

Abstract
Multiple system atrophy (MSA) is a neurodegenerative disease particularly affecting the basal ganglia, brainstem, cerebellum, and intermediolateral cell columns of the spinal cord but also the cerebral cortex. Clinically, cerebellar (MSA-C) and parkinsonian variants of MSA (MSA-P) are distinguished. We investigated 14 MSA patients (10 MSA-C, 4 MSA-P, men/women: 7/7, age: 61.1±3.3 years) and 14 matched controls (men/women: 7/7, age 58.6±5.1 years) with voxel-based morphometry (VBM) to analyze gray and white matter differences both at baseline and at follow-up, one year later.
Baseline comparisons between patients and controls confirmed significantly less gray matter in MSA in the cerebellum and cerebral cortex, and significantly less white matter in the cerebellar peduncles and brainstem. Comparisons of tissue-loss profiles (i.e., baseline versus follow-up) between patients and controls, revealed white matter reduction in MSA along the middle cerebellar peduncles, reflecting degeneration of the ponto-cerebellar tract as a particularly prominent and progressive morphological alteration in MSA. Comparisons between baseline and follow-up, separately performed in patients and controls, revealed additional white matter reduction in MSA along the corpus callosum at follow-up. This was replicated through additional shape-based analyses indicating a reduced callosal thickness in the anterior and posterior midbody, extending posteriorly into the isthmus. Callosal atrophy may reflect a possibly disease-specific pattern of neurodegeneration and cortical atrophy respectively, fitting well with the predominant impairment of motor functions in MSA patients.
Keywords: MSA, MSA-C, MSA-P, VBM, corpus callosum, follow-up
Introduction
Multiple system atrophy (MSA) is a sporadic, adult-onset disease characterized by progressive neurodegeneration in various parts of the central nervous system involving in particular the basal ganglia, brainstem, cerebellum, and intermediolateral cell columns of the spinal cord but also the cerebral cortex.1,2 Neuropathological hallmark of MSA are α-synuclein-positive oligodendroglial cytoplasmic inclusions (GCIs).3 Clinically, MSA patients present with various combinations of parkinsonism, cerebellar ataxia, and autonomic failure, most notably orthostatic hypotension and urinary incontinence. According to the clinical presentation, a parkinsonian (MSA-P) and a cerebellar variant of MSA (MSA-C) are distinguished.4,5 MSA shows a rapid clinical progression: Many patients are wheelchair-bound after five years of disease duration and often die only a few years later.5–7 Currently neither protective nor curative treatments are available. Future clinical trials may test the efficacy of treatments that aim to reduce the rate of progression of this fatal disease. Thus biomarkers to reliably quantify disease progression during lifetime are essential.
Magnetic resonance imaging (MRI) has been extensively used to study brain morphology in MSA patients. Different MRI analysis techniques, such as ROI-based analysis of T1-weighted and diffusion-weighted images (DWI), voxel-based-morphometry (VBM), voxel-based relaxometry (VBR), diffusion tensor imaging (DTI), or magnetisation transfer imaging (MTI) are useful for detecting disease-related morphological changes in the cerebellum, middle cerebellar peduncles, brainstem, striatum, corpus callosum, and cortical regions in vivo.7–13 The progression of atrophy over time has only recently been studied using different MRI methods detecting regionally varying annual rates of atrophy or signal alterations, partly correlating with clinical progression, and evidence of widespread (sub-) cortical gray matter loss during follow-up. Those studies have focused on predefined regions or were restricted to analyzing gray matter changes and most of them applied only one morphometric method respectively.9,11,14–16 However, applying diverse morphometric methods within one study can be advantageous, as it allows a direct comparison of methods, thus improving the validity of the results.
The aim of the present study was to investigate the progression of both gray and white matter reduction during a one-year follow-up period in 14 MSA patients of both clinical subtypes, to identify structural parameters that might be useful for measuring disease progression. For this purpose, we combined the application of two different methods, VBM and a computational surface-based morphometric method to specifically evaluate the thickness of the corpus callosum.
Methods
Subjects
The study was performed in 14 MSA patients (10 MSA-C, 4 MSA-P, males/females: 7/7, age: 61.1±3.3 years, disease duration: 3.4±1.6 years) and 14 healthy controls without history of neurological or psychiatric disorder and with a normal neurological examination (men/women: 7/7, age 58.6±5.1 years). Diagnosis of MSA was made according to established criteria.17 Patients and healthy controls underwent MRI imaging and neurological examination twice in a time interval of one year (time span in between: 14.6±3.0 years (MSA group) and 16.7±6.1 (control group)).
The study was approved by the ethics committee of the Medical Faculty of the University of Bonn. Informed, written consent was obtained from all participants.
Data Acquisition
Magnetic resonance imaging was performed using a 1.5T scanner (Siemens Symphony, Siemens AG, Erlangen, Germany) with the standard head coil. The MRI protocol comprised a sagittal T1-weighted (MPRAGE) sequence (TR 11.08 ms, TE 4.3 ms, FA 15°, FOV 230 mm, 256×256 acquisition matrix) yielding 200 sagittal slices and a voxel size of 0.9 × 0.9 × 0.9 mm3.
