissue debris. The supernatant was subsequently removed and centrifuged at 10,000×g for 10 minutes creating a sediment consisting of heavy organelles (i.e. mitochondria) and a supernatant termed the post-mitochondrial fraction (PMF). This fraction contains microsomes common in many in vitro experiments as well as soluble cell components. The PMFs were used for the metabolism experiments. The biochemical reactions are expected to be caused by cytochrome P450 (CYP450) enzymes carbonyl reductase and NADPH - P450 reductase and NAD(P)H: quinone oxidoreductase; cofactors are added accordingly.15–17 Magnesium chloride (5 mM), NADP (0.25 mM), glucose-6-phosphate (2.5 mM), and doxorubicin (50 μM) were added to final concentrations denoted by parentheses. The start of the reaction time was defined as the moment in which glucose-6-phosphate dehydrogenase (2.0 units) was added to the reaction mixture. The reaction mixture (final volume of 1.1 mL) was incubated at 37° C in a heated mixer (Eppendorf Thermomixer) and samples were removed at time intervals for analysis.
Extraction procedure
An extraction procedure previously shown to result in 94.7 – 99.9% recovery of doxorubicin14 was used to extract the compounds of interest from the biological matrix. The extraction solvent was prepared by mixing 14 μL of concentrated perchloric acid with 60 μL of HPLC mobile phase (67% water: 33% acetonitrile) in a 600 μL microcentrifuge tube. At a selected reaction time, 40 μL of the reaction mixture were removed and pipetted into the extraction mixture. The tube was vortexed for 30 seconds and then centrifuged at 3000×g for 3 minutes. The supernatant was used directly for analysis.
HPLC-LIF-MS
An Agilent (Santa Clara, CA) 1100 capillary HPLC system was used in this analysis. A sample volume of 0.5 μL was injected into the HPLC apparatus and separated on a C18 column (0.3 × 150 mm, 3 μm particles, ACE11115003, Mac-Mod Analytical, Inc., Chadds Ford, PA). The mobile phase consisted of 67% water (0.1% formic acid) and 33% acetonitrile. The flow rate was set at 3.5 μL/min.
The HPLC column was connected to the electrospray ionization chamber of the mass spectrometer using 30 cm of 50 μm I.D. fused silica capillary (Figure 1). Teflon sleeves (Upchurch, F-242X) were used to adapt the outer diameter of the capillary to fit standard size PEEK finger-tight fittings and reduce dead volume. For LIF detection, a 473 nm diode-pumped solid state laser (OnPoint Lasers, LTD, Eden Prairie, MN) was used for excitation. An approximately 5 mm section of the polyimide coating of the fused-silica capillary was burned off creating a detection window. A fluorescence flow-through cell (SpectrAlliance, Inc., St Louis, MO) equipped with fiber optics to deliver incident light and collect fluorescent light was used as an on-column detector. The collected fluorescence was filtered with a 580BP45 filter (Omega Optics) and monitored using a photomultiplier tube (Hamamatsu, biased at 800 V). Data from the photomultiplier tube was digitized (10 Hz) using a National Instruments I/O board (PCI-6034E) run with a LabVIEW 5.1.1 (National Instruments, Austin, TX) program created in house. Using standards of doxorubicin, the alignment of the fluorescence detection window was properly placed in the detection zone of the flow-through device to achieve the maximum signal. A 10 μM solution of doxorubicin in methanol was passed through the capillary using a syringe adapter and a 100 μL syringe operated by a syringe pump at a flow rate of 3.5 μL/min. The capillary was secured with the nut on the flow-through device. The limit of detection was 2.0 μM or 1.0 × 10−12 moles of doxorubicin.
Figure 1.
