

Abstract
BACKGROUND AND PURPOSE
The conversion of clopidogrel to its active metabolite, R-130964, is a two-step cytochrome P450 (CYP)-dependent process. The current investigations were performed to characterize in vitro the effects of different CYP inhibitors on the biotransformation and on the antiplatelet effect of clopidogrel.
EXPERIMENTAL APPROACH
Clopidogrel biotransformation was studied using human liver microsomes (HLM) or specific CYPs and platelet aggregation using human platelets activated with ADP.
KEY RESULTS
Experiments using HLM or specific CYPs (3A4, 2C19) revealed that at clopidogrel concentrations >10 µM, CYP3A4 was primarily responsible for clopidogrel biotransformation. At a clopidogrel concentration of 40 µM, ketoconazole showed the strongest inhibitory effect on clopidogrel biotransformation and clopidogrel-associated inhibition of platelet aggregation with IC50 values of 0.03 ± 0.07 µM and 0.55 ± 0.06 µM respectively. Clarithromycin, another CYP3A4 inhibitor, impaired clopidogrel biotransformation and antiplatelet activity almost as effectively as ketoconazole. The CYP3A4 substrates atorvastatin and simvastatin both inhibited clopidogrel biotransformation and antiplatelet activity, less potently than ketoconazole. In contrast, pravastatin showed no inhibitory effect. As clopidogrel itself inhibited CYP2C19 at concentrations >10 µM, the CYP2C19 inhibitor lansozprazole affected clopidogrel biotransformation only at clopidogrel concentrations ≤10 µM. The carboxylate metabolite of clopidogrel was not a CYP substrate and did not affect platelet aggregation.
CONCLUSIONS AND IMPLICATIONS
At clopidogrel concentrations >10 µM, CYP3A4 is mainly responsible for clopidogrel biotransformation, whereas CYP2C19 contributes only at clopidogrel concentrations ≤10 µM. CYP2C19 inhibition by clopidogrel at concentrations >10 µM may explain the conflicting results between in vitro and in vivo investigations regarding drug interactions with clopidogrel.
Keywords: clopidogrel, carboxylate metabolite of clopidogrel, CYP3A4, CYP2C19, drug interactions, R-130964
Introduction
The drugs currently used for the inhibition of platelet aggregation (IPA) include acetylsalicylic acid, glycoprotein IIb/IIIa receptor antagonists and the thienopyridine derivatives. The thienopyridines, in particular clopidogrel, have become standard drugs for the management of patients following percutaneous coronary intervention (PCI) and stent placement (Schulman, 2004). In addition, clopidogrel is also used in patients after acute coronary syndromes without PCI and in aspirin intolerant patients.
Clopidogrel is a pro-drug requiring hepatic biotransformation for pharmacological activity (Pereillo et al., 2002). The active metabolite of clopidogrel (R-130964) contains a thiol group, which binds irreversibly to a free cysteine in the P2Y12 receptor and blocks activation by ADP (Ding et al., 2003). In humans, >85% of clopidogrel is metabolized by esterases to a carboxylic acid metabolite (clopidogrel carboxylate, SR 26334) (Caplain et al., 1999; Lins et al., 1999; Reist et al., 2000; Taubert et al., 2004), which is considered to be inactive. The remainder is converted to the active metabolite (R-130964) in a two-step, cytochrome P450 (CYP)-dependent process, proceeding via the formation of 2-oxo-clopidogrel. Initial studies suggested that CYP3A4 plays a prominent role in these metabolic steps, with lesser involvement of CYP2C19, CYP2B6, CYP1A2 and CYP2C9 (Savi et al., 2000; Clarke and Waskell, 2003; Hulot et al., 2006; Farid et al., 2007; nomenclature follows Alexander et al., 2009). A more recent in vitro study indicated that CYP2C19 was the most important CYP for the conversion of clopidogrel to 2-oxo-clopidogrel and CYP3A4 for the conversion of 2-oxo-clopidogrel to the active metabolite R-130964 (Kazui et al., 2010).
Studies conducted in vitro (Clarke and Waskell, 2003) and ex vivo (Lau et al., 2003; Neubauer et al., 2003) suggested that the CYP3A4 substrate atorvastatin may attenuate the platelet inhibitory effect of clopidogrel due to interference with clopidogrel biotransformation by CYP3A4. These observations resulted in an intense debate about the clinical relevance of drug-drug interactions by lipophilic statins, drugs prescribed frequently to patients taking clopidogrel (Ratz Bravo et al., 2005). This controversy remains currently unresolved, as subsequent studies investigating effects of atorvastatin and/or simvastatin on anti-platelet effects of clopidogrel failed to confirm the initial findings (Muller et al., 2003; Gorchakova et al., 2004; Mitsios et al., 2004; Serebruany et al., 2004; Vinholt et al., 2005; Saw et al., 2007). More recent in vivo studies suggest an important role for both CYP3A4 and CYP2C19 regarding biotransformation and pharmacological activity of clopidogrel. Both concomitant treatment with strong CYP3A4 inhibitors (Suh et al., 2006; Farid et al., 2007; Siller-Matula et al., 2008) as well as a reduced activity of CYP2C19 in patients with CYP2C19 single nucleotide polymorphisms were associated with an impaired pharmacological activity of clopidogrel (Kim et al., 2008; Mega et al., 2009; Simon et al., 2009). Several clinical studies with ex vivo determination of residual platelet aggregation demonstrated that CYP2C19 inhibitors such as proton pump inhibitors (PPIs) decrease the pharmacological effect of clopidogrel (Gilard et al., 2008; O'Donoghue et al., 2009; Price et al., 2009; Zuern et al., 2010). These findings are supported by two retrospective studies with clinical endpoints (death, re-hospitalization and/or re-infarction) (Ho et al., 2009; Juurlink et al., 2009), whereas a third clinical study failed to show an increased cardiovascular risk for patients with concomitant ingestion of clopidogrel and a PPI (O'Donoghue et al., 2009).
Considering the uncertainties regarding drug interactions with clopidogrel, we undertook the current study to determine concentration-dependent effects of inhibitors and/or substrates of various CYPs on in vitro biotransformation and antiplatelet activity of clopidogrel.