Voxel-based Morphometry (VBM)
The data were processed according to our previously described protocol13 using statistical parametric mapping (SPM), only differing by the use of the updated version SPM5 instead of SPM2 (SPM5, http://www.fil.ion.ucl.ac.uk/spm/software/spm5). This protocol comprises a series of pre-processing steps, which were verified separately for each subject. As a first step, the anterior commissure was set in each image as the origin of the individual stereotaxic space. Further processing was performed according to the protocol originally described by Good et al.18 and adapted for the use with SPM5. This revised procedure optimizes the normalization and segmentation for the tissue type examined, using a unified segmentation method.19 In short, tissue probability maps, provided by the SPM5 software, are overlaid by nonlinear deformations onto the individual anatomy and used as priors to segment the original image into the three tissue classes, gray matter, white matter, and cerebro-spinal fluid (CSF). The procedure also includes a bias correction of the data to compensate for inhomogeneities in the distribution of voxel intensities across each tissue class. The resulting normalized and segmented images were resampled to a voxel-size of 1.5 × 1.5 × 1.5 mm and smoothed with a 12 mm Gaussian kernel.
Using the smoothed tissue segments, the following statistical comparisons were performed, separately, for gray and white matter: (1) two-sample t-tests between MSA patients and controls at baseline and at follow-up; (2) paired t-tests comparing MRI images at baseline and at follow-up, separately for MSA patients and controls; and (3) two-sample t-tests of difference-maps comparing MSA patients and controls, where individual difference maps were created by subtracting gray and white matter maps at follow-up from baseline (mapbaseline – mapfollow-up). The results were explored at an FDR-corrected threshold of p < 0.05 as voxel-wise threshold and an FWE-corrected threshold of p < 0.05 for the extent of the respective cluster. All statistical comparisons were controlled for global differences by including the overall brain volume as a confounding covariate in the design matrix.
Callosal Thickness Analysis
The corpus callosum was outlined in normalized data after applying 6-parameter (rigid-body) transformations. An overview of the basic steps for measuring callosal thickness is provided elsewhere.20 Briefly, one rater (E.L.) manually outlined upper and lower callosal boundaries in the midsagittal section of each brain. Subsequently, the spatial average from 100 equidistant surface points representing the upper and lower traces was calculated by creating a new midline segment, also consisting of 100 equidistant points. Finally, distances between the midline segment and the 100 corresponding surface points of the upper and lower callosal segments were quantified. These regional distances indicate callosal thickness with a high spatial resolution (i.e., at 100 locations distributed evenly over the callosal surface).
Using the callosal distance values, the following statistical comparisons were performed: (1) two-sample t-tests between MSA patients and controls at baseline and at follow-up; and (2) paired t-tests comparing MRI images at baseline and at follow-up, separately for MSA patients and controls. As statistical tests were made at hundreds of callosal surface points and adjacent data points are highly correlated, statistical results were corrected for multiple comparisons using the False Discovery Rate (FDR) method at p < 0.05.21 Both uncorrected and corrected maps were generated and displayed.
Results
Gray matter
Group comparison at baseline revealed that MSA patients had less gray matter than controls in cerebellar hemispheres, left cingulum and in several cortical regions, including bifrontal and left insular regions. At follow-up, we found a more pronounced reduction of gray matter in MSA patients in initially affected regions and additional gray matter reduction in thalamic and caudate nuclei.
Comparison of the follow-up and baseline images with paired t-tests detected gray matter reduction in initially affected brain regions and additionally in the right and left caudate nucleus, right thalamus and right cingulum. In contrast, two-sample t-tests of difference-maps comparing MSA and control group did not depict any gray matter reduction during the follow-up period (Table 1).
TABLE 1.
