

Abstract
A critical component in regulating cardiac and skeletal muscle contractility is the release of Ca2+ via ryanodine receptor (RyR) Ca2+ release channels in the sarcoplasmic reticulum (SR). In heart failure and myopathy, the RyR has been found to be excessively phosphorylated or nitrosylated and depleted of the RyR-stabilizing protein calstabin (FK506 binding protein 12/12.6). This remodeling of the RyR channel complex results in an intracellular SR Ca2+ leak and impaired contractility. Despite recent advances in heart failure treatment, there are still devastatingly high mortality rates with this disease. Moreover, pharmacological treatment for muscle weakness and myopathy is nearly nonexistent. A novel class of RyR-stabilizing drugs, rycals, which reduce Ca2+ leak by stabilizing the RyR channels due to preservation of the RyR-calstabin interaction, have recently been shown to improve contractile function in both heart and skeletal muscle. This opens up a novel therapeutic strategy for the treatment of contractile failure in the cardiac and skeletal muscle.
Introduction
Contractility is the central property of all types of muscle. This feature enables the heart to produce the power necessary for its pump function and the skeletal muscles to cause movement. Impaired contractility is the key phenomenon in muscular diseases such as heart failure, cardiac arrhythmias and myopathies. Heart failure is a gravely debilitating condition with a mortality rate higher than many forms of cancer[1]; a majority of patients with advanced cardiac failure die within the first year after diagnosis[2]. The incidence of heart failure increases progressively with age and approaches 10 per 1000 after 65 years of age[3]. With a growing proportion of elderly in Western societies, the future problems associated with failing heart function are expected to increase. Cardiac arrhythmias are a common cause of death in heart failure and ventricular tachycardia underlies half of the deaths in heart failure. Moreover, skeletal muscle function is also weakened in chronic heart failure and the impaired contractility seen in these two organs have common mechanisms, centered around defective cellular Ca2+ handling[2]. Interestingly, myopathies such as Duchenne muscular dystrophy, have been shown to share similar Ca2+ handling defects[4]. Despite progress in the therapeutic management of heart failure, major clinical problems and high mortality rates persist. Moreover, the absence of an effective treatment for myopathies shows that novel pharmacological regimens for contractile disorders are needed.
Stress signals in health and disease
The body is regularly exposed to different types of stress that can result in physiological responses leading to enhanced muscle function. For example, the evolutionary conserved neuro-hormonal-mediated fight-or-flight response activates many organs as manifested by increased heart rate and contractility and enhanced survival of the organism in acutely threatening situations. Furthermore, stress signals in endurance training improve heart and skeletal muscle function and this constitutes an example of beneficial adaptations to stress. Prolonged increases in the stress level, on the other hand, have harmful effects on cardiac and skeletal muscle. For example, sustained increase in catecholamine levels and adrenergic stimulation can manifest as deleterious stress and lead to impaired myocyte function. Chronically increased sympathetic activity and over-activation of the cardiac β-adrenergic receptor is linked to the deterioration of heart function and development of heart failure[5–8]. This has prompted the use of β-adrenergic receptor blockers in the management of cardiovascular disease, including heart failure and arrhythmias[9,10]. Moreover, skeletal muscle function is interlaced with beta-adrenergic signaling. Activation of the sympathetic nervous system and elevated plasma catecholamine levels during exercise constitutes the key adaptive signal that couples skeletal and cardiac muscle work in vivo. Moreover, both patients and animal models of heart failure suffer from reduced muscle fatigue resistance[11]. In fact, quality of life and prognosis of patients with heart failure is severely decreased due to skeletal muscle dysfunction (e.g. shortness of breath due to diaphragmatic weakness, and exercise intolerance due to limb skeletal muscle fatigue)[12].
