

Abstract

A new thiourea catalyst is reported for the enantioselective cationic polycyclization of hydroxylactams. Both the yield and enantioselectivity of this transformation were found to vary strongly with the identity of a single aromatic residue on a common catalyst framework, with more expansive and polarizable arenes proving optimal. Evidence is presented for a mechanism in which stabilizing cation-π interactions are a principal determinant of enantioselectivity.
Advances in cyclase enzymology have provided strong evidence that cation-π interactions play an essential catalytic role in biosynthetic polyene cyclizations.1,2 Structural, kinetic, and site-directed mutagenesis studies all suggest that the cationic intermediates and transition states accessed in these transformations are stabilized through a series of attractive interactions with aromatic residues that line the cyclase active site.3 This mechanistic insight suggests the intriguing possibility that analogous stabilizing cation-π interactions might also be engineered into selective small-molecule catalysts.4
We became interested in developing an asymmetric polycyclization jointly predicated on this biosynthetic model and our recent work in anion binding thiourea catalysis.5,6 The ability of arenes to stabilize cations offers a logical complement to the anion binding properties of thioureas.7 As such, an appropriate bifunctional catalyst would be capable of electrostatically stabilizing both poles of a reactive ion pair in a spatially resolved manner, increasing the probability of strong binding to the enantioselectivity-determining transition state structure (Scheme 1).8 Herein, we report the development of a new thiourea catalyst for the enantioselective bicyclization of hydroxylactams, together with evidence that stabilizing cation-π interactions play a principal role in asymmetric induction.
Scheme 1.

Proposal for enantioselective polycyclization
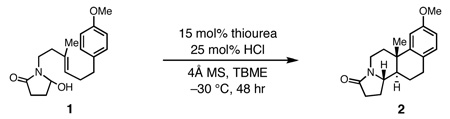
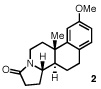
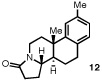
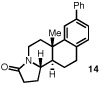
Our efforts focused on developing an enantioselective variant of a polycyclization originally reported by Speckamp that proceeds through an N-acyliminium ion intermediate (Table 1).9 In evaluating the bicyclization of hydroxylactam 1, a preliminary survey of thioureas, Brønsted acids and solvents produced a lead result with thiourea 3, with tetracycle 2 generated in 10% yield and 9% ee upon treatment with 0.25 equivalents of HCl in TBME containing 4Å molecular sieves at −30 °C (Table 1). Catalyst 4, a conformationally constrained analog of 3 bearing a 2-phenylpyrrolidine ring, afforded a modest increase in enantioselectivity.10 Modification of the electronic properties of the aromatic group of 4 by introduction of simple substituents had little effect on catalyst performance. In contrast, 2-aryl pyrrolidine catalysts bearing larger aromatic groups proved substantially more reactive and enantioselective. The naphthyl-substituted catalysts 5 and 6 both provided 2 in greater than 60% ee, while 9-phenanthryl-derived catalyst 7 furnished product 2 in 52% yield and 87% ee. Spurred by the apparent correlation between the expanse of the pyrrolidine arene and catalytic performance, we prepared and evaluated 4-pyrenyl-substituted thiourea derivative 8. This proved to be the optimal catalyst, providing 2 in 78% yield and 95% ee. Under the action of all the catalysts described in Table 1, tetracycle 2 was formed as a single detectable diastereomer, the relative stereochemistry of which was secured by X-ray crystallography (supporting information).11,12 Notably, reactions performed in the absence of a thiourea catalyst provided none of the desired bicyclization product.13
Table 1.