Gray matter reduction at baseline and during the follow-up period in 14 MSA patients in comparision with matched control group
| Cluster level | Voxel level | Coordinates | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P(Corr) | Size | P(uncorr) | P(FDR-cor) | T-Value | Z-Value | P(uncorr) | X | Y | Z | Side | Area |
| Gray matter reduction at baseline (compared with controls) | |||||||||||
| 0.000 | 39,833 | 0.000 | 0.000 | 7.73 | 5.52 | 0.000 | 0 | −38 | −20 | Midline | Cerebellum (Vermis) |
| 0.000 | 5.85 | 4.63 | 0.000 | −30 | −68 | −24 | Left | Cerebellum | |||
| 0.001 | 5.54 | 4.46 | 0.000 | −26 | 44 | −18 | Left | Middle orbito-frontal gyrus | |||
| 0.001 | 5.44 | 4.40 | 0.000 | −14 | −58 | −2 | Left | Lingnal gyrus | |||
| 0.001 | 5.38 | 4.37 | 0.000 | 22 | 62 | 28 | Right | Middle frontal gyrus | |||
| 0.001 | 5.33 | 4.34 | 0.000 | −12 | −20 | 50 | Left | Cingulam (ext. into supplemental motor area) | |||
| 0.001 | 5.30 | 4.33 | 0.000 | 18 | 64 | 26 | Right | Superior frontal gyrus | |||
| 0.001 | 5.26 | 4.30 | 0.000 | 30 | −40 | −18 | Right | Fusiforn gyrus | |||
| 0.001 | 5.10 | 4.21 | 0.000 | 32 | −68 | −22 | Right | Cerebellum | |||
| 0.001 | 5.01 | 4.15 | 0.000 | −24 | 54 | −18 | Left | Middle orbito-frontal gyrus | |||
| 0.001 | 4.67 | 3.95 | 0.000 | −38 | −66 | 50 | Left | Angular gyrus | |||
| 0.001 | 4.65 | 3.93 | 0.000 | 24 | 68 | −8 | Right | Middle orbito-frontal gyrus | |||
| 0.002 | 4.61 | 3.90 | 0.000 | −14 | 68 | −12 | Left | Superior orbito-frontal gyrus | |||
| 0.002 | 4.59 | 3.90 | 0.000 | −10 | 68 | −10 | Left | Midian orbito-frontal gyrus | |||
| Grey matter reduction during the follow-up (compared with controls) | |||||||||||
| 0.000 | 61,842 | 0.000 | 0.000 | 11.58 | 6.82 | 0.000 | 2 | −36 | −18 | Right | Cerbellum (vermis) |
| 0.000 | 7.05 | 5.23 | 0.000 | −20 | −58 | −56 | Left | Cerebellum | |||
| 0.000 | 6.90 | 5.16 | 0.000 | 28 | −64 | −54 | Right | Cerebellum | |||
| 0.000 | 6.74 | 5.08 | 0.000 | 26 | −64 | −14 | Right | Fusiform gyrus | |||
| 0.000 | 6.15 | 4.79 | 0.000 | −12 | −20 | 50 | Left | Cingulum (ext. into supplemental motor area) | |||
| 0.000 | 5.10 | 4.21 | 0.000 | −16 | −20 | −24 | Left | Parahippocampal gyrus | |||
| 0.000 | 5.09 | 4.20 | 0.000 | 30 | 20 | 46 | Right | Middle frontal gyrus | |||
| 0.000 | 5.08 | 4.19 | 0.000 | 18 | 64 | 26 | Right | Superior frontal gyrus | |||
| 0.000 | 5.07 | 4.19 | 0.000 | 22 | 68 | −8 | Right | Middle orbito-frontal gyrus | |||
| 0.001 | 4.79 | 4.02 | 0.000 | −14 | 68 | −12 | Left | Middle orbito-frontal gyrus | |||
| 0.001 | 4.56 | 3.87 | 0.000 | −40 | 6 | 6 | Left | Insullar gyrus | |||
| Grey matter reduction during the follow-up period (MSA group) | |||||||||||
| 0.000 | 22,318 | 0.000 | 0.011 | 7.98 | 4.73 | 0.000 | −34 | −48 | −22 | Left | Cerebellum |
| 0.011 | 7.85 | 4.69 | 0.000 | 8 | 54 | 34 | Right | Superior media-frontal gyrus | |||
| 0.011 | 6.52 | 4.27 | 0.000 | 0 | 6 | −10 | Midline | Olfactory gyrus | |||
| 0.011 | 6.42 | 4.24 | 0.000 | −18 | −12 | −22 | Left | Parahippocampus | |||
| 0.011 | 6.13 | 4.13 | 0.000 | 24 | −44 | −20 | Right | Cerebellum | |||
| 0.011 | 6.12 | 4.13 | 0.000 | −44 | −32 | −20 | Left | Interior temporal gyrus | |||
| 0.012 | 6.11 | 4.12 | 0.000 | −4 | 8 | −12 | Left | Olfactory gyrus | |||
| 0.012 | 6.09 | 4.12 | 0.000 | −40 | −30 | −20 | Left | Fusiform gyrus | |||
| 0.012 | 5.94 | 4.06 | 0.000 | 6 | 12 | 4 | Right | Caudate nucleus (ext. to left) | |||
| 0.012 | 5.87 | 4.03 | 0.000 | −6 | −40 | −14 | Left | Cerebellum (vermis) | |||
| 0.013 | 5.69 | 3.96 | 0.000 | −8 | 6 | −16 | Left | Rectus gyrus | |||
| 0.013 | 5.67 | 3.96 | 0.000 | −10 | 52 | 36 | Left | Superior media-frontal gyrus | |||
| 0.013 | 5.57 | 3.92 | 0.000 | −52 | 12 | 0 | Left | Insula | |||
| 0.013 | 5.47 | 3.87 | 0.000 | 6 | 16 | 44 | Right | Cingulum | |||
| 0.013 | 5.46 | 3.87 | 0.000 | 8 | −40 | −10 | Right | Cerebellum (vermis) | |||
| Grey matter reduction during the follow-up period (compared with controls) | |||||||||||
| No significant clusters | |||||||||||
Voxel size is 1.5 × 1.5 × 1.5 mm3
White matter
Group comparisons at baseline revealed that MSA patients had less white matter than controls along the middle cerebellar peduncles and brainstem (corticospinal tract) bilaterally. At follow-up, we found more pronounced reduction in previously affected regions and additional white matter reduction along the corticospinal tract bilaterally.
