

Abstract
BACKGROUND AND PURPOSE
Interest in non-selective cation channels has increased recently following the discovery of transient receptor potential (TRP) proteins, which constitute many of these channels.
EXPERIMENTAL APPROACH
We used the whole-cell patch-clamp technique on isolated ventricular myocytes to investigate the effect of flufenamic acid (FFA) and related drugs on membrane ion currents.
KEY RESULTS
With voltage-dependent and other ion channels inhibited, cells that were exposed to FFA, N-(p-amylcinnamoyl)anthranilic acid (ACA), ONO-RS-082 or niflumic acid (NFA) responded with an increase in currents. The induced current reversed at +38 mV, was unaffected by lowering extracellular Cl- concentration or by the removal of extracellular Ca2+ and Mg2+, and its inward but not outward component was suppressed in Na+-free extracellular conditions. The current was suppressed by Gd3+ but was resistant to 2-aminoethoxydiphenyl borate (2-APB) and to amiloride. It could not be induced by the structurally related non-fenamate anti-inflammatory drug diclofenac, nor by the phospholipase-A2 inhibitors bromoenol lactone and bromophenacyl bromide. Muscarinic or α-adrenoceptor activation or application of diacylglycerol failed to induce or modulate the current.
CONCLUSIONS AND IMPLICATIONS
Flufenamic acid and related drugs activate a novel channel conductance, where Na+ is likely to be the major charge carrier. The identity of the channel remains unclear, but it is unlikely to be due to Ca2+-activated (e.g. TRPM4/5), Mg2+-sensitive (e.g. TRPM7) or divalent cation-selective TRPs.
Keywords: cardiac, channel, cation, TRP
Introduction
Besides the classic selective ion channels, non-selective ion channels are expressed in many cells and may play important roles in normal cellular physiology and in pathology. In cardiac cells, the best known non-selective ion channels are the If or HCN channels, which conduct Na+ and K+ and play a major role in determining pacemaker activity (DiFrancesco, 1986), and the Ca2+-activated channels (Colquhoun et al., 1981; Ehara et al., 1988). Additional non-selective channels are present (see Discussion) but, generally, most such channels have remained difficult to identify using electrophysiological techniques not only in cardiac but also in other cell types. This is due to technical difficulties in distinguishing currents carried by non-selective channels from leak flows via non-channel pathways such as the seal between recording pipette and cell membrane, and to problems in separating currents carried by different non-selective channels co-expressed in the same cell, as specific inhibitors are still lacking. The identification of non-selective channels remains nevertheless important given their potential roles and the possibility that they may constitute targets for therapeutic agents. In recent years, interest in non-selective channels has increased significantly following the discovery of transient receptor potential (TRP) proteins, of which many have been shown to underlie various such channels in different types of cells (Clapham, 2003; Nilius, 2007).
In cardiac cells, molecular biology and immunostaining methods have demonstrated the expression of a variety of TRP channels, including those of the canonical (TRPC), melastatin (TRPM) and polycystin (TRPP) subfamilies (channel nomenclature follows Alexander et al., 2009). Only TRPC1 (Seth et al., 2009), TRPC3 (Onohara et al., 2006), a few TRPM (TRPM4, TRPM5, TRPM7) (Guinamard et al., 2004; 2006a,b; Gwanyanya et al., 2004) and putative TRPP channels (Volk et al., 2003), have been characterized electrophysiologically in the heart. The cardiac TRP channels thus far identified have been reported to be implicated in such diverse processes as pacemaking (Demion et al., 2007; Guinamard and Bois, 2007), Mg2+ transport (Gwanyanya et al., 2004), myocardial hypertrophy (Kuwahara et al., 2006; Onohara et al., 2006; Guinamard and Bois, 2007; Shan et al., 2008; Watanabe et al., 2008; Seth et al., 2009; Watanabe et al., 2009), arrhythmogenesis (Guinamard and Bois, 2007; Alvarez et al., 2008) and response to stretch (Dyachenko et al., 2009). Despite this now well-recognized importance of TRPs it remains possible that other, non-TRP proteins may underlie some cation non-selective channels. Molecular biology techniques have been used successfully to study ion channels, but when such tools cannot be utilized to identify, separate or modulate ion channels because the underlying proteins are not known, a combination of electrophysiology and pharmacological tools remains a powerful experimental approach.