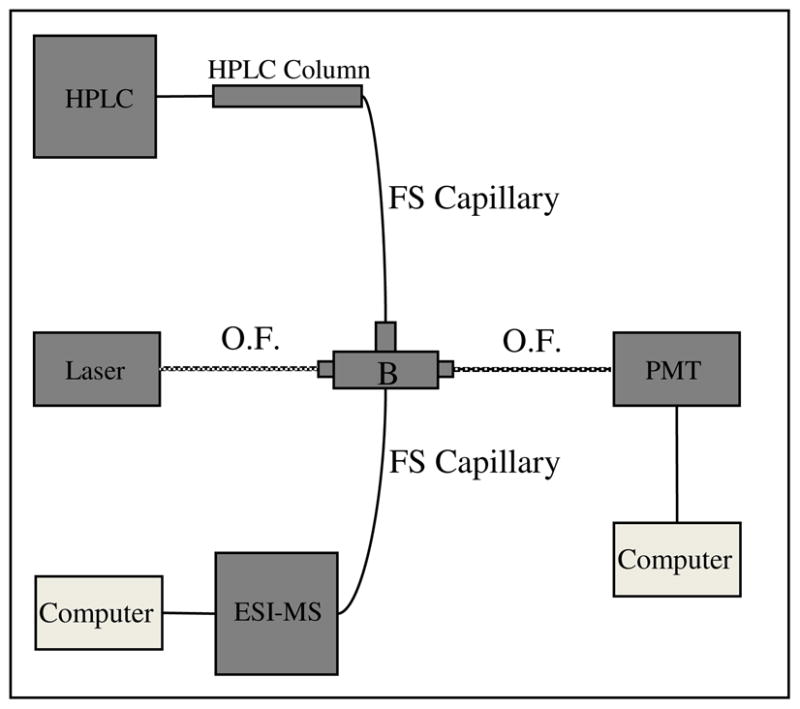
Diagram of HPLC-LIF-MS setup. The fused silica (FS) capillary (365 μm O.D., 50 μm I.D.) connecting the HPLC column and the ESI source was 30 cm long. The detection window was formed by burning an approximately 1 cm window in the polyimide coating. The interface between the fused-silica capillary and electrospray source was accomplished using a Teflon sleeve (Upchurch, F-242X) and a PEEK finger-tight fitting (Upchurch, F-300X). The ESI-MS instrument was a Bruker MicrOTOFQ mass spectrometer. The fluorescence cell (B) was obtained from SpectrAlliance and guides incident laser light (473nm) and fluorescent light via fiber optics (O.F., 1 m in length). Optical fibers, internal to the cell, guide incident light and fluorescent light at a 90° angle. PMT is a photomultiplier tube biased at 800 V; fluorescence was selected with a 580BP45 optical filter in front of the PMT; the data acquisition rate was 10 Hz.
Mass spectral data were collected using a Bruker MicrOTOFQ mass spectrometer (Bruker Daltonics Inc., Billerica, MA) capable of high accuracy MS and MS/MS data collection (10,000 to 15,000 mass resolution for m/z 350–800). Helium was used as the collision gas because doxorubicin was found to fragment extensively when using Argon. Positive ions were detected. Calibration of the mass spectrometer was conducted using a dilute solution of sodium formate as the calibrant. The calibration solution was infused using a syringe pump at a flow rate of 3.5 μL per minute. Six m/z values (158.9641, 362.9263, 498.9012, 566.8886, 634.8760, and 770.8509) were used to conduct an enhanced quadratic calibration in the mass range of expected (i.e. doxorubicinol, doxorubicinone and 7-deoxydoxorubicinone).18 The parameters (funnel 1 and 2 RF, hexapole RF and collision RF) were adjusted to maximize the total ion current in the 300 to 850 m/z range. The limits of detection was ~ 10−12 moles of doxorubicin).