Comparison of the follow-up and baseline images with paired t-tests revealed white matter reduction in initially affected brain regions and additionally along the corticospinal tracts (left and right internal capsule, subcortical to left precentral gyrus) and along the entire corpus callosum. The two-sample t-tests comparing difference-maps between MSA patients and controls similarly showed white matter reduction during the follow-up period in cerebellum, middle cerebellar peduncles and subcortical to left precentral gyrus, but did not depict changes of corticospinal tracts at further levels or the corpus callosum (Table 2).
TABLE 2.
White matter reduction at baseline and during the follow-up period in 14 MSA patients in comparison with a matched control group
| Cluster level | Voxel level | Coordinates | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P(corr) | Size | P(uncorr) | P(FDR-cor) | T-value | Z-vaule | P(uncorr) | X | Y | Z | Side | Area |
| White matter reduction at baseline (compared with controls) | |||||||||||
| 0.001 | 751 | 0.000 | 0.000 | 7.11 | 5.25 | 0.000 | 12 | −20 | −34 | Right | MCP, CST |
| 0.000 | 6.62 | 5.03 | 0.000 | −14 | −22 | −32 | Left | MCP, CST | |||
| White matter reduction at follow-up (compared with controls) | |||||||||||
| 0.000 | 4.251 | 0.000 | 0.000 | 7.98 | 5.62 | 0.000 | −16 | −34 | −36 | Left | MCP (extending to the right MCP) |
| 0.035 | 484 | 0.004 | 0.003 | 4.43 | 3.79 | 0.000 | −6 | −8 | −6 | Left | CST |
| 0.003 | 4.40 | 3.77 | 0.000 | 4 | −14 | −8 | Right | CST | |||
| White matter reduction during the follow-up period (MSA group) | |||||||||||
| 0.000 | 21,151 | 0.000 | 0.000 | 16.03 | 6.19 | 0.000 | 20 | −44 | −44 | Right | MCP |
| 0.000 | 10.59 | 5.34 | 0.000 | −16 | −44 | −42 | Left | MCP | |||
| 0.001 | 7.79 | 4.67 | 0.000 | 6 | 16 | 20 | Right | Corpus callosum | |||
| 0.001 | 7.71 | 4.65 | 0.000 | −14 | −24 | −24 | Left | MCP + CST | |||
| 0.003 | 5.80 | 4.01 | 0.000 | −8 | −2 | 8 | Left | Internal capsule | |||
| 0.004 | 5.57 | 3.91 | 0.000 | 30 | −36 | 14 | Right | Subcortical to superior temporl gyrus | |||
| 0.005 | 5.51 | 3.89 | 0.000 | −6 | −6 | 24 | Left | Corpus callosum | |||
| 0.005 | 5.50 | 3.88 | 0.000 | −4 | −42 | 16 | Left | Corpus callosum (forceps major) | |||
| 0.005 | 5.47 | 3.87 | 0.000 | 16 | −34 | −42 | Right | MCP + CST | |||
| 0.005 | 5.40 | 3.84 | 0.000 | 4 | −42 | 14 | Right | Corpus callosum (forceps major) | |||
| 0.005 | 5.32 | 3.81 | 0.000 | 16 | −12 | −24 | Right | CST | |||
| 0.006 | 5.27 | 3.79 | 0.000 | 36 | −44 | 6 | Right | Subcortical to middle temporal gyrus | |||
| 0.006 | 5.23 | 3.77 | 0.000 | 18 | −22 | 22 | Right | Corona radiata | |||
| 0.006 | 5.19 | 3.75 | 0.000 | 8 | 0 | 6 | Right | Internal capsule | |||
| White matter reduction during the follow-up period (compared with controls) | |||||||||||
| 0.000 | 645 | 0.000 | 0.001 | 8.01 | 5.64 | 0.000 | 18 | −44 | −44 | Right | Cerebellum, MCP |
| 0.000 | 662 | 0.000 | 0.003 | 6.41 | 4.92 | 0.000 | −18 | −42 | −42 | Left | Cerebellum, MCP |
| 0.028 | 186 | 0.009 | 0.026 | 3.94 | 3.46 | 0.000 | −30 | −8 | 30 | Left | Subcortical to precentral gyrus |
Voxel size 1.5 × 1.5 × 1.5 mm3.
MCP, middle cerebellar peduncle; CST, corticospinal tract.
Callosal thickness
As shown in Figure 1 (top panels), callosal regions were thinner in MSA patients compared to healthy controls. The difference was more pronounced at follow-up (panel B) than at baseline (panel A). At baseline, the corpora callosa of MSA patients were thinner mainly across the anterior and posterior body. At follow-up, the isthmus was additionally affected. We did not detect any region in which callosal thickness was thicker in MSA patients compared to controls.