Flufenamic acid (FFA) and other fenamates are phenylaminobenzoic (or phenylanthranilic) acid derivatives generally known for their anti-inflammatory and ion channel-blocking properties. The anti-inflammatory action is due to the inhibition of cyclo-oxygenase, that is, to the suppression of prostaglandin synthesis from arachidonic acid. The blocking action of fenamates is exerted on a variety of channels, including Cl- channels, voltage-dependent Na+ or Ca2+ channels and TRP channels (Kraft and Harteneck, 2005). They also inhibit other non-channel transporters, for example, the Na+-dicarboxylate transporter (Pajor and Randolph, 2007). Similar inhibitory actions have been reported for the related cinnamoyl-anthranilic derivatives (Harteneck et al., 2007). In contrast to these inhibitory effects, stimulatory actions of fenamates on channels have also been increasingly reported. Examples include a stimulation of Ca2+-activated large conductance K+ (or maxi KCa) channels (Liu et al., 2005) and the activation by FFA of some TRP channels, such as TRPC6 (Jung et al., 2002; Foster et al., 2009) or TRPA1 (Hu et al., 2010). In our attempts to test for a blocking effect of FFA and related drugs on TRPM7 channel currents in cardiac myocytes, we discovered that they induce a current with novel characteristics, different from those of other, well-characterized, channels.
Methods
All animal care and experimental procedures in this study were according to the institutional guidelines for laboratory animal care and approved by the Ethical Commission on Animal Experiments of the University of Leuven.
Cell isolation and electrophysiological techniques
We used left ventricular myocytes dissociated from pig and sheep hearts by enzymatic digestion during Langendorff perfusion at 36°C, as described in detail before (Macianskiene et al., 2002; Gwanyanya et al., 2004; 2006). Experiments were performed either at room temperature (22–23°C; for ion current measurements) or at 36–37°C (for action potential recordings). Voltage clamp experiments testing bromoenol lactone (BEL) effects were also carried out at 36°C (Smani et al., 2003).
The pClamp 8.1 software was used to generate stimuli protocols and to record data via a Digidata 1322A acquisition system (Axon instruments, Union City, CA, USA). The voltage clamp protocols usually consisted of 4 s symmetrical ramps from −120 mV to +80 mV and back to −120 mV, applied every 10 s. During the ascending limb of the ramp, the slow rate of depolarization (0.1 V·s−1) allowed activation and inactivation of the voltage-dependent Na+ current. The currents of interest were measured during the descending limb of the voltage ramp. In a few experiments, 600 ms voltage steps were also given to various potentials from a holding of −40 mV. In most experiments currents through L-type Ca2+ (CaV1.2) or via rapid delayed rectifier K+ (KV11.1 or IKr) channels were blocked by nifedipine (10 µM) and E-4031 (5 µM) respectively. Action potentials were elicited under current clamp in ruptured or perforated patch recording modes. The stimulus consisted of 2 ms rectangular current pulse applied at 1 Hz via the patch electrode. Action potential durations were measured at 90% repolarization (APD90).