Catalyst optimization
 |
|---|
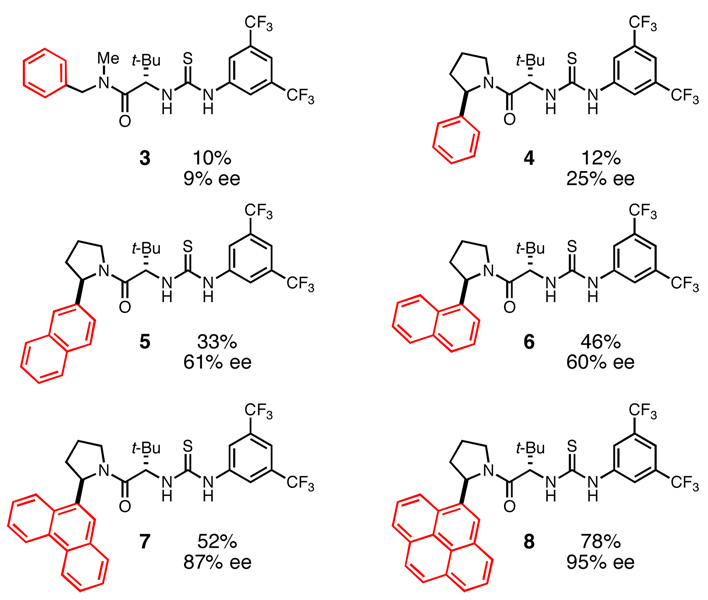 |
Optimization reactions performed on 0.033 mmol scale.
Yields determined by GC analysis relative to an internal standard.
Enantiomeric excess determined by SFC analysis on commercial chiral columns.
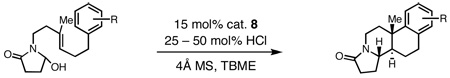
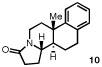
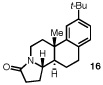
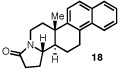
Having identified a selective catalyst and suitable reaction conditions, we evaluated the substrate scope of this bicyclization protocol. A variety of aromatic groups were found to be efficient and selective terminating nucleophiles (Table 2). In addition to the model substrate 1, the unsubstituted phenyl substrate 9 (entry 1), a number of electronically and sterically diverse para-substituted phenyl derivatives (entries 2–5), an extended naphthyl-containing substrate 17 (entry 6), as well as a chlorinated thiophene 19 (entry 7) all underwent cyclization with high enantioselectivity under the action of catalyst 8. Furthermore, in each case the bicyclization products were formed as single diastereomers, as judged by NMR analysis. Unfortunately, alteration of the non-aromatic portions of the substrate led to significant losses in reactivity and enantioselectivity (supporting information).
Table 2.
Substrate scope
 | ||||
|---|---|---|---|---|
| entry | substrate | product | yield (%)a | ee (%)b |
| 1 |  |
 |
51 | 89 |
| 2 | 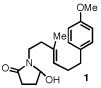 |
 |
72 | 94 |
| 3 | 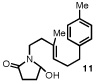 |
 |
62 | 91 |
| 4c | 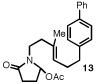 |
 |
54 | 91 |
| 5 | 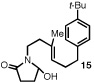 |
 |
71 | 91 |
| 6 | 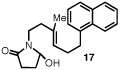 |
 |
75 | 92 |
| 7d | 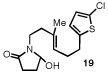 |
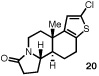 |
77 | 91 |
Reactions performed on 0.25 mmol scale.
Isolated yields after chromatography on silica gel.
Enantiomeric excess determined by SFC analysis on commercial chiral columns.
Reaction run with acetoxylactam and 2.0 equivalents of TMSCl.
The structure and absolute configuration of 20 was established by X-ray crystallography and the stereochemistry of all other products was assigned by analogy.
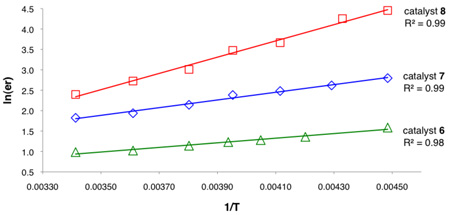
The fact that the enantioselectivity observed in these polycyclizations is highly dependent upon the expanse of the catalyst arene, taken together with the cationic nature of the reaction, raises the intriguing possibility that stabilizing cation-π interactions may play a key role in asymmetric induction. To evaluate this hypothesis, an Eyring analysis of enantioselectivity was conducted for catalysts 6, 7, and 8 in the bicyclization of substrate 1. All three catalysts displayed linear correlations between ln(er) and reciprocal temperature over a 70 °C range (Table 3). Evaluation of the differential activation parameters derived from these plots revealed that enantioselectivity was enthalpically-controlled in all cases, and that the magnitude of the differential enthalpy increased markedly as the catalyst arene increased in size. In fact, this term roughly doubles in magnitude with the addition of each new aromatic ring, reaching nearly 4 kcal/mol for the optimal catalyst 8. The effect of this increase was attenuated slightly by a compensating increase in the differential entropy terms across the series.