Figure 1. Differences in callosal thickness.
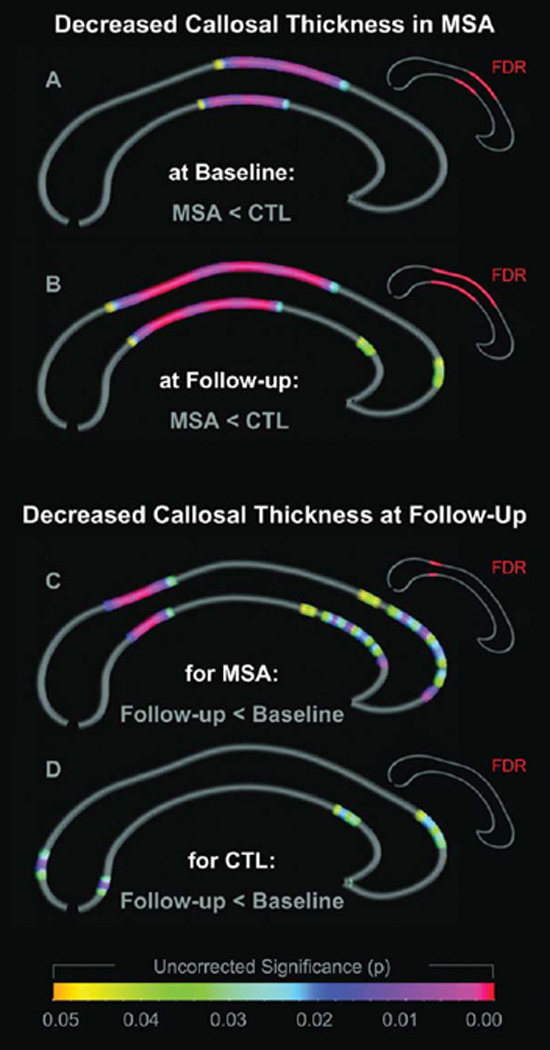
The two top panels illustrate the decreased callosal thickness in MSA patients compared to controls at baseline (panel A) and at follow-up (panel B). The two bottom panels illustrate the decreased callosal thickness at follow-up compared to baseline in MSA patients (panel C) and in controls (panel D). Group and time effects are color-coded and projected onto the average callosal surface map. Spectral colors indicate uncorrected significance (large callosal maps); red indicates FDR-corrected significance (small callosal maps).
As further shown in Figure 1 (bottom panels), callosal thickness declined over time. The decline was more pronounced in MSA patients (panel C) than in controls (panel D). More specifically, the thickness of the corpora callosa in MSA patients mainly decreased in the isthmus. The thickness of the corpora callosa of controls slightly declined in the splenium and in the anterior third, but this decline did not reach the level of statistical significance. We did not detect any region, in MSA patients or in controls, in which callosal thickness increased over time.
Discussion
We performed a one-year longitudinal MRI study of 14 MSA patients to investigate the progression of brain tissue loss in MSA. Comparison of MSA patients with controls confirmed a reduction of gray matter in the cerebellum and certain cortical areas and a reduction of white matter in the cerebellum, cerebellar peduncles and brainstem, already detectable at baseline. Analysis of the follow-up MRIs revealed further progression of brain matter loss.
The MSA within-group comparison between baseline and follow-up as well as the comparison of MSA patients and controls at follow-up showed progressive gray and white matter reduction mainly in regions that were already affected at baseline. Interestingly, these areas show partly resemblance with areas normally affected by age-related shrinkage (i.e., in healthy subjects). For example, the progressive reduction of the brainstem and cerebellum is well documented to occur during normal aging and is most prominent after the age of 50 years.22,23 This might also explain why the comparison of difference maps between MSA and controls did not reveal progressive tissue loss in all those regions, but only confirmed progressive white matter reduction of the middle cerebellar peduncles. As the latter approach (comparison of the difference maps) takes into account normal age-related decline of the brain, it may be more appropriate to depict disease-specific changes.
White matter tissue reduction along the middle cerebellar peduncles likely reflects a degeneration of the pontocerebellar tract as a particularly prominent and progressive morphological alteration in MSA brains. This finding is in line with the results of recent morphometric studies investigating tissue-specific properties such as diffusivity (diffusion-weighted images, diffusion tensor imaging) instead of MRI-signal alterations (VBM). These studies similarly identified degeneration of the middle cerebellar peduncles as a MSA-specific feature.12,24–28 Therefore, atrophy of the middle cerebellar peduncles seems to be the most robust, aging-independent, and disease-specific atrophy in MSA.