Regulation of muscle contractility
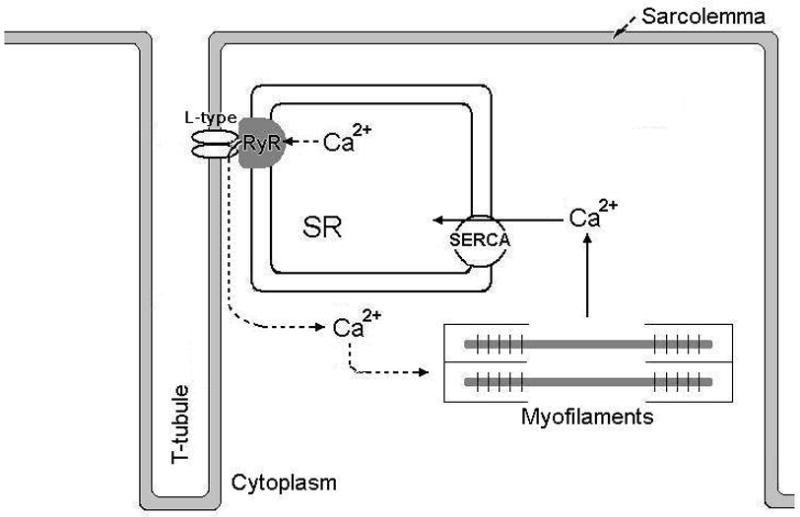
At the cellular level muscle contraction occurs through a process referred to as excitation-contraction (E-C) coupling. This process starts with an action potential (AP) in the cell membrane (sarcolemma). Then, a series of steps occur that couple the initial excitation to commencement of contractile work. Central to the E-C coupling process is an increase in cytoplasmic free Ca2+. Cellular Ca2+ handling is a highly controlled process that involves ion exchange systems, ion pumps and specialized compartments for Ca2+ storage within the cell (Fig 1A–B).
Figure 1. Excitation-contraction coupling in cardiac and skeletal muscle.
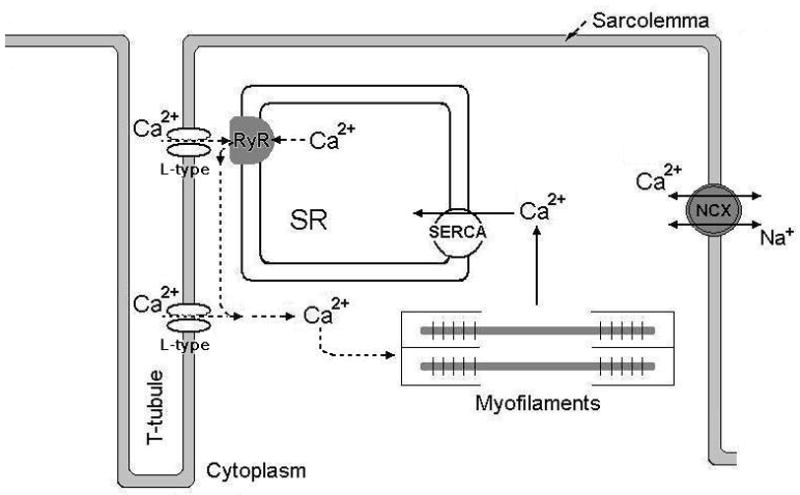
A) Figure showing the principals of Ca2+ cycling in excitation-contraction (E-C) coupling of the cardiomyocyte. Upon depolarization of the sarcolemma, L-type Ca2+ channels open to allow Ca2+ to enter the cell. This Ca2+ will in turn activate the juxtaposed RyR and more Ca2+ will be released. The bulk of cytoplasmic [Ca2+] increase is due to RyR-mediated release of Ca2+ from the SR. Cytoplasmic Ca2+ will trigger myofilament activity so that contraction can occur. To relax, cytoplasmic Ca2+ is pumped back into the SR via the SR Ca2+ ATPase (SERCA2a) and extruded out of the cell through the sarcolemmal Na+-Ca2+ exchanger (NCX).
B) Depicts Ca2+ handling in skeletal muscle E-C coupling. L-type Ca2+ channels at the sarcolemma and RyR1 in the SR membrane are in close vicinity with each other. Depolarization of the sarcolemma causes the L-type Ca2+ channel to interact with and activate the RyR independent of entry of extracellular Ca2+. Virtually all of the increase in cytoplasmic [Ca2+] that is needed for myofilament activation and force generation comes from SR Ca2+ release. The Ca2+ is cleared from the cytoplasm via the SERCA. The absence of transsarcolemmal Ca2+ fluxes underscores the importance of RyR1 Ca2+ release function in skeletal muscle ECC.