Methods
Kinetic studies of clopidogrel or the carboxylate metabolite of clopidogrel with human liver microsomes (HLM) or rhCYP
The incubation mixture (final volume 250 µL) contained varying concentrations of clopidogrel (5–100 µM), incubation buffer (0.1 M potassium phosphate, pH 7.4), reduced glutathione (GSH; 5 mM), NADPH-regenerating system containing MgCl2 (3.3 mM), NADP+ (1.3 mM), glucose-6-phosphate (3.3 mM) and glucose-6-phosphate dehydrogenase (0.4 U·mL−1) and either HLM, rhCYP3A4 or rhCYP2C19. Preliminary studies were performed to determine the incubation time and protein concentrations producing a linear rate. For HLM, 10 min incubation and 0.25 mg·mL−1 protein were selected, and for rhCYP3A4 and rhCYP2C19 10 min incubation and 10 pmol CYP450·mL−1. The concentrations of the substrates (clopidogrel or clopidogrel carboxylate) are given in the figures. The final volume of methanol (solvent for clopidogrel) did not exceed 1.0% of the total incubation volume and was identical in all incubations including controls. Each reaction mixture was equilibrated for 4 min at 37°C in a shaking thermomixer. The reaction was initiated by adding the NADPH-regenerating system and the system incubated for the respective time at 37°C. Reactions were stopped by addition of 100 µL of chilled acetonitrile (containing 6.5 µM naproxen as internal standard) and cooled on ice for 10 min. Precipitated proteins were removed by centrifugation at 10 000 g for 10 min and supernatants were analysed by HPLC as described below. The fraction of substrate metabolized was calculated as the difference between the measured and initial clopidogrel or clopidogrel carboxylate concentration expressed as a percentage of the initial concentration.
In vitro inhibition of clopidogrel metabolism
Cytochrome P450 inhibition studies were performed in the presence of the respective inhibitors or substrates following the same incubation procedure as described for the kinetic experiments. Stock solutions containing inhibitors (see figures) were prepared in methanol or in water. The final volume of methanol did not exceed 1.0% of the total incubation volume and was identical in all incubations including controls. The inhibitor concentrations are given in the figures. IC50 values were calculated by non-linear regression analysis using the software program GraphPad Prism version 4.00 (San Diego, CA, USA).
HPLC analysis of clopidogrel and the clopidogrel carboxylate
Clopidogrel concentrations were determined using a LaChrom® high performance liquid chromatography (HPLC) system equipped with an UV detector operating at a wavelength of 235 nm, a column oven, a quaternary pump and an autosampler. The column temperature was maintained at 32°C and the injection volume was 30 µL. Separation was performed on a Nucleosil 50-5-C18 column equipped with a corresponding guard column using a gradient of solvent A (sodium phosphate 0.01 M, pH 3.0:acetonitrile; 50:50 v·v−1) and solvent B (sodium phosphate 0.01 M, pH 3.0:acetonitrile; 20:80 v·v−1). The gradient started at 80% A and 20% B for 2.5 min, changed to 100% B for 3.5 min and finally returned to the starting conditions for 4 min. The flow rate was 1 mL·min−1 and the total run time 10 min. The variability of the method was <10% at high and low clopidogrel concentrations. Calibration curves were performed in a concentration range of 1.0–43.0 µg·mL−1.
The carboxylate metabolite of clopidogrel was determined using the same HPLC system as described above, but with a different mobile phase. The mobile phase consisted of a gradient of solvent A (sodium phosphate 0.01 M, pH 3.0:acetonitrile; 50:50 v·v−1), solvent B (sodium phosphate 0.01 M, pH 3.0:acetonitrile; 20:80 v·v−1) and solvent C (sodium phosphate 0.01 M, pH 3.0:acetonitrile; 80:20 v·v−1). The gradient started at 20% A and 80% C for 2.5 min, changed to 100% B for 3.5 min and returned to the starting conditions for 4 min. The flow rate was 1 mL·min−1 and the total run time 10 min. Clopidogrel carboxylate was quantified by comparison with a standard curve. Variability and calibration curve range were identical to clopidogrel.
Ex vivo IPA by activated clopidogrel
Isolation of platelet-rich plasma (PRP) and platelet aggregation experiments were performed according to Born and Cross (1963). The platelet count in PRP samples was adjusted with platelet-poor plasma to 200–250 x109 platelets per L. Clopidogrel or clopidogrel carboxylate were activated in a mixture containing clopidogrel, HLM (0.25 mg·mL−1), incubation buffer (0.1 M potassium phosphate, pH 7.4) and NADPH-regenerating system for different periods of time. Inhibitors (dissolved in methanol; concentrations in figures) were evaporated to dryness at 37°C before addition of the same biotransformation mixture as above containing clopidogrel or clopidogrel carboxylate. In order to test the effect of GSH, biotransformation experiments were performed also in the presence of 5 mM GSH.
To assess platelet aggregation, 120 µL incubation mixture (activated clopidogrel or clopidogrel metabolite) was added to the same volume of platelets and preincubated at 37°C for 15 min. Platelet aggregation was stimulated with ADP (final concentration 2.5 µM) and recorded with an APACT4 aggregometer (LABiTec, Ahrensburg, Germany) as the maximal percentage in light transmittance of the reaction mixture. The percentage of IPA was calculated from the observed maximum platelet aggregation (MPA) as follows (Farid et al., 2007):
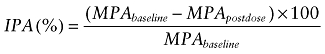 |
Molecular modelling studies
Molecular modelling was performed on a structure of human cytochrome 3A4 (PDBcode: 1W0G). All calculations were performed on a Dell Precision 670 workstation using the program Moloc (http://www.moloc.ch). Clopidogrel was docked manually into the active site of the enzyme which was previously modified such that an oxygen atom was placed at the position completing the octahedral geometry of the central Fe2+ of the haem. Multiple positions of clopidogrel were tried and subsequently optimized with the force field integrated in Moloc. All atoms of the structure were considered for calculations but only substrate atoms were allowed to move. An analogous procedure was applied for the more hydrophilic clopidogrel carboxylate.
Kinetic analysis and statistics
Kinetic parameters of clopidogrel biotransformation were calculated according to the Michaelis-Menten equation using nonlinear regression (GraphPad Prism version 4.00; San Diego, CA, USA):
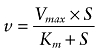 |
v and S are biotransformation rate and substrate concentration, respectively, Vmax the maximal biotransformation rate and Km the Michaelis-Menten constant. IC50 values for inhibitors were calculated by non-linear regression analysis (GraphPad Prism version 4.00; San Diego, CA, USA). P-values were calculated using one-way analysis of variance (ANOVA) with Dunnet's multiple comparison test for post hoc analysis. Data are presented as mean ± SEM. A P-value <0.05 was considered to be significant.
Materials
Clopidogrel hydrogen sulphate was isolated from commercially available tablets (Plavix®, Sanofi Aventis, Geneva, Switzerland) and the carboxylate metabolite of clopidogrel was obtained by saponification of clopidogrel (ReseaChem life science, Burgdorf, Switzerland). The purity was >99% for both substances as assessed by nuclear magnetic resonance spectroscopy (NMR). Atorvastatin was obtained from Sequoia Research Products Ldt. (Pangbourne, UK). NADPH regeneration system, pooled HLM (same batch was used for all experiments), recombinant human CYP3A4 supersomes (rhCYP3A4) and rhCYP2C19 were from BD Biosciences Gentest (Woburn, MA, USA). Acetonitrile LiChrosolv for HPLC use was obtained from Merck (Darmstadt, Germany). All other chemicals used were purchased from Sigma or Fluka (Buchs, Switzerland).