Table 3.
Eyring analysis of enantioselectivity
 | ||||
|---|---|---|---|---|
| entry | catalyst | ΔΔH‡ (kcal/mol) | ΔS‡ (cal/mol•K) | |
| 1 | 6 | 1.12 ± 0.07 | 2.0 ± 0.3 | |
| 2 | 7 | 1.87 ± 0.08 | 2.8 ± 0.3 | |
| 3 | 8 | 3.95 ± 0.17 | 8.9 ± 0.7 | |
The energetic benefits of increasing the strength of a non-covalent binding interaction are typically manifested enthalpically.14 As such, the increasing magnitude of the differential enthalpy in catalysts with more extended arenes is consistent in principle with a progressively more stabilizing cation-π interaction in the dominant transition state structure and with the fact polycyclic aromatic hydrocarbons are known to bind cations more strongly as they increase in size.15 However, these data do not rule out the possibility that increasing the expanse of the arene energetically destabilizes the minor transition state assembly, presumably through steric interactions. In order to ascertain whether the extended aromatic system is stabilizing the transition state leading to the major enantiomer or destabilizing the minor pathway, we investigated whether correlations existed between the degree of observed enantioinduction and the physical properties that underlie cation-π interactions. The strength with which a given arene interacts with a positive charge in a transition state is primarily a function of electrostatic and dispersion forces.16,17 As such, if the strength of a cation-π interaction is a determinant in enantioselectivity, there may be a correlation between the degree of asymmetric induction observed and the quadrupole moment and polarizability of the arene involved. Conversely, if the effect were largely steric and repulsive in origin, no significant correlation with these physical properties would be expected. The enantioselectivity observed with catalysts 4, 6, 7, and 8 under standard conditions was found to correlate strongly with both the polarizability and the quadrupole moment of the arenes found in each catalyst (R2 = 0.99, 0.97 respectively, see supporting information).18,19 Taken altogether, these data provide compelling evidence for a mechanism incorporating a selectivity-determining cation-π interaction.
In conclusion, the enantioselective cationic polycyclization reactions catalyzed by 8 appear to engage stabilizing cation-π interactions as a principal element of enantioselectivity. In this respect, these findings emulate the particularly striking role cation-π interactions play in the catalysis of biosynthetic polyene cyclizations and provides clear support for the notion that these interactions can dictate stereocontrol in small-molecule catalysis contexts as well. Moreover, this work further advances the view that stabilization of the dominant transition state structure through non-covalent interactions is a viable means of achieving high levels of enantioselectivity in counter ion catalysis.
Supplementary Material
Acknowledgement
This work was supported by the NIGMS (PO1 GM-69721 and RO1 GM-43214) and by a postdoctoral fellowship to R.R.K. from the NIH. We thank Dr. Shao-Liang Zheng for crystal structure determinations and Dr. Kristine Nolin for the synthesis and use of catalysts.
Footnotes
Supporting Information Available: Full experimental procedures, characterization data for all new compounds, NMR spectra for bicyclization products, SFC traces for scalemic bicyclization products, data sets for Eyring analysis and correlations with arene properties, and crystal structure analysis for compounds 2, 18, and 20 (57 pages). This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.For the first suggestion of this interaction in cyclases, see: Shi Z, Buntel CJ, Griffin JH. Proc. Nat. Acad. Sci. U.S.A. 1994;91:7370–7374. doi: 10.1073/pnas.91.15.7370. Poralla K, Hewelt A, Prestwich GD, Abe I, Reipen I, Sprenger G. Trends. Biochem. Sci. 1994;19:157–158. doi: 10.1016/0968-0004(94)90276-3. Dougherty DA. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163.