MSA within-group comparison between baseline and follow-up revealed not only reduction of white matter in typically affected regions like middle cerebellar peduncles and corticospinal tract but also across the entire corpus callosum. VBM - primarily developed to study gray matter - generates smoothed probability maps of white matter for the final statistical comparison. Due to methodological constraints, anatomical information contained within those maps may have only a limited precision regarding the exact anatomical localisation of affected callosal subregions. The prominent involvement of the corpus callosum was therefore independently validated by a second morphometric method to explore the regional pattern of callosal atrophy in MSA patients, in great detail. For this purpose, we investigated corpus callosum thickness at 100 points across the callosal surface. At baseline, we found a deficit in callosal thickness in MSA patients compared to controls in the anterior and posterior body, extending even further posteriorly at follow-up. According to recent DTI-based study of callosal connectivity, the anterior midbody connects cortical premotor and supplementary motor regions between both hemispheres.29 The posterior midbody contains the highest density of fibres with a large diameter30 and is linked to the primary motor cortex.29 Thus, the observed pattern of callosal atrophy agrees closely with the predominant impairment of motor functions in MSA patients. As we investigated a mixed patient sample including MSA-C and MSA-P patients, further studies will need to determine whether callosal atrophy is equally associated with both clinical subtypes.
So far, little attention has been paid to callosal atrophy in MSA, although glial cytoplasmic inclusions have been observed in the corpus callosum as well.3,31 Watanabe et al. used a ROI-based MRI approach and detected callosal atrophy in a subgroup of MSA patients, not correlating with disease duration.7 Since they also found signs of cortical atrophy in MSA patients with callosal atrophy, the authors suggested a link between cortical and callosal atrophy. Another study found callosal atrophy only in MSA-P but not in MSA-C patients.32
Cerebellar atrophy, especially of the middle cerebellar peduncles, is one of the most prominent findings in MSA, but its degree depends on the clinical subtype and is highly variable, whereas the degree of cortical atrophy was more similar in both subtypes.10,33,34 To validate new therapies it would be advantageous to have morphological markers of disease progression independently of the clinically dominant symptom. If callosal atrophy occurs as a result of Wallerian degeneration, it can serve as an indirect neuroanatomical marker for cortical atrophy. Especially neuronal loss in the third cortical layer results in axonal degeneration and consecutive callosal atrophy and has been described in neurodegenerative disorders like Alzheimer’s disease, frontotemporal dementia, progressive supranuclear palsy and corticobasal degeneration.35–37 Separation of age-related from disease-related callosal atrophy is critical and essential for its use as morphological marker in follow-up investigations. However, regionally circumscribed callosal atrophy as described here for MSA may possibly reflect a disease-specific pattern of neurodegeneration and cortical atrophy respectively.
Acknowledgement
We gratefully acknowledge the participation of all patients and healthy controls investigated in this study. This study was supported by the National Institutes of Health through the NIH Roadmap for Medical Research, Grant U54 RR021813 entitled Center for Computational Biology (CCB). Information on the National Centers for Biomedical Computing can be obtained from <http://nihroadmap.nih.gov/bioinformatics>. Additional support was provided by NIH grants P41 RR013642, M01 RR000865, R01 EB007813, R01 EB008281, R01 EB008432, R01 HD050735, and R01 AG020098.
Footnotes
Financial disclosure/Conflict of interest concerning the research related to the manuscript:
There is no conflict of interest.
- Research project: A. Conception, B. Organization C. Execution
- Statistical Analysis: A. Design, B. Execution, C. Review and Critique
- Manuscript: A. Writing of the first draft, B. Review and Critique
Martina Minnerop: 1A, 1B, 1C, 2A, 2B, 3A
Eileen Luders: 1B, 2A, 2B, 2C, 3A, 3B
Karsten Specht: 1B, 2A, 2C, 3B
Jürgen Ruhlmann: 1B, 1C, 2C, 3B
Nicole Schimke: 1B, 2C, 3B
Paul M. Thompson: 1B, 2C, 3B
Yi-Yu Chou: 1B, 2B, 3B
Arthur W. Toga: 1B, 2C, 3B
Michael Abele: 1A, 2C, 3B
Ullrich Wüllner: 1A, 1B, 2C, 3B
Thomas Klockgether: 1A, 1B, 2A, 2C, 3B
Full Financial Disclosures of all Authors for the Past Year:
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment Research Centre Juelich |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants none |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment University of California, Los Angeles (UCLA) |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants none |
Other none |