Results
In vitro metabolism of clopidogrel and clopidogrel carboxylate by HLM and rhCYP
We used HLM and the recombinant human enzymes CYP3A4 (rhCYP3A4) and CYP2C19 (rhCYP2C19) for this purpose. Clopidogrel was metabolized in a concentration-dependent fashion following Michaelis-Menten kinetics by both HLM and rhCYPs. In the presence of HLM, the apparent Km was 23.1 ± 3.7 µM and the Vmax 34.1 ± 2.1 nmoles·min−1·mg−1 protein (Figure 1A). In the presence of rhCYP3A4, the corresponding values were 45.9 ± 12.4 µM (Km) and 0.62 ± 0.09 nmoles·min−1·pmol−1 P450 (Vmax) (Figure 1B).
Figure 1.
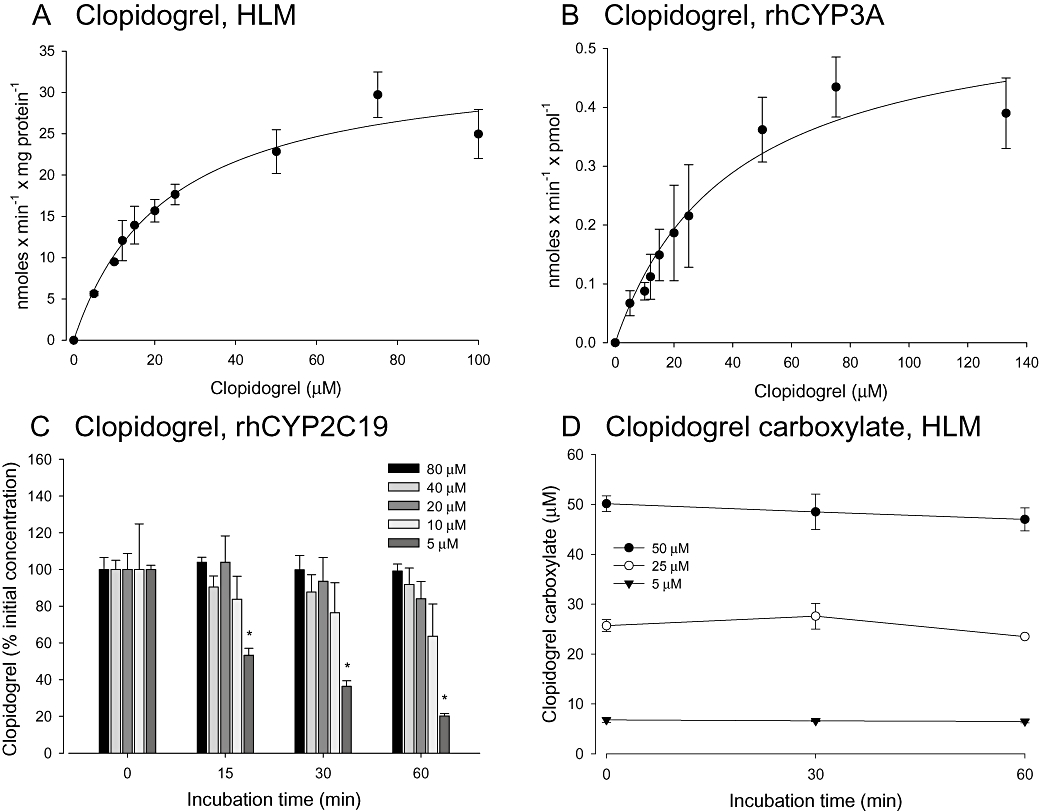
Biotransformation of clopidogrel and its carboxylate metabolite. Kinetic parameters for clopidogrel metabolism were determined in the presence of human liver microsomes (HLM) or supersomes (rhCYP3A4 or rhCYP2C19). Where possible, data were described using the Michaelis-Menten model. Increasing concentrations of clopidogrel (A, B, C) or of its carboxylate derivative (D) were incubated with HLM, rhCYP3A4 or rhCYP2C19 and analysed by HPLC. The biotransformation of clopidogrel in the presence of CYP3A4 showed a clear saturation and could be described by Michaelis-Menten kinetics (A, B). CYP2C19 activates clopidogrel only at concentrations <20 µM (C). The carboxylate metabolite of clopidogrel is not metabolized by HLM (D).
As shown in Figure 1C, clopidogrel biotransformation by rhCYP2C19 was observed only at clopidogrel concentrations <20 µM, indicating that clopidogrel is an inhibitor of CYP2C19 at higher concentrations. Its conversion rate at 10 µM clopidogrel was estimated to be 0.07 nmoles·min−1·pmol−1 P450, a value approximately 10 times lower than the Vmax obtained for rhCYP3A4.
To the best of our knowledge, there are no published data regarding the possible biotransformation of clopidogrel carboxylate, the main metabolite of clopidogrel (Caplain et al., 1999; Taubert et al., 2004). Concentrations of clopidogrel carboxylate did not decrease measurably during incubation, in either of the two in vitro systems tested, indicating that clopidogrel carboxylate is metabolized neither by HLM (Figure 1D), nor by rhCYP3A4 (data not shown).
Effect of specific CYP inhibitors and CYP substrates on clopidogrel biotransformation
The inhibition of clopidogrel (40 µM) metabolism by various CYP inhibitors and substrates was investigated using HLM (Figure 2). The oxidation of clopidogrel was significantly impaired by the CYP3A4 inhibitors ketoconazole and clarithromycin at concentrations in the nanomolar range. Ciprofloxacin, a strong inhibitor of CYP1A2 and a weak inhibitor of CYP3A4 (McLellan et al., 1996), reduced clopidogrel oxidation by 35% at 500 µM, but not at lower concentrations. In contrast, inhibitors of CYP2C9 (sulphaphenazole), CYP2D6 (quinidine), CYP2B6 (N,N′,N″-triethylenethiophosphoramide, thioTEPA) and CYP2C19 (omeprazole) revealed no significant effect on clopidogrel biotransformation by HML. For amiodarone, we found no significant inhibition of clopidogrel biotransformation in a concentration range of 1 to 100 µM (data only partially shown).
Figure 2.