In the heart, generation of an AP normally occurs in the specialized pacemaker cells of the sino-atrial (sinus) node, from which a depolarization wave propagates cell to cell via gap junctions (intercalated discs) and thereby linking the cardiac cells electrically with each other. On the cardiomyocyte level, the AP propagates over the plasma membrane and into the cell via the transverse (t)-tubule system, where it activates voltage gated L-type Ca2+ channels (also referred to as dihydropyridine receptors, DHPRs; Cav1.2). These channels allow Ca2+ to pass through the sarcolemma and enter the cell. The trans-sarcolemmal Ca2+ influx occurs down a ~10,000 fold concentration gradient, with extracellular [Ca2+] being ~3–5 mM and resting cytoplasmic [Ca2+] ~100 nM. A narrow junction between the t-tubules and the SR places the cardiac L-type Ca2+ channels in close proximity to the SR Ca2+ release channels (RyRs). At rest, the SR lumen contains ~1–2 mM Ca2+. The L-type Ca2+ channel-mediated trans-sarcolemmal Ca2+-influx triggers RyR2 opening, which then permits SR Ca2+ release. This process is referred to as Ca2+-induced Ca2+ release[13]. Importantly, the Ca2+ released from the SR contributes substantially more to the total increase in cytoplasmic Ca2+ than the L-type Ca2+ influx. In humans, ~70% of the total increase in cytoplasmic Ca2+ can be attributed to SR Ca2+ release[14]. The increase in cytosolic [Ca2+] permits binding of Ca2+ to troponin C, a myofilament regulatory protein which in turn facilitates interaction of myosin and actin filaments leading to sarcomere shortening which generates contraction (systole). To allow relaxation of the heart (diastole), cytoplasmic Ca2+ is reduced mainly by uptake into the SR via an energy-requiring process dependent on the cardiac SR Ca2+-ATPase (SERCA2a) [15]. However in the steady state condition, an efflux of Ca2+ over the sarcolemma must also be present to balance the L-type Ca2+ influx (otherwise there would be a net build-up of total cellular Ca2+). This efflux is mainly effectuated by the Na/Ca exchanger (NCX)[14].
The general principles of E-C coupling in cardiac and skeletal muscle are similar. However, there are some important differences regarding the regulation of cytoplasmic Ca2+. Similar to E-C coupling in the heart, action potential-mediated depolarization of the skeletal myocyte plasma membrane causes activation of L-type Ca2+ channels (Cav1.1). However in the skeletal muscle, the L-type Ca2+ channel does not conduct any Ca2+ current that is of importance to E-C coupling. Upon activation, the skeletal muscle L-type channel interacts directly with the juxtaposed ryanodine receptors (RyR1) that open and allow Ca2+ to be released of from the SR into the cytoplasm so that contraction can occur. To relax the muscle, Ca2+ is pumped back into the SR by the skeletal muscle SERCAs. Thus, in skeletal muscle, cellular Ca2+ is cycled between the SR and cytoplasm with little or no exchange with the extracellular environment[16].
The magnitude of cardiomyocyte contractility is in large part graded by the amount of Ca2+ released into the cytosol. Thus, increased contractility is associated with mechanisms leading to increased [Ca2+]cyt. Cardiac Ca2+ handling can be influenced by a multitude of factors, including redox dependent modifications and phosphorylation of proteins involved in E-C coupling [17,18]. An important stimulus that influences Ca2+ handling is mediated by catecholamines (adrenalin and noradrenalin) from the sympathetic adrenergic system. In the heart, catecholamines bind to the β-adrenergic receptors (β-receptor). Activation of these receptors increase heart rate (chronotropy), relaxation speed (lusitropy), and contractility (inotropy). The β-receptor-induced increase in relaxation speed and contractility are considered to be mainly the effect of cAMP-dependent protein kinase (PKA)-mediated phosphorylation of proteins involved in cardiomyocyte Ca2+ handling [19]. Three important Ca2+ handling proteins that are targeted by PKA-mediated phosphorylation are the L-type Ca2+ channel, phospholamban and the RyR2. When phosphorylated, the influx of Ca2+ through the L-type Ca2+ channel is increased[14,20]. Phosphorylation of phospholamban leads to increased activity of SERCA2a, which accelerates SR Ca2+ uptake[15]. Moreover, the RyR2 can be PKA-phosphorylated which leads to increased channel open probability and facilitated SR Ca2+ release[18,21]. β-adrenergic signaling is therefore of central importance in the acute regulation of heart function and cardiovascular homeostasis. Nevertheless, chronically sustained adrenergic stress can be deleterious for cardiac function and promotes the development of heart failure and cardiac arrhythmias[5,7,22].