Solutions
The composition of the standard Tyrode solution used during cell isolation and during action potential measurements was (in mM): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 0.9 MgCl2, 0.33 NaH2PO4, 10 HEPES (pH 7.4, adjusted with NaOH) and 10 glucose. The solution was bubbled with 100% O2 during cell dissociation. During ion current measurements, cells were superfused with a solution of similar composition except that K+ was replaced by Cs+. When desired, divalent cations were omitted or N-methyl-D-glucamine (NMDG) used to replace Na+. The standard pipette solution contained (in mM): 130 Cs glutamate, 25 CsCl or tetraethyl ammonium chloride (TEA Cl), 5 Na2ATP, 5.5 MgCl2 (0.8 mM free Mg2+), 1 EGTA, 0.1 Na2GTP, 5 HEPES (pH 7.25; adjusted with CsOH). This internal solution was modified in a few experiments, to increase Ca2+ buffering (1 EGTA replaced by 10 EDTA), to remove Na+ (5.5 MgCl2 and 5 Na2ATP replaced by 5.5 MgATP) or to increase free intracellular Mg2+ to 4.6 mM (10 MgCl2 and 5.5 MgATP). The pipette solution for current clamp measurements was similar to the above standard internal solution, with Cs+ replaced by K+. For perforated patch recordings, 325 µM amphotericin B and 1.8 mM CaCl2 (to cause cell death in case of patch rupture) were added to this pipette solution.
Data and statistical analyses
Data were analysed using Clampfit 8.2 (Axon Instruments) and Origin 7 (Microcal, Northampton, MA, USA). The following Hill equation was used to fit the drug (D) effects as a function of concentration:
 |
where ID is the current induced at a given drug concentration [D], Imax the maximum drug-induced current, K0.5 the drug concentration for 50% effect and nHill the Hill coefficient. Average data are expressed as mean ± standard error of the mean, with n indicating the number of cells studied. Means were compared using the two-tailed t-test, or anova followed by Bonferroni's post-test. P≤ 0.05 was taken as threshold for statistical significance.
Materials
The chemical structures of FFA and related drugs tested in the present study are displayed in Figure 1. FFA and diclofenac were obtained from Sigma-Aldrich (Bornem, Belgium), whereas N-(p-amylcinnamoyl)anthranilic acid (ACA) and 2-(p-amylcinnamoyl)amino-4-chlorobenzoic acid (ONO-RS-082) were from Biomol (Tebu-Bio, Boechout, Belgium; or Enzo Life Sciences, Zandhoven, Belgium). Oleoyl-acetyl-glycerol (OAG) and nifedipine were also from Biomol. All other drugs or chemicals were from Sigma-Aldrich or Merck (Darmstadt, Germany). FFA, ACA, diclofenac, BEL and bromophenacyl bromide were prepared as stock solutions in DMSO. Nifedipine was prepared in ethanol. Other chemicals were dissolved in water.
Figure 1.
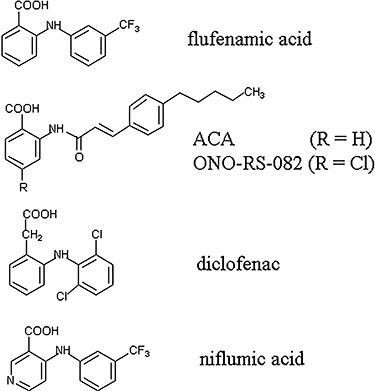
Chemical structures of flufenamic acid and related drugs. Flufenamic (2-{[3-(trifluoromethyl)phenyl]amino}benzoic) acid is a typical fenamate, containing a phenyl ring attached, via an amino group, to benzoic acid (the aminobenzoic acid group is also called anthranilic acid). ACA (N-(p-amylcinnamoyl)anthranilic acid and ONO-RS-082 (2-(p-amylcinnamoyl)amino-4-chloro benzoic acid) contain the aminobenzoic acid moiety but with a cinnamoyl, instead of a phenyl group attached to the amino group. The two drugs differ by the substituents (H vs. Cl) in position 4 of the benzoic moiety. Diclofenac (2-[2-(2,6-dichlorophenyl)aminophenyl]acetic acid) differs from flufenamic acid by having a phenylacetyl instead of the benzoyl (phenylcarboxyl) group, and by the substitutions in the second phenyl group. Niflumic acid (2-{[3-(trifluoromethyl)phenyl]amino}nicotinic acid) is similar to flufenamic acid but its benzoic group is replaced by a nicotinic group.