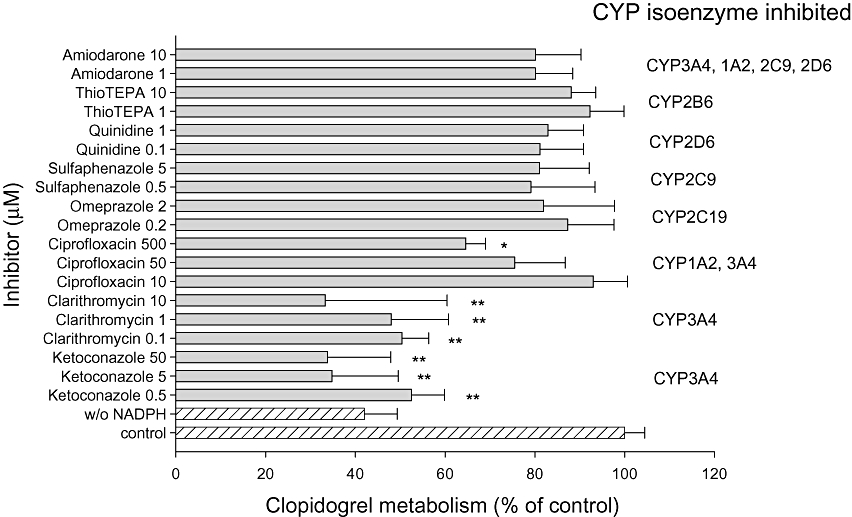
Effect of various CYP450 inhibitors on clopidogrel biotransformation. Clopidogrel (40 µM) was co-incubated in the presence of human liver microsomes (HLM) with different concentrations of CYP inhibitors. Data are expressed as the percentage of clopidogrel activated in the presence of the inhibitor compared with biotransformation without inhibitor (100%). Data points consist of five individual determinations. Data are presented as mean ± SEM. *P < 0.05, **P < 0.001 versus control incubations.
Due to the importance of CYP2C19 for clopidogrel biotransformation (Kim et al., 2008; Mega et al., 2009; Simon et al., 2009), CYP2C19 inhibitors were investigated in more detail (Figure 3). At a clopidogrel concentration of 40 µM, neither the PPIs omeprazole (up to 100 µM) and lansoprazole (up to 100 µM), nor ticlopidine, inhibited clopidogrel biotransformation by HLM. In contrast, at 5 µM clopidogrel, lansoprazole affected clopidogrel biotransformation in a concentration-dependent manner, reaching significance at 100 µM.
Figure 3.
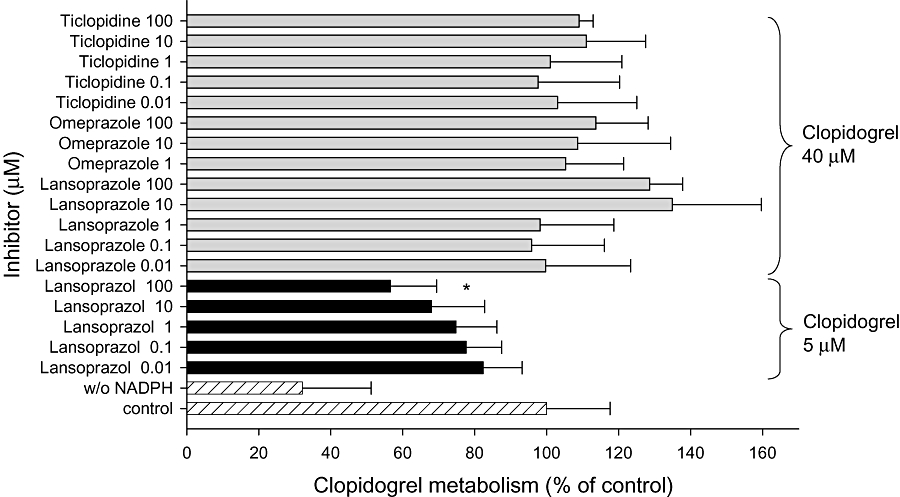
Effect of CYP2C19 inhibitors on clopidogrel biotransformation. Clopidogrel (40 or 5 µM) was co-incubated with different concentrations of CYP2C19 inhibitors in the presence of human liver microsomes (HLM). Data are expressed as the percentage clopidogrel biotransformation in the presence of the inhibitor compared with biotransformation without inhibitor (100%). Data points consist of three individual determinations. Data are presented as mean ± SEM. **P < 0.001 versus control incubations.
As atorvastatin may impair clopidogrel biotransformation (Lau et al., 2003), we investigated the impact of the CYP3A4 substrates atorvastatin and simvastatin on clopidogrel biotransformation by HLM. Both statins significantly inhibited clopidogrel biotransformation at the maximal concentration tested (10 µM). In contrast, pravastatin (not a CYP3A4 substrate) showed no inhibitory effect at this concentration (Figure 4).
Figure 4.
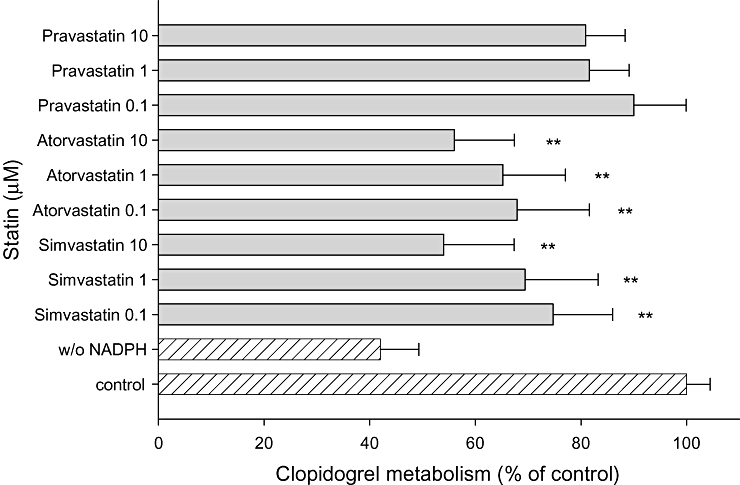
Effect of statins on clopidogrel biotransformation. Clopidogrel (40 µM) was co-incubated with different concentrations of statins in the presence of human liver microsomes (HLM). Data are expressed as the percentage clopidogrel biotransformation compared with the biotransformation without inhibitor (100%). Data points consist of five individual determinations. Data are presented as mean ± SEM. *P < 0.05, **P < 0.001 versus control incubations.
Inhibition of clopidogrel biotransformation by CYP inhibitors or substrates was confirmed by the determination of the corresponding IC50 values (Table 1). Ketoconazole showed a slightly stronger inhibitory effect (IC50 0.03 µM) than clarithromycin (IC50 0.33 µM). An IC50 for ciprofloxacin could not be determined, as 50% inhibition were not reached up to 500 µM. Simvastatin and atorvastatin both revealed dose-dependent inhibition of clopidogrel biotransformation with IC50 values of 1.28 µM and 16.9 µM respectively.
Table 1.
IC50 (µM) values for inhibition of clopidogrel biotransformation by human liver microsomes (HLM) and for the antiplatelet effect of clopidogrel in the presence of CYP3A4 inhibitors or substrates
| IC50 for biotransformation by HLM (µM) | IC50 for antiplatelet effect (µM) | |
|---|---|---|
| Ketoconazole | 0.03 ± 0.07 | 0.55 ± 0.06 |
| Clarithromycin | 0.33 ± 0.09 | 0.95 ± 0.04 |
| Simvastatin | 1.28 ± 0.09 | 1.29 ± 0.02 |
| Atorvastatin | 16.9 ± 0.3 | 3.83 ± 0.07 |
The assay conditions are described in Methods. The clopidogrel concentration was 40 µM for all incubations. Data are expressed as mean ± SEM of n = 4–8 experiments.