Separation and Detection Methods
In order to develop the separation and detection methods, standards of doxorubicin and two commonly reported metabolites (doxorubicinol and doxorubicinone) were used. These standards were prepared as stock solutions in methanol (1 mM) and diluted to obtain the desired concentrations. It was found that a C18 column was able to completely separate all components under isocratic conditions of 67% water and 33% acetonitrile. Due to the fact that an isocratic separation proved to be adequate, a gradient was not used for the separation. Using an isocratic method decreases the analysis time by eliminating an equilibration step of the HPLC instrument between separations. Using standard solutions and extracted tissue solutions there were no observed peaks by LIF or MS detection after a 20 minute separation time window. Therefore, carryover of peaks between samples was not of concern.
Data Analysis
MS detection of an eluting peak lagged 29±1 seconds relative to fluorescence detection. This time difference was used to associate MS and LIF signals. MS data were analyzed using DataAnalysis 4.0 supplied by Bruker Daltonics. Fluorescence data were analyzed using in house developed Igor Pro procedures (WaveMetrics, Inc., Portland, OR).
The fluorescence quantum yields of doxorubicinol and 7-deoxydoxorubicinone relative to doxorubicin were determined by comparing total fluorescence intensities of equimolar (10 μM) solutions in HPLC mobile phase. The total fluorescence intensity was measured with a spectrofluorometer (Jasco FP-6200, 465 nm excitation) over the 557–602 nm range, which matches the transmission range of the 580BP45 optical filter in the LIF detector, The relative quantum yield of 7-deoxydoxorubicinone to doxorubicin was 1.0±0.1 (n=3). The relative quantum yield of doxorubicinol to doxorubicin was 1.7±0.1 (n=3). Since doxorubicin and the aglycone 7-deoxydoxorubicinone (i.e. doxorubicin without the daunosamine moiety) have the same quantum yield, we assumed that doxorubicinol and 7-deoxydoxorubicinolone (i.e. doxorubicinol without the daunosamine moiety) have also a similar fluorescence quantum yield.
The tranformation of doxorubicin into metabolites was expressed in terms mole fractions using the expression
| (Equation 1) |
in which ni is the mole fraction corresponding to compound (i) (i.e. doxorubicin, doxorubicinone, 7-deoxydoxorubicinone, or 7-deoxydoxorubicinolone), and Ci is the concentration of compound (i). Each Ci was determined using the expression
| (Equation 2) |
where F is the LIF detector response factor relating doxorubicin concentration to total fluorescence peak area in the sample at t=0 minutes of incubation determined for each data set, φF is the fluorescence quantum yield, and Ai is the area of the peak. Both doxorubicin and doxorubicinone peaks must be included in the F factor because they are detected at the first time point (i.e. t=0 min) and represent 50 μM doxorubicin used at the onset of the reaction. It is worth noticing that for each sample taken at different reaction times ΣCi varies from 35 to 54 μM. Deviation of ΣCi from the expected value of 50 μM does not influence the results because the the abundance of each compound is represented as a mole fraction (c.f. equation 1).
Chemical compositions were calculated from the acquired MS data using the Bruker Data Analysis software. The MicrOTOFQ routinely collects spectra with 10,000 to 11,000 mass resolution and ~0.2 mDa precision, and tolerances ~ 10 ppm, which is adequate to generate an initial list of possible chemical compositions. Unfortunately, this list still has many possible chemical formulas for each observed mass ion. For example, an ion with m/z of 400 detected in an instrument with 10 ppm mass tolerance would be a match to 50–60 elemental compositions containing C, H, N, O and S.21 In this study, the calculated elemental compositions in combination with prior knowledge of the parent drug and likely biochemical transformations, allowed the selection of the most likely candidate molecules. In this study, the two restrictions used were: (i) the maximum number of nitrogen atoms in the chemical formula was set to two, and (ii) the mass tolerance was 20 ppm. Using these restrictions the maximum number of chemical formulas for each compound detected was five. Among these formulas, the selection of the best match was strongly dependent on its sigma value, which is an index of the deviations of observed from the predicted masses and intensities of monoisotopic peaks corresponding to a given chemical formula. When using standard compounds, the formula with the lowest sigma value was the correct chemical composition; this was not always the case for the metabolism samples, but prior knowledge on doxorubicin metabolism, comparison of retention times with standards, and very low sigma values increased the confidence of the compounds’ identifications.