- 2.For recent reviews on synthetic and enzymatic polycyclizations that discuss the role of cation-π interactions, see: Wendt KU, Schulz GE, Corey EJ, Liu DR. Angew. Chem., Int. Ed. 2000;39:2812–2833. Yoder RA, Johnston JN. Chem. Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. Hoshino T, Sato T. Chem. Commun. 2002:291–301. doi: 10.1039/b108995c.
- 3.Wendt KU, Poralla K, Shultz GE. Science. 1997;277:1811–1815. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]; (b) Wendt KU, Lenhart A, Schultz GE. J. Mol. Bio. 1999;286:175–187. doi: 10.1006/jmbi.1998.2470. [DOI] [PubMed] [Google Scholar]; (c) Thoma R, Schulz-Gasch T, D’Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M, Ruf A. Nature. 2004;432:118–122. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]; (d) Hoshino T, Sato T. Chem. Commun. 1999:2005–2006. [Google Scholar]; (e) Merkofer T, Pale-Grosdemange C, Wendt KU, Rohmer M, Poralla K. Tetrahedron Lett. 1999;40:2121–2124. [Google Scholar]; (f) Hoshino T, Kouda M, Abe T, Sato T. Chem. Commun. 2000:1485–1486. [Google Scholar]; (g) Morikubo N, Fukuda Y, Ohtake K, Shinya N, Kiga D, Sakamoto K, Asanuma M, Hirota H, Yokoyama S, Hoshino T. J. Am. Chem. Soc. 2006;128:13184–13194. doi: 10.1021/ja063358p. [DOI] [PubMed] [Google Scholar]
- 4.For an antibody-catalyzed polycyclization where cation-π interactions play a key role, see: Paschall CM, Hasserodt J, Jones T, Lerner RA, Janda KD, Christianson DW. Angew. Chem., Int. Ed. 1999;38:1743–1747. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1743::AID-ANIE1743>3.0.CO;2-3.
- 5.For seminal work in the development of enantioselective polyene cyclizations, see: Ishihara K, Nakamura S, Yamamoto H. J. Am. Chem. Soc. 1999;121:4906–4907. Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2001;123:1505–1506. Ishibashi H, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2004;126:11122–11123. doi: 10.1021/ja0472026. Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. Mullen CA, Campbell MA, Gagné MR. Angew. Chem., Int. Ed. 2008;147:6011–6014. doi: 10.1002/anie.200801423.
- 6. Taylor MS, Jacobsen EN. J. Am. Chem. Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p. Taylor MS, Tokunaga N, Jacobsen EN. Angew. Chem., Int. Ed. 2005;44:6700–6704. doi: 10.1002/anie.200502277. Raheem IT, Thiara PS, Peterson EA, Jacobsen EN. J. Am. Chem. Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w. Raheem IT, Thiara PS, Jacobsen EN. Org. Lett. 2008;10:1577–1580. doi: 10.1021/ol800256j. Reisman SE, Doyle AG, Jacobsen EN. J. Am. Chem. Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. Klausen RS, Jacobsen EN. Org. Lett. 2009;11:887–890. doi: 10.1021/ol802887h. Zuend SJ, Jacobsen EN. J. Am. Chem. Soc. 2009;131:15358–15374. doi: 10.1021/ja9058958. Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN. Science. doi: 10.1126/science.1182826. in press. De CK, Klauber EG, Seidel D. J. Am. Chem. Soc. 2009;131:17060–17061. doi: 10.1021/ja9079435. For a recent review, see: Zhang Z, Schreiner PR. Chem. Soc. Rev. 2009;38:1187–1198. doi: 10.1039/b801793j.
- 7.For reviews discussing general aspects of cation-π and anion binding respectively, see: Ma J, Dougherty DA. Chem. Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. Schmidtchen FP, Berger M. Chem. Rev. 1997;97:1609–1646. doi: 10.1021/cr9603845. (c) see reference 6k.