IPA by activated clopidogrel
Further, we developed a test system for analysing platelet aggregation in incubations containing HLM, the drugs investigated and human platelets (Figure 5A). Clopidogrel inhibited platelet aggregation concentration-dependently, reaching 67% at 200 µM. In contrast, clopidogrel without biotransformation by HLM showed no significant IPA. To demonstrate the formation of an active metabolite, we investigated the antiplatelet effect of clopidogrel in the presence of 5 mM GSH. As GSH is known to affect formation of and breaking of disulfide bonds in cells (Dickinson and Forman, 2002), we hypothesized that it could trap the newly formed thiol group of activated clopidogrel (Ding et al., 2003). As expected, addition of GSH significantly decreased the effect of clopidogrel on platelet aggregation (Figure 5B).
Figure 5.
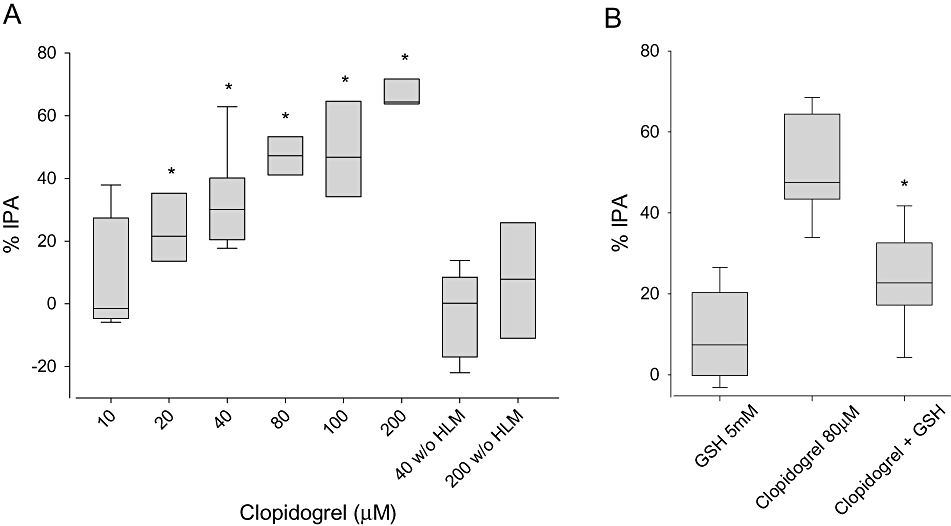
Effect of activated clopidogrel on platelet aggregation. (A) Increasing concentrations of clopidogrel (10–200 µM) activated by human liver microsomes (HLM) were incubated with platelet-rich plasma. Platelet aggregation was determined in response to 2.5 µM ADP by light transmittance aggregometry. Clopidogrel incubated in the absence of HLM served as a negative control. (B) In the presence of 5 mM glutathione, the effect of clopidogrel on platelet aggregation was significantly decreased. GSH itself had no effect on platelet aggregation. Results are expressed as a percentage inhibition of platelet aggregation (IPA) calculated from the maximum platelet aggregation in the presence of the solvent. Data are presented as box-plots with the median indicated by the line within the box (n = 8 to 12). *P < 0.001 versus clopidogrel 40 µM without (w/o) HLM (A) or clopidogrel 80 µM (B).
Clopidogrel carboxylate has no antiplatelet effect
Clopidogrel and clopidogrel carboxylate were incubated individually with HLM for 5, 15, 30 and 60 min prior to ADP-induced platelet aggregation. As expected, clopidogrel showed a time-dependent inhibitory effect on platelet aggregation, while 80 and 200 µM clopidogrel carboxylate completely failed to inhibit platelet aggregation after incubation with HLM (Figure 6A and 200 µM not shown).
Figure 6.
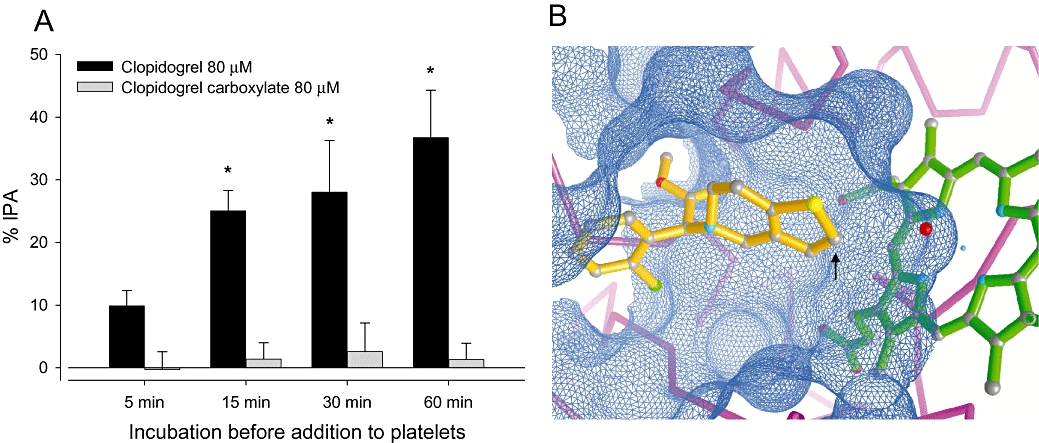
Time course of the effect of clopidogrel or clopidogrel carboxylate (80 µM) on platelet aggregation and interaction of clopidogrel with CYP3A4. (A) Clopidogrel or clopidogrel carboxylate were incubated with human liver microsomes (HLM) for 5, 15, 30 or 60 min. At the indicated times, aliquots were incubated with platelet-rich plasma and platelet aggregation determined in response to 2.5 µM ADP. Clopidogrel inhibited platelet biotransformation over time whereas the carboxylate derivative did not affect platelet aggregation. Results are expressed as the percentage inhibition of platelet aggregation (% IPA), calculated from the maximum platelet aggregation obtained in the presence of the vehicle (1% methanol v·v−1). Data are presented as mean ± SEM (n = 4). *P < 0.05, **P < 0.01 clopidogrel or clopidogrel carboxylate versus incubations containing no clopidogrel (not shown). (B) Clopidogrel (orange) interacts with the active site (blue net) of CYP3A4 (backbone, magenta). A haem molecule with the Fe2+ (cyan dot in centre of haem) is shown in green. Note that the activated oxygen (red ball) above the Fe2+ of the haem is placed in an ideal position to interact with the 2-carbon of clopidogrel (arrow). In contrast, the clopidogrel carboxylate (not shown) carries a polar and solvated carboxylate group preventing it from bringing the 2-carbon close enough to the activated oxygen and the Fe2+ of the haem, which are crucial conditions for its subsequent biotransformation.