Results and Discussion
Recovery after extraction
Initially, we attempted to recover doxorubicin metabolites by solid-phase extraction (SPE) using a C18 SPE cartridge (Waters).10 While extraction of standards from buffer solutions recovered 99% of doxorubicin and the standard metabolites, the approach failed to recover reproducibly the same standards from a biological matrix (i.e. homogenized liver tissue). A second extraction method based on previous work14 that uses perchloric acid treatment provided the best reproducibility and recovery. This procedure recovered 98±1% of doxorubicin, doxorubicinol and doxorubicinone standards from liver tissue homogenate.
Validation of tandem LIF-MS detection system
LIF was adequate to detect chromatographically separated standards of doxorubicin, doxorubicinol, and doxorubicinone. The linear range for peak area versus concentration was between 2 μM and 50 μM doxorubicin (the range tested). The separation efficiency was ~ 17,000 theoretical plates for doxorubicin (Table 1).
Table 1.
Resolution and separation efficiencies for both detectors. N1 and t1 are the number of theoretical plates and retention time, respectively, of Peak 1, doxorubicin. R1,2 is the resolution between peaks 1 and 2. See Figure 2 for peak assignments. Values reported are mean values and standard deviations (n = 4 replicates).
| Detector | N | R1,2 | t1 (s) |
|---|---|---|---|
| LIF | 17000±2000 | 6.1±1.2 | 360±30 |
| MS | 4100±300 | 3.1±0.5 | 390±30 |
MS Detection was delayed by 29±1 seconds compared to the LIF detection due to the extra distance of travel through the tubing connecting the HPLC column to the ESI source. The separation resolution and the number of theoretical plates were reduced in MS relative to LIF detection (Table 1). Diffusion, which explains only 0.0015% of MS peak broadening, does not explain the degradation of the separation occurring between the LIF and MS detectors (Supplementary Material, Page S-5).19 Peak tailing, due to adsorption of doxorubicin and the metabolites to the silica surface20 is similar in both LIF and MS peaks. Thus, adsorption cannot explain the lower separation performance at the MS detector. The most likely contributor to peak broadening is the ESI interface to the mass spectrometer. The inner diameter of the sprayer needle is 90–100 μm, which is larger than the 50 μm capillary connecting to the MS ionization chamber. This can cause significant peak broadening as the analytes reach this connection point.20 Although it would be desirable to re-design the interface to the mass spectrometer to maintain a good separation performance, a more relevant issue was that the peak area response from the MS detector was erratic. For example, the relative standard deviation of the peak area of doxorubicin standards in MS chromatograms was 78%. Therefore, LIF data was used for quantification purposes.
Separation of doxorubicin and its metabolites
The tandem LIF and MS detector was adequate to monitor biotransformation of doxorubicin in the PMF from a rat liver. Figure 2 shows a separation of the PMF of the liver tissue incubated with doxorubicin for 10 minutes then extracted and analyzed by HPLC-LIF-MS. The LIF chromatogram (lower trace) and the corresponding MS chromatogram (upper trace) are comprised of doxorubicin (Peak 1) and possible metabolites (Peaks 2, 3 and 4). For LIF detection, an efficiency of 15250 theoretical plates is achieved for doxorubicin, 7200 theoretical plates for Peak 2, and 8600 for Peak 4. Resolutions for each peak are greater than 1.0 except for Peaks 2 and 3 (Resolution ~ 0.8). However, Peaks 2 and 3 can be distinguished from each other by examining the mass spectra. Furthermore, while all observed signals detected by LIF have corresponding MS signals, there are components that are only present in the MS chromatogram. These components correspond to biological matrix because they are also present in the absence of doxorubicin treatments (See Figure S-1, Supplementary Material). Identifying these components is outside the scope of this work.