RyR Ca2+ release channel
The key cellular signal that controls muscle contractility is cytoplasmic Ca2+ released from the SR. Regulation of SR Ca2+ release channel RyR is therefore of pivotal interest in the control of muscle cell performance[18]. Three isoforms of RyR (RyR1, RyR2, RyR3) exists in mammals. Although the RyRs are found in an array of tissue types (e.g. muscle, neurons, testis, thymus, pancreas and ovaries)[23], RyR1 and RyR2 are predominantly expressed in striated muscle. In skeletal muscle, RyR1 is the isoform responsible for ECC, whereas RyR2 mediates Ca2+ release in the heart[18]. The RyRs are homotetrameric channels that are located in the SR membrane. Each monomer of the RyR consists of a ~560-kDa molecule, with a massive N-terminal domain protruding into the cytosol, and a smaller C-terminal domain that contains the transmembrane segments and the pore region[18].
In its intracellular environment, the RyR forms the centerpiece of a large macromolecular complex[18]. The cytoplasmic N-terminal domain of the RyR serves as a scaffold for allosteric modulators which regulate the function of the C-terminal Ca2+-conducting pore region. These modulators include cyclic AMP dependent protein kinase A (PKA)[5], phosphatases such as protein phosphatase 1 and 2A[24], calcium dependent calmodulin kinase II (CaMKII)[25,26]. Small molecules and ions such as ATP, Ca2+, Mg2+ and the pharmacological substance caffeine are other modulators of RyR function[18]. The peptidyl-propyl-cis-trans isomerases calstabin1 (FK506 binding protein 12 or FKBP12) and calstabin2 (FKBP12.6) are proteins with important regulatory function on SR Ca2+ release. Calstabins bind to the RyR via amphiphilic β-sheet structures and effectuate their modulatory function through protein-protein interactions with the SR Ca2+ release channel. Each RyR monomer binds one calstabin and under normal conditions an RyR tetramer is associated with four calstabin proteins[27]. Calstabin1 associates predominantly with the skeletal muscle RyR1. In contrast, the cardiac muscle RyR2 have the highest affinity for calstabin2[28]. The calstabins function as RyR channel-stabilizing proteins. In a resting myocyte, the calstabins increase the probability of the channel to be in its closed state[29,30]. This is an important mechanism to prevent spontaneous SR Ca2+ release due to a pathologic leak via RyR channels [31–33].
Maladaptations of RyR and Ca2+ leak– a mechanism of contractile failure
Heart failure is characterized by an inadequate cardiac pump function that does not meet the metabolic requirements of the organs for blood flow. This feature can be a consequence of several cardiovascular pathologies that directly and indirectly compromise cardiac function, e.g. cardiac infarction, hypertension, inherited and acquired cardiomyopathies. Initially, the failing cardiac function can be offset by increased sympathetic and catecholaminergic signaling which will trigger cell surface β-adrenergic receptors and cause downstream activation of PKA that phosphorylates important Ca2+ handling proteins, including the RyR2. This leads to increased SR Ca2+ release, hence increased cytoplasmic [Ca2+] and stronger contractions. However chronic adrenergic activation, as seen in heart failure or during prolonged strenuous exercise, is associated with maladaptations in the SR Ca2+ release function. One such adaptation, shown by our laboratory, is PKA hyperphosphorylation of the RyR[5,34,35]. This term refers to a situation where the RyR is chronically PKA phosphorylated at 3 to 4 of the 4 PKA sites per tetrameric channel (1 per RyR monomer) and depleted of 3 to 4 of the 4 calstabins bound to each channel [5,35]. Under these circumstances the RyRs from either heart or skeletal muscle exhibit a pathologic increase in the open probability under resting conditions (i.e. low activating cytosolic calcium). The RyR channels are leaky and Ca2+ leaks out from the SR. Over time, this can lead to reduction in SR Ca2+ content, with less Ca2+ available for release and consequently weaker muscle contractions (Fig. 2).