Interaction of clopidogrel with CYP3A4
In order to investigate the reason for the different binding affinity of neutral clopidogrel and its much more hydrophilic carboxylate derivative to CYP3A4, we manually docked both compounds into the active site of CYP3A4. As shown in Figure 6B, the neutral and hydrophobic clopidogrel fits smoothly into the hydrophobic active site of CYP3A4, which is optimized to recognize and bind hydrophobic substances. In contrast, the slightly smaller carboxylate metabolite contains a polar and solvated carboxylate functionality that does not bind in a productive way to the hydrophobic catalytic site of CYP3A4 (not shown).
Effect of CYP3A4 inhibitors and/or substrates on clopidogrel-associated IPA
Finally, we addressed the question whether a diminished clopidogrel biotransformation by CYP3A4 inhibitors or substrates is associated with an IPA. As shown in Table 1, ketoconazole turned out to be the most potent inhibitor with an IC50 of 0.55 µM, confirming our results obtained in the biotransformation experiments. The inhibitory effect of clarithromycin was comparable (IC50 0.95 µM), whereas the statins were less effective inhibitors.
Discussion and conclusions
In our studies, clopidogrel was metabolized in a concentration-dependent manner in all incubations containing CYP3A4 (Figure 1A,B), whereas supersomes containing rhCYP2C19 metabolized clopidogrel only at substrate concentrations ≤10 µM. Regarding the inhibition of CYP2C19 by clopidogrel, our data are in accordance with recent studies (Hagihara et al., 2008; Nishiya et al., 2009), showing that clopidogrel is a mechanism-based inhibitor of CYP2C19 with an IC50 in the low micromolar range. Clopidogrel is rapidly and efficiently absorbed from the GI tract (Caplain et al., 1999), but more than 85% of the drug is converted to its carboxylate metabolite during the first passage across intestine and liver (Taubert et al., 2004). Assuming that the maximal concentration of the carboxylate derivative of clopidogrel in plasma approximately corresponds to the maximal clopidogrel concentration in the liver (no data on the clopidogrel concentration in the liver are available), a maximal concentration of 5–20 µmol·L−1 is reached in hepatocytes after ingestion of 75 mg clopidogrel (Caplain et al., 1999; Taubert et al., 2004). Taking into account that clopidogrel inhibits CYP2C19 at concentrations >10 µM without affecting CYP3A4 and that, after oral ingestion of 75 mg, the hepatocellular clopidogrel concentration will drop below 10 µM with time, it can be expected that in vivo both CYP3A4 and CYP2C19 contribute to clopidogrel biotransformation. These considerations therefore help to explain why both, strong inhibitors of CYP3A4 (Suh et al., 2006; Farid et al., 2007; Siller-Matula et al., 2008) and genetic variants of CYP2C19 (Kim et al., 2008; Mega et al., 2009; Simon et al., 2009), are associated with impaired antiplatelet activity of clopidogrel.
In contrast to the recent study of Kazui et al. (2010), in the current study, clopidogrel was biotransformed not only by HML, but also by rhCYP3A4, indicating that CYP3A4 can also perform the conversion of clopidogrel to 2-oxo-clopidogrel. Our data agree in this point with those of Clarke and Waskell (2003), who found that CYP3A4 and CYP3A5 are the most important CYPs for the biotransformation of clopidogrel. In the study of Kazui et al. (2010), CYP2C19, CYP1A2 and CYP2B6 were the most important CYPs regarding the formation of 2-oxo-clopidogrel from clopidogrel.
The good correlation between the inhibition of clopidogrel biotransformation and the antiplatelet effect by clopidogrel suggests that our systems are able to predict drug interactions with clopidogrel. In our study, ketoconazole, a potent inhibitor of CYP3A4, showed the strongest inhibitory effect on clopidogrel biotransformation and IPA. These data are in good agreement with a clinical study demonstrating that ketoconazole not only decreases clopidogrel biotransformation but also its antiplatelet activity (Farid et al., 2007). Based on our data, the macrolide antibiotic clarithromycin and possibly other CYP3A4 inhibitors may have similar in vivo effects as ketoconazole.
Amiodarone is a drug prescribed often with clopidogrel and possibly interferes with its biotransformation (Lau et al., 2004). It is mainly metabolized by CYP3A4 in humans and is an inhibitor of CYP2C9, CYP2D6 and CYP3A4 (Ohyama et al., 2000). Surprisingly, amiodarone did not affect clopidogrel biotransformation in our in vitro system up to concentrations of 100 µM. This finding may be explained by the fact that the in vivo generated desethylamiodarone, the major metabolite of amiodarone, is a more potent inhibitor of human CYPs than amiodarone itself (Ohyama et al., 2000). A prolonged incubation time would possibly have been necessary to generate this metabolite and detect inhibitory effects of amiodarone in our system.
Ciprofloxacin, a well-known CYP1A2 inhibitor, significantly inhibited clopidogrel biotransformation at a concentration of 500 µM, but not at lower concentrations. In the studies demonstrating inhibition of CYP1A2, ciprofloxacin concentrations in the range of 10–200 µM were used (Cherstniakova et al., 2001; Karjalainen et al., 2008). On the other hand, McLellan et al. (1996) reported that ciprofloxacin significantly decreased the activity of CYP3A4 when used at high concentrations (∼2 mM), which is accordance with our findings.
Our investigations with statins are in good agreement with results presented by Lau et al. (2003), Neubauer et al. (2003) and by Clarke and Waskell (2003). Atorvastatin and simvastatin are both metabolized by CYP3A4 and significantly inhibited clopidogrel biotransformation in vitro, whereas pravastatin showed no such inhibitory effect (Figure 4). Additionally, we could demonstrate that the inhibitory effect of atorvastatin and simvastatin on clopidogrel biotransformation resulted in an impaired antiplatelet effect of clopidogrel (Table 1). In agreement with our findings, Lau et al. demonstrated in their ex vivo study a dose-dependent attenuation of the clopidogrel-associated antiplatelet effects by atorvastatin (Lau et al., 2003). The results of another small ex vivo analysis using simvastatin confirmed the occurrence of a clopidogrel-statin interaction (Neubauer et al., 2003). In contrast, other studies investigating the influence of CYP3A4-metabolized statins on antiplatelet effects of clopidogrel failed to confirm these findings (Muller et al., 2003; Gorchakova et al., 2004; Mitsios et al., 2004; Serebruany et al., 2004; Vinholt et al., 2005; Saw et al., 2007). Taking into account our findings, the exact time-points when clopidogrel and reversible CYP inhibitors such as statins are ingested may play a crucial role for the occurrence and possible manifestation of drug-drug interactions.