Figure 2.

Mass and LIF chromatograms of a liver PMF treated with doxorubicin. The PMF was treated with 50μM doxorubicin, extracted after 10 minutes, and processed as described in the experimental. A volume of 0.5 μL was injected into the HPLC system. Separation was conducted in a 150×0.3 mm C18 column (ACE11115003, Mac-Mod) under isocratic conditions (67% water, 0.1% formic acid: 33% acetonitrile). The detection conditions are described in Figure 1.
Analysis of mass spectrometry data
The sigma factor of the mass spectrometry software, deviations for the predicted mass, and comparison of retention time with those of standards were used to determine the identities of doxorubicin (Figure 3A) and compounds resulting from the doxorubicin treatment of the PMF fractions (Figures 3B-D). The latter figures show that the predominant m/z values for compounds derived from doxorubicin are 401.1182, 397.0873 and 399.1022. Table 2 is a summary of identities, calculated masses, observed masses and errors.
Figure 3.

Mass spectra of compounds detected compounds. See Figure 2. The mass spectra were averaged across chromatographic peaks.
Table 2.
Identities of the peaks observed in MS chromatograms. See Figure 2 for peak assignments.
| Peak | Identity | Measured Mass (m/z) | Calculated Mass (m/z) | Mean Error (ppm) | Retention Time (min) |
|---|---|---|---|---|---|
| 1 | Doxorubicin | 544.1824 | 544.1813 | 1.9 | 6.2 |
| 2 | 7-deoxydoxorubicinolone | 401.1182 | 401.1194 | 6.9 | 8.1 |
| 3 | Doxorubicinone | 397.0873 | 397.0923 | 6.8 | 8.7 |
| 4 | 7-deoxydoxorubicinone | 399.1022 | 399.1079 | 10.7 | 15.3 |
Doxorubicin (Peak 1 in Figure 2, Figure 3A, Figure 4) has an observed characteristic m/z value of 544.1824 (theoretical m/z 544.1813 for (M+H)+) with a minor fragment observed at 397.0873 m/z (theoretical m/z 397.0923).
Figure 4.

Proposed doxorubicin metabolism by the rat liver PMF. Compound detection (Figure 2) and identification (Figure 3) indicate that 7-deoxydoxorubicinone and 7-deoxydoxorubicinolone are present. Doxorubicinol was not detected in this study. Doxorubicinone forming due to acid hydrolysis or metabolic transformation is indistinguishable in this study. Question marks indicate metabolic pathway that cannot be elucidated with the current information.29
From the mass spectra in Figure 3D, Peak 4 in Figure 2 was identified as 7-deoxydoxorubicinone (Figure 4). This aglycone metabolite has a molecular mass of 398.3628 Da, The protonated ion form has an m/z value of 399.1079 Da which is within a 10.7 ppm mass deviation of 399.1022 m/z, the observed mass. Furthermore, the retention times of the observed compound (Peak 4, Figure 2) and 7-deoxydoxorubicinone standard were both 15.3 minutes.
Similarly, Peak 2 in Figure 2 was identified as 7-deoxydoxorubicinolone (Figure 4). The m/z value of 401.1182 (Figure 3B) is within 6.8 ppm error of the calculated mass of 7-deoxydoxorubicinolone, 401.1194 Da. The accurate mass measurements were particularly useful in ruling out a second match for Peak 2. This false match is the demethylated form of doxorubicinone that has a calculated m/z of 401.0867 for the (M+H)+ ion. The mass of the demethylated metabolite, however, is a 78.5 ppm mass deviation from the observed value (401.7782) and is therefore a much less likely candidate than 7-deoxydoxorubicinolone.