Figure 2. Dissociation of calstabin (FKBP12/FKBP12.6) from the RyR macromolecular complex underlies SR Ca2+ leak. A–B).

Under normal conditions, the interaction between calstabin and the RyR subunits stabilize the channel function and minimizes SR Ca2+ leak when the cell is in resting condition(A). B) Upon activation of the RyR (e.g. by increased [Ca2+]or via interaction with the DHPR), the channel opens and Ca2+ is released from the sarcoplasmic reticulum (SR) into the cytosol, driven by the large SR-cytosolic concentration gradient. C–D) The RyR is a target for stress signals (e.g. β-adrenergic receptor-mediated PKA-dependent phosphorylation of serine residues, nitrosylation of cysteines and oxidative modifications of the RyR). Chronic stress signaling can lead to dissociation of calstabin from the Ca2+ release channel that then becomes leaky, Ca2+ oozes out through the channel and the SR Ca2+ load is diminished (C). Under such conditions, the driving force for Ca2+ across the SR membrane is reduced and upon activation of the RyR less Ca2+ will be released to the cytosol.
About half the deaths in heart failure are due to sudden cardiac death and spontaneous openings of the RyR2 in cardiac diastole can cause triggered ventricular arrhythmias. The increase in stochastic openings of the calstabin2 depleted RyR2, e.g. as seen in heart failure, links cardiac contractile failure with the increased propensity for ventricular arrhythmias to a common mechanism. Spontaneous release of SR Ca2+ during diastole activates the forward mode of the NCX which provides a depolarizing transient inward current. If the threshold to activate Na+ channels is reached an action potential will be triggered and an aberrant contraction will occur[36].
In skeletal muscle, sustained β-adrenergic signaling and depletion of calstabin1 from RyR1 plays a role in the mechanism of contractile failure and muscle fatigue during strenuous exercise[37]. This has recently been shown in both an animal model as well as in exercising humans[35]. More recently it was shown that impaired Ca2+ regulation is also present in the severely debilitating human myopathy, Duchenne muscular dystrophy (DMD). In DMD a null mutation in the dystrophin gene leads to disruption of the important extracellular-cytoskeleton spanning dystroglycan complex and patients present with overt muscle weakness and cardiomyopathy[4,38]. RyR1 from the DMD mouse model (the mdx mouse) was shown to be excessively cysteine nitrosylated and this was coupled to depletion of calstabin1 from RyR1, increased spontaneous RyR1 openings (calcium sparks), and reduced specific muscle force[4]. Moreover in the mdx mouse, RyR2 in the heart is also nitrosylated, depleted from calstabin2 and these mice display increased frequency of calcium sparks and ventricular arrhythmias [38]. In a mouse model of the inherited arrhythmogenic disorder catecholaminergic polymorphic ventricular tachycardia (CPVT) that harbors a heterozygous RyR2-R2474S mutation, RyR2 are calstabin2 depleted and both cardiac and neuronal cells display Ca2+ leak. These mice present a global phenotype with cardiac arrhythmias and seizures[39]. This identifies calstabin dissociation and SR Ca2+ leak as mechanisms of importance, not only in diseases characterized by contractile dysfunction, but also in a more general pathophysiological setting.
Reducing RyR leak as a treatment for contractile dysfunction
Reducing SR Ca2+ leak by restoring RyR-calstabin interaction is a promising therapeutic strategy for conditions with failing contractile function (e.g. heart failure and myopathies)[32,40]. The 1,4-benzothiazepine derivatives JTV519 and S107 are novel small molecule drugs that have the effectively enhance RyR-calstabin binding and stabilizing the closed state of the RyR. These drugs are often referred to as Rycals due to their stabilizing effect on the RyR Ca2+ release function. Haploinsufficient calstabin-2+/−, but not calstabin-2−/− knockout mice that were treated with JTV519 were rescued from pacing-induced ventricular arrhythmias [41], indicating that JTV519 exerts its effects through facilitating RyR-calstabin binding. In dogs with pacing-induced heart failure, where cardiac RyR2 was hyperphosphorylated and depleted from calstabin2, JTV519 restored the RyR2/calstabin2 interaction to normal levels, reduced Ca2+ leak and improved left ventricular function[42]. In addition, JTV519 was shown to enhance RyR1-calstabin1 binding, restore skeletal muscle RyR1 channel function and decrease muscle fatigue in mice after myocardial infarction[43]. These results suggest that rycals might be an effective treatment not only to prevent heart failure and ventricular arrhythmias after myocardial infarction but also to reduce concomitant skeletal muscle symptoms such as muscle weakness and fatigability.