As clopidogrel inhibits CYP2C19 at concentrations >10 µM (this study and studies by Hagihara et al. (Hagihara et al., 2008) and by Nishiya et al. (Nishiya et al., 2009)), it could be expected that CYP2C19 inhibitors would not affect clopidogrel activation at high clopidogrel concentrations. Accordingly, the CYP2C19 inhibitors tested in the current study (ticlopidine, omeprazole and lansoprazole) revealed no inhibition of clopidogrel biotransformation at 40 µM clopidogrel. Lansoprazole, a strong inhibitor of CYP2C19 (Li et al., 2004), impaired clopidogrel biotransformation only at 5 µM clopidogrel. As discussed above, in the liver of patients treated with clopidogrel, the clopidogrel concentration can be assumed to drop to levels at which inhibition of clopidogrel biotransformation by PPIs becomes potentially significant. This assumption is supported by studies showing that PPIs diminish the pharmacological effect of clopidogrel on platelet aggregation determined ex vivo (Gilard et al., 2008; O'Donoghue et al., 2009; Price et al., 2009; Zuern et al., 2010). Accordingly, in two retrospective studies with clinical endpoints, coadministration of PPIs was associated with an impaired outcome (Ho et al., 2009; Juurlink et al., 2009). Since a third retrospective, clinical study failed to show such an effect of PPIs (O'Donoghue et al., 2009), the clinical relevance of the inhibition of clopidogrel biotransformation by PPIs is actually unclear, however. Nevertheless, PPIs with a strong inhibition of CYP2C19 such as lansoprazole, esomeprazole and omeprazole (Li et al., 2004) should best be avoided in patients treated with clopidogrel, especially when they are treated also with CYP3A4 inhibitors.
In conclusion, we could demonstrate that CYP3A4 is the most important CYP isoenzyme for clopidogrel biotransformation at clopidogrel concentrations >10 µM, as clopidogrel inhibits CYP2C19 at high concentrations. At concentrations ≤10 µM, CYP2C19 starts to contribute to clopidogrel biotransformation and the clopidogrel biotransformation can be inhibited by PPIs. The concentration-dependent interaction pattern between CYP inhibitors, clopidogrel and CYP3A4 and CYP2C19 helps to explain the often diverging results regarding clopidogrel biotransformation and pharmacological activity between studies conducted in vitro and in vivo.
Acknowledgments
The study was supported by a grant from the Swiss National Science Foundation to S.K. (31003A-112483).
Glossary
Abbreviations
- GSH
glutathione
- HLM
human liver microsomes
- IPA
inhibition of platelet aggregation
- MPA
maximum platelet aggregation
- PCI
percutaneous coronary intervention
- PPI
proton pump inhibitor
- PRP
platelet-rich plasma
- (rh)CYP
(recombinant human) cytochrome P450
Conflicts of interest
None of the authors declares any conflict of interest regarding this manuscript.
Supplemental material
Supporting Information: Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born GV, Cross MJ. The aggregation of blood platelets. J Physiol. 1963;168:178–195. doi: 10.1113/jphysiol.1963.sp007185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplain H, Donat F, Gaud C, Necciari J. Pharmacokinetics of clopidogrel. Semin Thromb Hemost. 1999;25(Suppl. 2):25–28. [PubMed] [Google Scholar]
- Cherstniakova SA, Bi D, Fuller DR, Mojsiak JZ, Collins JM, Cantilena LR. Metabolism of vanoxerine, 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine, by human cytochrome P450 enzymes. Drug Metab Dispos. 2001;29:1216–1220. [PubMed] [Google Scholar]
- Clarke TA, Waskell LA. The metabolism of clopidogrel is catalyzed by human cytochrome P450 3A and is inhibited by atorvastatin. Drug Metab Dispos. 2003;31:53–59. doi: 10.1124/dmd.31.1.53. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochem Pharmacol. 2002;64:1019–1026. doi: 10.1016/s0006-2952(02)01172-3. [DOI] [PubMed] [Google Scholar]
- Ding Z, Kim S, Dorsam RT, Jin J, Kunapuli SP. Inactivation of the human P2Y12 receptor by thiol reagents requires interaction with both extracellular cysteine residues, Cys17 and Cys270. Blood. 2003;101:3908–3914. doi: 10.1182/blood-2002-10-3027. [DOI] [PubMed] [Google Scholar]
- Farid NA, Payne CD, Small DS, Winters KJ, Ernest CS, 2nd, Brandt JT, et al. Cytochrome P450 3A inhibition by ketoconazole affects prasugrel and clopidogrel pharmacokinetics and pharmacodynamics differently. Clin Pharmacol Ther. 2007;81:735–741. doi: 10.1038/sj.clpt.6100139. [DOI] [PubMed] [Google Scholar]
- Gilard M, Arnaud B, Cornily JC, Le Gal G, Lacut K, Le Calvez G, et al. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol. 2008;51:256–260. doi: 10.1016/j.jacc.2007.06.064. [DOI] [PubMed] [Google Scholar]
- Gorchakova O, von Beckerath N, Gawaz M, Mocz A, Joost A, Schomig A, et al. Antiplatelet effects of a 600 mg loading dose of clopidogrel are not attenuated in patients receiving atorvastatin or simvastatin for at least 4 weeks prior to coronary artery stenting. Eur Heart J. 2004;25:1898–1902. doi: 10.1016/j.ehj.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Hagihara K, Nishiya Y, Kurihara A, Kazui M, Farid NA, Ikeda T. Comparison of human cytochrome P450 inhibition by the thienopyridines prasugrel, clopidogrel, and ticlopidine. Drug Metab Pharmacokinet. 2008;23:412–420. doi: 10.2133/dmpk.23.412. [DOI] [PubMed] [Google Scholar]
- Ho PM, Maddox TM, Wang L, Fihn SD, Jesse RL, Peterson ED, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA. 2009;301:937–944. doi: 10.1001/jama.2009.261. [DOI] [PubMed] [Google Scholar]
- Hulot JS, Bura A, Villard E, Azizi M, Remones V, Goyenvalle C, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108:2244–2247. doi: 10.1182/blood-2006-04-013052. [DOI] [PubMed] [Google Scholar]
- Juurlink DN, Gomes T, Ko DT, Szmitko PE, Austin PC, Tu JV, et al. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. CMAJ. 2009;180:713–718. doi: 10.1503/cmaj.082001. Epub 28 January 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karjalainen MJ, Neuvonen PJ, Backman JT. In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin Pharmacol Toxicol. 2008;103:157–165. doi: 10.1111/j.1742-7843.2008.00252.x. [DOI] [PubMed] [Google Scholar]
- Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38:92–99. doi: 10.1124/dmd.109.029132. [DOI] [PubMed] [Google Scholar]
- Kim KA, Park PW, Hong SJ, Park JY. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: a possible mechanism for clopidogrel resistance. Clin Pharmacol Ther. 2008;84:236–242. doi: 10.1038/clpt.2008.20. [DOI] [PubMed] [Google Scholar]