Peak 3 in Figure 2 was identified as doxorubicinone (Figure 4). While the molecular mass of doxorubicinone is 414.3622 Da, the observed value is 397.0873 m/z value (Figure 3C), which appears to be caused by protonation and loss of one water molecule resulting from collision induced dissociation. This ion is the same as a fragment observed in the doxorubicin mass spectrum (c.f. Figure 3A). However, doxorubicinone is clearly identified because the retention time of Peak 3 in Figure 2 (i.e. 8.7 minutes) matches that of doxorubicinone standards (i.e. 8.6 minutes).
Doxorubicinone is an aglycone metabolite of doxorubicin that results from oxidative cleavage of the daunosamine sugar,18 but also form as a result of acid catalyzed cleavage of the daunosamine sugar during the sample preparation. In fact, a control using only a doxorubicin standard in incubation buffer followed by perchloric acid extraction also resulted in doxorubicinone detection after 20 minutes of exposure to the acid (data not shown). Therefore, it is not possible to determine the separate relative contributions of metabolic or acid catalyzed reactions to doxorubicinone formation.
The m/z value for doxorubicinol (one of the most commonly reported metabolites),8–10, 18 546 m/z, was not observed in any experiments conducted here. Doxorubicinol standard is easily detectable in both LIF and MS when extracted from biological material and analyzed using this procedure (See Figure S-2, Supplementary Material), which rules out an inadequate extraction and detection scheme. While it is possible that carbonyl reductase, the enzyme responsible for the conversion of doxorubicin to doxorubicinol, has low activity in the PMF of rat liver, a more likely explanation is that any doxorubicinol that formed is rapidly transformed into doxorubicinol by carbonyl reductase17 and then quickly transformed into 7-deoxydoxorubicinolone (Figure 4), which is in agreement with the scheme proposed by Takanashi, et al.22 Based on this argument, both doxorubicin and doxorubicinol are converted further by NADPH – cytochrome P450 reductase and NAD(P)H: quinone oxidoreductase producing the aglycone metabolites 7-deoxydoxorubicinone and 7-deoxydoxorubicinolone (Figure 4).17
The lack of formation of doxorubicinol in this report may be reconciled with the observed formation of this metabolite in other studies, by considering the enzymatic composition of the in vitro systems under investigation. In other studies, the in vitro systems are cytosolic fractions without microsomes and the enzymes (cytochrome P450s) responsible for cleaving the daunosamine sugar of doxorubicin.9 In our in vitro system, the PMF includes microsomes, thereby providing a different metabolic profile for rat liver.
Biotransformation dynamics monitored by LIF
The quantitative power of the LIF detector was suitable to monitor the dynamics of the in vitro metabolism of doxorubicin in four different animals (Figures 5A-D). Doxorubicin and its metabolites were expressed as mole fractions (c.f. Equations 1 and 2), which avoided complications caused differences in the fluorescence quantum yields of the various compounds. As observed in Figure 5, while doxorubicin (diamonds) was consumed, 7-deoxydoxorubicinolone (Peak 2, squares) and 7-deoxydoxorubicinone (Peak 4, triangles) increased. After ~10 minutes, little or not change was noticed. Based on equation 1, metabolite concentrations (at 30 minutes) were 29 μM 7-deoxydoxorubicinolone and 11 μM 7-deoxydoxorubicinone. It is important to note that MS detection was not suitable to monitor the dynamics of the in vitro metabolism (Figure S-3, Supplementary Material).
Figure 5.

Doxorubicin biotransformation dynamics in a rat liver PMF. Mole fractions were calculated using equation 1. Diamonds represent doxorubicin (peak 1); squares represent 7-deoxydoxorubicinolone (peak 2); and triangles represent 7-deoxydoxorubicinone (peak 4). Parts A through D correspond to different animals. Other conditions are given in Figure 2.