Because JTV519 is also known to affect other ion channels (e.g. L-type Ca2+ and Na+ channels)[44] and thereby potentially interfere with cardiac electrophysiological properties, another rycal, S107, with more specific RyR stabilizing properties has been developed[4,35,39]. In a screening procedure, S107 was found to have no activity against hundreds of enzymes, channels and signal transduction molecules[35]. Nonetheless, S107 facilitates the binding of calstabin1/2 to RyR1/2. Via this mechanism, S107 prevented depletion of calstabin1 from RyR1 and improved skeletal muscle fatigue resistance in mice subjected to severe exercise[35]. In the Duchenne muscular dystrophy model, the mdx mouse, treatment with S107 reduced signs of muscle damage and improved skeletal muscle function[4]. Interestingly, the mdx mice also develop cardiomyopathy with a Ca2+ leak phenotype and S107 treatment inhibited the Ca2+ leak and prevented arrhythmias in these mice[38]. Moreover, CPVT mice that harbor a heterozygous RyR2-R2474S mutation were protected against stress-induced ventricular arrhythmias and displayed an increased threshold for seizures after treatment with S107[39]. Thus, several lines of experimental evidence show that rycals can be used to stabilize RyR Ca2+ release function in pathological conditions where SR Ca2+ leak is present.
Summary and conclusions
Stress-induced maladaptations of the RyR and dissociation of calstabin are linked to defective Ca2+ handling and impaired contractility in diseases affecting both cardiac and skeletal muscle. In heart failure, impaired Ca2+ homeostasis is a key cellular mechanism that contributes to reduced cardiac output as seen in these patients. Despite improved pharmacological and device-based therapies of heart failure in recent years, a devastating mortality rate is still apparent in heart failure patients. Moreover, skeletal muscle dysfunction, as seen in heart failure or inherited muscular dystrophies, is virtually without effective pharmacological treatment. Thus, there is a strong need for novel therapeutic regimens that combat contractile dysfunction. Drugs that restore RyR Ca2+ release function, like the rycals JTV519 and S107, are therefore promising candidates. Rycal treatment would be ideal in conditions with co-morbidity between cardiac and skeletal muscle, as seen in cardiomyopathy and muscular dystrophy. Speculatively, there could also be benefits of rycal treatment in other conditions where cardiac and/or skeletal function is impaired. With a growing proportion of elderly in the society, aging-dependent dysfunction of the heart and skeletal muscle would be particularly important to study with respect to RyR Ca2+ leak and potential benefits of rycal treatment.
In summary, restoring RyR-calstabin binding and inhibiting Ca2+ leak through rycal treatment holds promise as a novel pharmacological tool in combating contractile failure of the heart and skeletal muscle.
Acknowledgments
We thank Dr. Maria Armiento for valuable assistance with figure editing.