| Stock Ownership in medically-related fields NordicNeuroLab AS |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment University of Bergen |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants Bergen Research Foundation, International Neuroinformatics Coordinating Facility |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment Medizin Center Bonn, MVZ |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants none |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment University Hospital of Marburg |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants none |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment University of California, Los Angeles (UCLA) |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants National Institutes of Health |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment University of California, Los Angeles (UCLA) |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants none |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment University of California, Los Angeles (UCLA) |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants National Institutes of Health |
Other none |
| Stock Ownership in medically-related fields none |
Intellectual Property Rights none |
| Consultancies none |
Expert Testimony none |
| Advisory Boards none |
Employment Free-lancing |
| Partnerships none |
Contracts none |
| Honoraria none |
Royalties none |
| Grants none |
Other none |
| Stock Ownership in medically-related fields None |
Intellectual Property Rights None |
| Consultancies Boehringer-Ingelheim, Lifescience KG, Teva, UCB Pharma |
Expert Testimony None |
| Advisory Boards Boehringer-Ingelheim, Lifescience KG, Teva, UCB Pharma |
Employment University of Bonn |
| Partnerships None |
Contracts None |
| Honoraria None |
Royalties None |
| Grants Deutsche Forschungsgemeinschaft, Bundesministerium für Forschung und Technologie, National Ataxia Foundation, deutsche Parkinson-Vereinigung e.V. |
Other None |
| Stock Ownership in medically-related fields Bayer |
Intellectual Property Rights None |
| Consultancies Lundbeck (Rater Training) |
Expert Testimony None |
| Advisory Boards Parkinsonism & Related Disorders |
Employment University of Bonn |
| Partnerships None |
Contracts None |
| Honoraria Lectures (Pfizer, Neurosearch) |
Royalties Thieme, Marcel Dekker |
| Grants Deutsche Forschungsgemeinschaft, Bundesministerium für Forschung und Technologie, European Community, Santhera |
Othe None |
References
- 1.Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1969;32:28–34. doi: 10.1136/jnnp.32.1.28. 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinn N. Multiple system atrophy – the nature of the beast. J Neurol Neurosurg Psychiatry. 1989 Suppl:78–89. doi: 10.1136/jnnp.52.suppl.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain. 1994;117:235–243. doi: 10.1093/brain/117.2.235. [DOI] [PubMed] [Google Scholar]
- 4.Schulz JB, Klockgether T, Petersen D, et al. Multiple system atrophy: natural history, MRI morphology, and dopamine receptor imaging with 123IBZM-SPECT. J Neurol Neurosurg Psychiatry. 1994;57:1047–1056. doi: 10.1136/jnnp.57.9.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenning GK, Ben Shlomo Y, Magalhaes M, Daniel SE, Quinn N. Clinical features and natural history of multiple system atrophy: an analysis of 100 cases. Brain. 1994;117:835–845. doi: 10.1093/brain/117.4.835. [DOI] [PubMed] [Google Scholar]
- 6.Klockgether T, Lüdtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain. 1998;121:589–600. doi: 10.1093/brain/121.4.589. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain. 2002;125:1070–1083. doi: 10.1093/brain/awf117. [DOI] [PubMed] [Google Scholar]
- 8.Eckert T, Sailer M, Kaufmann J, et al. Differentiation of idiopathic Parkinson's disease, multiple system atrophy, progressive supranuclear palsy, and healthy controls using magnetization transfer imaging. Neuroimage. 2004;21:229–235. doi: 10.1016/j.neuroimage.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 9.Hauser TK, Luft A, Skalej M, et al. Visualization and quantification of disease progression in multiple system atrophy. Mov Disord. 2006;21:1674–1681. doi: 10.1002/mds.21032. [DOI] [PubMed] [Google Scholar]
- 10.Minnerop M, Specht K, Ruhlmann J, et al. Voxel-based morphometry and voxel-based relaxometry in multiple system atrophy-a comparison between clinical subtypes and correlations with clinical parameters. Neuroimage. 2007;36:1086–1095. doi: 10.1016/j.neuroimage.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 11.Seppi K, Schocke MF, Mair KJ, et al. Progression of putaminal degeneration in multiple system atrophy: a serial diffusion MR study. Neuroimage. 2006;31:240–245. doi: 10.1016/j.neuroimage.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Shiga K, Yamada K, Yoshikawa K, Mizuno T, Nishimura T, Nakagawa M. Local tissue anisotropy decreases in cerebellopetal fibers and pyramidal tract in multiple system atrophy. J Neurol. 2005;252:589–596. doi: 10.1007/s00415-005-0708-0. [DOI] [PubMed] [Google Scholar]
- 13.Specht K, Minnerop M, Muller-Hubenthal J, Klockgether T. Voxel-based analysis of multiple-system atrophy of cerebellar type: complementary results by combining voxel-based morphometry and voxel-based relaxometry. Neuroimage. 2005;25:287–293. doi: 10.1016/j.neuroimage.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Horimoto Y, Aiba I, Yasuda T, et al. Longitudinal MRI study of multiple system atrophy - when do the findings appear, and what is the course? J Neurol. 2002;249:847–854. doi: 10.1007/s00415-002-0734-0. [DOI] [PubMed] [Google Scholar]