In two cases (i.e., Figure 5A and C), an increase in doxorubicin content is observed after reaching a plateau. Both biosynthesis of doxorubicin and variations in detector response are unlikely explanations. In expressing the concentrations of each compound as a mole fraction of doxorubicin, the sum of the concentrations of the four observed compounds is effectively normalized to the initial doxorubicin concentration (50 μM). Therefore, any increase in signal due to a variation in detector response would be eliminated since the sum of all concentrations is taken to be 50 μM in all samples. On the other hand, the difficulty in controlling the length of the extraction procedure because each sample is prepared separately may result in variable acid hydrolysis of doxorubicin to form doxorubicinone. In order to investigate this possibility, we replotted Figure 5C using combined the mole fractions of doxorubicin and doxorubicinone (Figure S-4, Supplementary Material). The apparent increase in doxorubicin disappears.
Not surprisingly, the PMF of each animal exhibited unique biotransformation dynamics, yet consistently forms 7-deoxydoxorubicinolone (squares, Figure 5) and 7-deoxydoxorubicinone (triangles, Figure 5). Clearly, these temporal biotransformation profiles are proof-of-principle of a method that could be easily extended to other fluorescent xenobiotics of biomedical or environmental relevance.
Concluding Remarks
The HPLC-LIF-MS setup described here was adequate to collect simultaneously both structural and quantitative data needed to identify and quantify in vitro metabolism products of doxorubicin from rat liver. The high resolution and high mass accuracy of the MicrOTOFQ narrowed down the number of possible structures of metabolites and revealed 7-deoxydoxorubicinolone and 7-deoxydoxorubicinone are formed, while doxorubicinol, considered a major metabolite in plasma from patients treated with doxorubicin,9, 23 is not detectable. The LIF detector made it possible to estimate concentrations of doxorubicin and its metabolites over the course of an in vitro metabolic study. Future applications of HPLC-LIF-MS systems may include the analysis subcellular factions and intracellular localization of doxorubicin metabolites in cytotoxicity studies,24–26 in vivo metabolism of other fluorescent xenobiotics, biotransformations in different tissue types,18, 27 or metabolic alterations associated with disease and human aging.28 Lastly, the tandem detection scheme described here may have wide applicability in the analysis of fluorescent compounds because it would aid in validating HPLC separations of complex samples prior to the implementation of such separations in instruments with only LIF detection. That is, once the identities of the compounds have been established and interferences have been ruled out, the methods could be directly transferred to instruments with only spectroscopic detection.4
Supplementary Material
Acknowledgments
We thank the support provided by Dr. D. Reed and Dr. J. Dalluge (Directors of the Mass Spectrometry Facility, Department of Chemistry, University of Minnesota) and Professor L. Thompson and her research group, primarily W. Torgerud, who handled animal protocols and procedures. J.B.K was supported by NIH Training grant T32-AG029796.
References
- 1.Crow FW, Cragun JD, Johnson KL, Ruiz MV, Posada De La Paz M, Naylor S. Biomed Chromatogr. 2002;16:311–318. doi: 10.1002/bmc.160. [DOI] [PubMed] [Google Scholar]
- 2.Akbay C, Rizvi SA, Shamsi SA. Anal Chem. 2005;77:1672–1683. doi: 10.1021/ac0401422. [DOI] [PubMed] [Google Scholar]