- 8.For examples of ditopic receptors for the molecular recognition of ion pairs, see: Deetz MJ, Shang M, Smith BD. J. Am. Chem. Soc. 2000;122:6201–6207. Mahoney JM, Beatty AM, Smith BD. J. Am. Chem. Soc. 2001;123:5847–5848. doi: 10.1021/ja0156082. Nabeshima T, Saiki T, Iwabuchi J, Akine S. J. Am. Chem. Soc. 2005;127:5507–5511. doi: 10.1021/ja042882y.
- 9.(a) Dijkink J, Speckamp N. Tetrahedron Lett. 1977;11:935–938. [Google Scholar]; (b) Dijkink J, Speckamp N. Tetrahedron. 1978;34:173–178. [Google Scholar]; (c) Romero AG, Leiby JA, Mizsak SA. J. Org. Chem. 1996;61:6974–6979. doi: 10.1021/jo960673t. [DOI] [PubMed] [Google Scholar]
- 10.For the synthesis of enantioenriched 2-arylpyrrolidines, see: Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C. J. Am. Chem. Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265.
- 11.For seminal work on the relative stereochemical outcomes of polycyclization reactions, see: Stork G, Burgstahler AW. J. Am. Chem. Soc. 1955;77:5068–5077. Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv. Chim. Acta. 1955;38:1890–1904. Johnson WS. Acc. Chem. Res. 1968;1:1–8.
- 12.For an alternative asymmetric synthesis of related trans-perhydroisoquinolone structures, see: Kamatani A, Overman LE. Org. Lett. 2001;3:1229–1232. doi: 10.1021/ol015696v.
- 13.In the absence of the thiourea catalyst, an HCl-catalyzed process forms a monocyclized product. This byproduct is also formed to some extent in the reactions performed with thiourea catalysts, but with very low enantioselectivity.
- 14.(a) Williams DH, Calderone CT. J. Am. Chem. Soc. 2001;123:6262–6267. doi: 10.1021/ja003016y. [DOI] [PubMed] [Google Scholar]; (b) Dunitz JD. Chem. Biol. 1995;2:709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]; (c) Westwell MS, Searle MS, Klein J, Williams DH. J. Phys. Chem. 1996;100:16000–16001. [Google Scholar]
- 15.For discussion of cation-π binding with polycyclic aromatic hydrocarbons, see: Vijay D, Sastry N. Phys. Chem. Chem. Phys. 2008;10:582–590. doi: 10.1039/b713703f. Ng KM, Ma NL, Tsang CW. Rapid Commun. Mass Spectrom. 1998;12:1679–1684. Gal J-F, Maria P-C, Decouzon M, Mo O, Yanez M, Abboud JLM. J. Am. Chem. Soc. 2003;125:10394–10401. doi: 10.1021/ja029843b. Amunugama R, Rodgers MT. Int. J. Mass Spec. 2003;227:1–20.
- 16.For a discussion of the importance of polarizability and dispersion forces in catalysis involving extended arenes and in cation-π binding, see: McCurdy A, Jimenez L, Stauffer DA, Dougherty DA. J. Am. Chem. Soc. 1992;114:10314–10321. Ngola SM, Dougherty DA. J. Org. Chem. 1996;61:4355–4360. doi: 10.1021/jo960521y. Kennan AJ, Whitlock HW. J. Am. Chem. Soc. 1996;118:3027–3028. Cubero E, Luque FJ, Orozco M. Proc. Nat. Acad. Sci. U.S.A. 1998;95:5976–5980. doi: 10.1073/pnas.95.11.5976.
- 17.For a discussion of the electrostatic component of cation-π binding, see: Mecozzi S, West AP, Dougherty DA. J. Am. Chem. Soc. 1996;118:2307–2308.
- 18.For calculated values of the quadrupole moments of extended arenes, see: Heard GL, Boyd RJ. J. Phys. Chem. A. 1997;101:5374–5377. (b) see reference 15b.
- 19.For the polarizabilities of extended arenes, see: Waite JM, Papadopoulos MG, Nicolaides CA. J. Chem. Phys. 1982;77:2536–2539. Alparone A, Librando V, Minniti Z. Chem. Phys. Lett. 2008;460:151–154.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.