- 15.Paviour DC, Price SL, Jahanshahi M, Lees AJ, Fox NC. Longitudinal MRI in progressive supranuclear palsy and multiple system atrophy: rates and regions of atrophy. Brain. 2006;129:1040–1049. doi: 10.1093/brain/awl021. [DOI] [PubMed] [Google Scholar]
- 16.Brenneis C, Egger K, Scherfler C, et al. Progression of brain atrophy in multiple system atrophy. A longitudinal VBM study. J Neurol. 2007;254:191–196. doi: 10.1007/s00415-006-0325-6. [DOI] [PubMed] [Google Scholar]
- 17.Gilman S, Low PA, Quinn N, et al. Consensus statement of the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 18.Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS. A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage. 2001;14:21–36. doi: 10.1006/nimg.2001.0786. [DOI] [PubMed] [Google Scholar]
- 19.Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–851. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 20.Luders E, Narr KL, Zaidel E, Thompson PM, Jancke L, Toga AW. Parasagittal asymmetries of the corpus callosum. Cereb Cortex. 2006;16:346–354. doi: 10.1093/cercor/bhi112. [DOI] [PubMed] [Google Scholar]
- 21.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Royal Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- 22.Luft AR, Skalej M, Schulz JB, et al. Patterns of age-related shrinkage in cerebellum and brainstem observed in vivo using three-dimensional MRI volumetry. Cereb Cortex. 1999;9:712–721. doi: 10.1093/cercor/9.7.712. [DOI] [PubMed] [Google Scholar]
- 23.Pieperhoff P, Hömke L, Schneider F, et al. Deformation field morphometry reveals age-related structural differences between the brains of adults up to 51 years. J Neurosci. 2008;28:828–842. doi: 10.1523/JNEUROSCI.3732-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito M, Watanabe H, Kawai Y, et al. Usefulness of combined fractional anisotropy and apparent diffusion coefficient values for detection of involvement in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2007;78:722–728. doi: 10.1136/jnnp.2006.104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taoka T, Kin T, Nakagawa H, et al. Diffusivity and diffusion anisotropy of cerebellar peduncles in cases of spinocerebellar degenerative disease. Neuroimage. 2007;37:387–393. doi: 10.1016/j.neuroimage.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Terajima K, Matsuzawa H, Shimohata T, Akazawa K, Nishizawa M, Nakada T. Tract-by-tract morphometric and diffusivity analyses in vivo of spinocerebellar degeneration. J Neuroimaging. 2009;19:220–226. doi: 10.1111/j.1552-6569.2008.00273.x. [DOI] [PubMed] [Google Scholar]
- 27.Oishi K, Konishi J, Mori S, et al. Reduced fractional anisotropy in early-stage cerebellar variant of multiple system atrophy. J Neuroimaging. 2009;19:127–131. doi: 10.1111/j.1552-6569.2008.00262.x. [DOI] [PubMed] [Google Scholar]
- 28.Prakash N, Hageman N, Hua X, Toga AW, Perlman SL, Salamon N. Patterns of fractional anisotropy changes in white matter of cerebellar peduncles distinguish spinocerebellar ataxia-1 from multiple system atrophy and other ataxia syndromes. Neuroimage. 2009;47 Suppl 2:T72–T81. doi: 10.1016/j.neuroimage.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Hofer S, Frahm J. Topography of the human corpus callosum revisited--comprehensive fiber tractography using diffusion tensor magnetic resonance imaging. Neuroimage. 2006;32:989–994. doi: 10.1016/j.neuroimage.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 30.Aboitiz F, Scheibel AB, Fisher RS, Zaidel E. Fiber composition of the human corpus callosum. Brain Res. 1992;598:143–513. doi: 10.1016/0006-8993(92)90178-c. [DOI] [PubMed] [Google Scholar]
- 31.Costa C, Duyckaerts C, Cervera P, Hauw JJ. Oligodendroglial inclusions, a marker of multisystemic atrophies. Rev Neurol (Paris) 1992;148:274–280. [PubMed] [Google Scholar]
- 32.Kamitani T, Kuroiwa Y, Wang L, et al. Visual event-related potential changes in two subtypes of multiple system atrophy, MSA-C and MSA-P. J Neurol. 2002;249:9975–9982. doi: 10.1007/s00415-002-0764-7. [DOI] [PubMed] [Google Scholar]
- 33.Brenneis C, Seppi K, Schocke MF, et al. Voxel-based morphometry detects cortical atrophy in the Parkinson variant of multiple system atrophy. Mov Disord. 2003;18:1132–1138. doi: 10.1002/mds.10502. [DOI] [PubMed] [Google Scholar]
- 34.Brenneis C, Boesch SM, Egger KE, et al. Cortical atrophy in the cerebellar variant of multiple system atrophy: a voxel-based morphometry study. Mov Disord. 2006;21:159–165. doi: 10.1002/mds.20656. [DOI] [PubMed] [Google Scholar]
- 35.Innocenti GM. General organization of callosal connections in the cerebral cortex. In: Jones EG, Peters A, editors. Cerebral cortex. 5th ed. New York: Plenum Press; 1986. pp. 291–353. [Google Scholar]
- 36.Wiltshire K, Foster S, Kaye JA, Small BJ, Camicioli R. Corpus callosum in neurodegenerative diseases: findings in Parkinson's disease. Dement Geriatr Cogn Disord. 2005;20:345–351. doi: 10.1159/000088526. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi H, Fukuyama H, Nagahama Y, et al. Comparison of the pattern of atrophy of the corpus callosum in frontotemporal dementia, progressive supranuclear palsy, and Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2000;69:623–629. doi: 10.1136/jnnp.69.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]