Footnotes
Conflict of interest: A.R. Marks is a consultant for a start-up company, ARMGO Pharma Inc., that is targeting RyR channels to treat heart disease and to improve exercise capacity in muscle diseases
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cowie MR, et al. Survival of patients with a new diagnosis of heart failure: a population based study. Heart. 2000;83 (5):505–510. doi: 10.1136/heart.83.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farr MA, Basson CT. Sparking the failing heart. N Engl J Med. 2004;351 (2):185–187. doi: 10.1056/NEJMcibr041466. [DOI] [PubMed] [Google Scholar]
- 3.Lloyd-Jones D, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 121(7):e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 4.Bellinger AM, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15 (3):325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101 (4):365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 6.Cohn JN, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. New England Journal of Medicine. 1984;311 (13):819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 7.Clark AL, Cleland JG. The control of adrenergic function in heart failure: therapeutic intervention. Heart Fail Rev. 2000;5 (1):101–114. doi: 10.1023/A:1009854325711. [DOI] [PubMed] [Google Scholar]
- 8.Kaye DM, et al. Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol. 1995;26(5):1257–1263. doi: 10.1016/0735-1097(95)00332-0. [DOI] [PubMed] [Google Scholar]
- 9.Jessup M, Brozena S. Heart failure. New England Journal of Medicine. 2003;348 (20 ):2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 10.Reiken S, et al. Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation. 2003;107 (19):2459–2466. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 11.Crimi E, et al. Mechanisms by which exercise training benefits patients with heart failure. Nat Rev Cardiol. 2009;6 (4):292–300. doi: 10.1038/nrcardio.2009.8. [DOI] [PubMed] [Google Scholar]
- 12.Harrington D, et al. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. Journal of the American College of Cardiology. 1997;30 (7):1758–1764. doi: 10.1016/s0735-1097(97)00381-1. [DOI] [PubMed] [Google Scholar]
- 13.Fabiato A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85 (2):291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 15.Maclennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4(7):566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 16.Allen DG, et al. Skeletal muscle fatigue: cellular mechanisms. Physiological Reviews. 2008;88 (1):287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- 17.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovascular Research. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Zalk R, et al. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- 19.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415 (6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 20.Tsien RW, et al. Mechanisms of calcium channel modulation by beta-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol. 1986;18(7):691–710. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- 21.Wehrens XH, et al. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103 (3):511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curran J, et al. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100 (3):391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 23.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82 (4):893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 24.Marx SO, et al. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. Journal of Cell Biology. 2001;153 (4):699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wehrens XH, et al. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94 (6):e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 26.Currie S, et al. Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochemical Journal. 2004;377 (Pt 2):357–366. doi: 10.1042/BJ20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayaraman T, et al. FK506 binding protein associated with the calcium release channel (ryanodine receptor) Journal of Biological Chemistry. 1992;267 (14):9474–9477. [PubMed] [Google Scholar]
- 28.Xin HB, et al. Affinity purification of the ryanodine receptor/calcium release channel from fast twitch skeletal muscle based on its tight association with FKBP12. Biochemical & Biophysical Research Communications. 1995;214 (1):263–270. doi: 10.1006/bbrc.1995.2283. [DOI] [PubMed] [Google Scholar]
- 29.Brillantes AB, et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77 (4):513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 30.Kaftan E, et al. Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ Res. 1996;78 (6):990–997. doi: 10.1161/01.res.78.6.990. [DOI] [PubMed] [Google Scholar]
- 31.McCall E, et al. Effects of FK-506 on contraction and Ca2+ transients in rat cardiac myocytes. Circ Res. 1996;79 (6):1110–1121. doi: 10.1161/01.res.79.6.1110. [DOI] [PubMed] [Google Scholar]
- 32.Wehrens XH, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113 (7):829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 33.Reiken S, et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. Journal of Cell Biology. 2003;160 (6):919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yano M, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102 (17):2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 35.Bellinger AM, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105 (6):2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wehrens XH, et al. Ryanodine receptor-targeted anti-arrhythmic therapy. Ann N Y Acad Sci. 2005;1047:366–375. doi: 10.1196/annals.1341.032. [DOI] [PubMed] [Google Scholar]
- 37.Bellinger AM, et al. Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest. 2008;118 (2):445–453. doi: 10.1172/JCI34006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fauconnier J, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 107(4):1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehnart SE, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118 (6):2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yano M, et al. Mechanisms of Disease: ryanodine receptor defects in heart failure and fatal arrhythmia. Nat Clin Pract Cardiovasc Med. 2006;3(1):43–52. doi: 10.1038/ncpcardio0419. [DOI] [PubMed] [Google Scholar]
- 41.Wehrens XH, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304 (5668):292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 42.Yano M, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107 (3):477–484. doi: 10.1161/01.cir.0000044917.74408.be. [DOI] [PubMed] [Google Scholar]
- 43.Wehrens XH, et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci U S A. 2005;102 (27):9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaneko N, et al. Pharmacological characteristics and clinical applications of K201. Curr Clin Pharmacol. 2009;4 (2):126–131. doi: 10.2174/157488409788184972. [DOI] [PMC free article] [PubMed] [Google Scholar]