- Lau WC, Waskell LA, Watkins PB, Neer CJ, Horowitz K, Hopp AS, et al. Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation: a new drug-drug interaction. Circulation. 2003;107:32–37. doi: 10.1161/01.cir.0000047060.60595.cc. [DOI] [PubMed] [Google Scholar]
- Lau WC, Gurbel PA, Watkins PB, Neer CJ, Hopp AS, Carville DG, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation. 2004;109:166–171. doi: 10.1161/01.CIR.0000112378.09325.F9. [DOI] [PubMed] [Google Scholar]
- Li XQ, Andersson TB, Ahlstrom M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos. 2004;32:821–827. doi: 10.1124/dmd.32.8.821. [DOI] [PubMed] [Google Scholar]
- Lins R, Broekhuysen J, Necciari J, Deroubaix X. Pharmacokinetic profile of 14C-labeled clopidogrel. Semin Thromb Hemost. 1999;25(Suppl. 2):29–33. [PubMed] [Google Scholar]
- McLellan RA, Drobitch RK, Monshouwer M, Renton KW. Fluoroquinolone antibiotics inhibit cytochrome P450-mediated microsomal drug metabolism in rat and human. Drug Metab Dispos. 1996;24:1134–1138. [PubMed] [Google Scholar]
- Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- Mitsios JV, Papathanasiou AI, Rodis FI, Elisaf M, Goudevenos JA, Tselepis AD. Atorvastatin does not affect the antiplatelet potency of clopidogrel when it is administered concomitantly for 5 weeks in patients with acute coronary syndromes. Circulation. 2004;109:1335–1338. doi: 10.1161/01.CIR.0000124581.18191.15. [DOI] [PubMed] [Google Scholar]
- Muller I, Besta F, Schulz C, Li Z, Massberg S, Gawaz M. Effects of statins on platelet inhibition by a high loading dose of clopidogrel. Circulation. 2003;108:2195–2197. doi: 10.1161/01.CIR.0000099507.32936.C0. [DOI] [PubMed] [Google Scholar]
- Neubauer H, Gunesdogan B, Hanefeld C, Spiecker M, Mugge A. Lipophilic statins interfere with the inhibitory effects of clopidogrel on platelet function – a flow cytometry study. Eur Heart J. 2003;24:1744–1749. doi: 10.1016/s0195-668x(03)00442-1. [DOI] [PubMed] [Google Scholar]
- Nishiya Y, Hagihara K, Kurihara A, Okudaira N, Farid NA, Okazaki O, et al. Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel, and prasugrel. Xenobiotica. 2009;39:836–843. doi: 10.3109/00498250903191427. [DOI] [PubMed] [Google Scholar]
- O'Donoghue ML, Braunwald E, Antman EM, Murphy SA, Bates ER, Rozenman Y, et al. Pharmacodynamic effect and clinical efficacy of clopidogrel and prasugrel with or without a proton-pump inhibitor: an analysis of two randomised trials. Lancet. 2009;374:989–997. doi: 10.1016/S0140-6736(09)61525-7. Epub 31 August 2009. [DOI] [PubMed] [Google Scholar]
- Ohyama K, Nakajima M, Suzuki M, Shimada N, Yamazaki H, Yokoi T. Inhibitory effects of amiodarone and its N-deethylated metabolite on human cytochrome P450 activities: prediction of in vivo drug interactions. Br J Clin Pharmacol. 2000;49:244–253. doi: 10.1046/j.1365-2125.2000.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereillo JM, Maftouh M, Andrieu A, Uzabiaga MF, Fedeli O, Savi P, et al. Structure and stereochemistry of the active metabolite of clopidogrel. Drug Metab Dispos. 2002;30:1288–1295. doi: 10.1124/dmd.30.11.1288. [DOI] [PubMed] [Google Scholar]
- Price MJ, Nayak KR, Barker CM, Kandzari DE, Teirstein PS. Predictors of heightened platelet reactivity despite dual-antiplatelet therapy in patients undergoing percutaneous coronary intervention. Am J Cardiol. 2009;103:1339–1343. doi: 10.1016/j.amjcard.2009.01.341. [DOI] [PubMed] [Google Scholar]
- Ratz Bravo AE, Tchambaz L, Krahenbuhl-Melcher A, Hess L, Schlienger RG, Krahenbuhl S. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263–275. doi: 10.2165/00002018-200528030-00007. [DOI] [PubMed] [Google Scholar]
- Reist M, Roy-de Vos M, Montseny JP, Mayer JM, Carrupt PA, Berger Y, et al. Very slow chiral inversion of clopidogrel in rats: a pharmacokinetic and mechanistic investigation. Drug Metab Dispos. 2000;28:1405–1410. [PubMed] [Google Scholar]
- Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP, et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost. 2000;84:891–896. [PubMed] [Google Scholar]
- Saw J, Brennan DM, Steinhubl SR, Bhatt DL, Mak KH, Fox K, et al. Lack of evidence of a clopidogrel-statin interaction in the CHARISMA trial. J Am Coll Cardiol. 2007;50:291–295. doi: 10.1016/j.jacc.2007.01.097. [DOI] [PubMed] [Google Scholar]
- Schulman SP. Antiplatelet therapy in non-ST-segment elevation acute coronary syndromes. JAMA. 2004;292:1875–1882. doi: 10.1001/jama.292.15.1875. [DOI] [PubMed] [Google Scholar]
- Serebruany VL, Midei MG, Malinin AI, Oshrine BR, Lowry DR, Sane DC, et al. Absence of interaction between atorvastatin or other statins and clopidogrel: results from the interaction study. Arch Intern Med. 2004;164:2051–2057. doi: 10.1001/archinte.164.18.2051. [DOI] [PubMed] [Google Scholar]
- Siller-Matula JM, Lang I, Christ G, Jilma B. Calcium-channel blockers reduce the antiplatelet effect of clopidogrel. J Am Coll Cardiol. 2008;52:1557–1563. doi: 10.1016/j.jacc.2008.07.055. [DOI] [PubMed] [Google Scholar]
- Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, Meneveau N, et al. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360:363–375. doi: 10.1056/NEJMoa0808227. [DOI] [PubMed] [Google Scholar]
- Suh JW, Koo BK, Zhang SY, Park KW, Cho JY, Jang IJ, et al. Increased risk of atherothrombotic events associated with cytochrome P450 3A5 polymorphism in patients taking clopidogrel. CMAJ. 2006;174:1715–1722. doi: 10.1503/cmaj.060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert D, Kastrati A, Harlfinger S, Gorchakova O, Lazar A, von Beckerath N, et al. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost. 2004;92:311–316. doi: 10.1160/TH04-02-0105. [DOI] [PubMed] [Google Scholar]
- Vinholt P, Poulsen TS, Korsholm L, Kristensen SR, Hallas J, Damkier P, et al. The antiplatelet effect of clopidogrel is not attenuated by statin treatment in stable patients with ischemic heart disease. Thromb Haemost. 2005;94:438–443. doi: 10.1160/TH05-01-0046. [DOI] [PubMed] [Google Scholar]
- Zuern CS, Geisler T, Lutilsky N, Winter S, Schwab M, Gawaz M. Effect of comedication with proton pump inhibitors (PPIs) on post-interventional residual platelet aggregation in patients undergoing coronary stenting treated by dual antiplatelet therapy. Thromb Res. 2010;125:e51–e54. doi: 10.1016/j.thromres.2009.08.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.