- 3.Huhn C, Neususs C, Pelzing M, Pyell U, Mannhardt J, Putz M. Electrophoresis. 2005;26:1389–1397. doi: 10.1002/elps.200410163. [DOI] [PubMed] [Google Scholar]
- 4.Gennaro LA, Salas-Solano O. Anal Chem. 2008;80:3838–3845. doi: 10.1021/ac800152h. [DOI] [PubMed] [Google Scholar]
- 5.Wieling J. Chromatographia. 2002;55:107–113. [Google Scholar]
- 6.Anderson AB, Arriaga EA. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;808:295–302. doi: 10.1016/j.jchromb.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 7.Andrews PA, Brenner DE, Chou FT, Kubo H, Bachur NR. Drug Metab Dispos. 1980;8:152–156. [PubMed] [Google Scholar]
- 8.Colombo T, Donelli MG, Urso R, Dallarda S, Bartosek I, Guaitani A. Exp Gerontol. 1989;24:159–171. doi: 10.1016/0531-5565(89)90026-0. [DOI] [PubMed] [Google Scholar]
- 9.Minotti G, Licata S, Saponiero A, Menna P, Calafiore AM, Di Giammarco G, Liberi G, Animati F, Cipollone A, Manzini S, Maggi CA. Chem Res Toxicol. 2000;13:1336–1341. doi: 10.1021/tx000143z. [DOI] [PubMed] [Google Scholar]
- 10.Lachatre F, Marquet P, Ragot S, Gaulier JM, Cardot P, Dupuy JL. J Chromatogr B Biomed Sci Appl. 2000;738:281–291. doi: 10.1016/s0378-4347(99)00529-0. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Yang Y, Liu X, Jiang T. Talanta. 2008;74:887–895. doi: 10.1016/j.talanta.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Anderson AB, Ciriacks CM, Fuller KM, Arriaga EA. Anal Chem. 2003;75:8–15. doi: 10.1021/ac020426r. [DOI] [PubMed] [Google Scholar]
- 13.Mross K, Maessen P, van der Vijgh WJ, Gall H, Boven E, Pinedo HM. J Clin Oncol. 1988;6:517–526. doi: 10.1200/JCO.1988.6.3.517. [DOI] [PubMed] [Google Scholar]
- 14.Urva SR, Shin BS, Yang VC, Balthasar JP. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:837–841. doi: 10.1016/j.jchromb.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 15.Coleman S, Liu S, Linderman R, Hodgson E, Rose RL. Chem Biol Interact. 1999;122:27–39. doi: 10.1016/s0009-2797(99)00107-6. [DOI] [PubMed] [Google Scholar]
- 16.Sheets JJ, Schmidt A, Samaritoni JG, Gifford JM. J Agric Food Chem. 1997;45:4826–4832. [Google Scholar]
- 17.Tani N, Yabuki M, Komuro S, Kanamaru H. Xenobiotica. 2005;35:1121–1133. doi: 10.1080/00498250500342746. [DOI] [PubMed] [Google Scholar]
- 18.Licata S, Saponiero A, Mordente A, Minotti G. Chem Res Toxicol. 2000;13:414–420. doi: 10.1021/tx000013q. [DOI] [PubMed] [Google Scholar]
- 19.Golay MJE. In: Gas Chromatography, 1958. Hesty DH, editor. Academic Press; New York: 1958. p. 36. [Google Scholar]
- 20.Sternberg JC. Advances in Chromatography. 1966;2:205–270. [Google Scholar]
- 21.Leslie AD, Volmer DA. Spectroscopy. 2007;22:32–39. [Google Scholar]
- 22.Takanashi S, Bachur NR. J Pharmacol Exp Ther. 1975;195:41–49. [PubMed] [Google Scholar]
- 23.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 24.Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Oncogene. 1999;18:6641–6646. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- 25.Sokolove PM. Arch Biochem Biophys. 1991;284:292–297. doi: 10.1016/0003-9861(91)90298-w. [DOI] [PubMed] [Google Scholar]
- 26.Sokolove PM. Int J Biochem. 1994;26:1341–1350. doi: 10.1016/0020-711x(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 27.Nolin TD, Naud J, Leblond FA, Pichette V. Clin Pharmacol Ther. 2008;83:898–903. doi: 10.1038/clpt.2008.59. [DOI] [PubMed] [Google Scholar]
- 28.Kiechel JR. Gerontology. 1982;28:101–112. doi: 10.1159/000212577. [DOI] [PubMed] [Google Scholar]
- 29.Takanashi S, Bachur NR. Drug Metab Dispos. 1976;4:79–87. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.