Results
Induction of a current by the fenamate FFA and by related compounds
Experiments were carried out on myocytes intracellularly dialysed with solutions containing Mg2+ (0.8–4.6 mM free ion concentration) to prevent the activation of the TRPM7 current (Zakharov et al., 2003; Gwanyanya et al., 2004; Demeuse et al., 2006). Under such conditions, baseline currents remained stable over time, that is, there was no increase of outward current with time, in contrast to what is observed when dialysing cells with low-Mg2+ or Mg2+-free solutions (Zakharov et al., 2003). The effect of applying the fenamate, FFA or the related compound ACA, on such cells is illustrated in Figure 2. In Figure 2A membrane current tracings obtained using a voltage ramp between −120 mV and +80 mV in control conditions, during application of 100 µM FFA and after drug washout are displayed. The drug caused an inward shift of the current–voltage relation at potentials below +38 mV and an outward shift at more positive potentials. The time evolution of inward currents measured at −120 mV and of outward currents measured at +80 mV in the same cell is illustrated in Figure 2B. Currents were stable before drug application but progressively increased during treatment with FFA. Figure 2C displays tracings obtained in another cell in control conditions, during application of 50 µM ACA and after drug washout, whereas Figure 2D shows the corresponding time evolution of currents measured at −120 mV and +80 mV during two repetitive ACA applications. The effects of ACA were qualitatively similar to those of FFA: inward current shift at potentials below +36 mV and outward shift at more positive potentials. The drug-induced current (not illustrated), defined as the difference current obtained by subtracting the tracing in basal conditions from the one in the presence of the drug, showed a reversal potential (Erev) of +38 ± 2.8 mV (n = 9) for FFA and +38 ± 1.2 mV (n = 32) for ACA.
Figure 2.
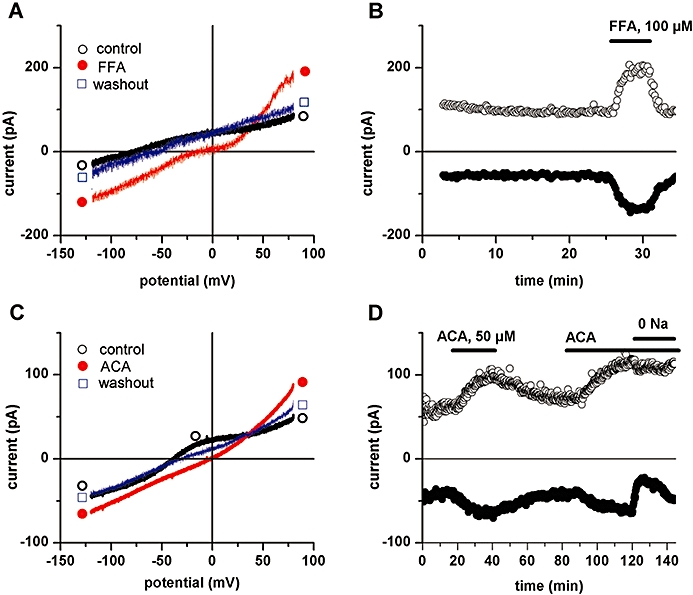
Flufenamic acid (FFA)- and N-(p-amylcinnamoyl)anthranilic acid (ACA)-induced current in ventricular myocytes. A, C: Effects of extracellular application of FFA (100 µM; A) or ACA (50 µM; C) on whole-cell membrane currents. Traces of current–voltage relationships obtained using voltage ramps from +80 mV to −120 mV, under control extracellular conditions (○), in the presence of either drug (•) and after drug washout (□). B, D: Time course of whole-cell currents measured at +80 mV and −120 mV in the same cells as in A and C respectively. The period of superfusion with drug is indicated by horizontal bar.
The effects of FFA or ACA developed progressively with time and reached steady state usually over several (>5) minutes. The effects of FFA were readily reversible upon drug washout (Figure 2B), as were those following short exposures of ACA (Figure 2D). In six cells where sufficient washout could be carried out following 30–50 µM ACA application, the currents induced at −120 mV was −0.5 ± 0.14 pA/pF when measured relative to baseline currents (before drug application), and −0.4 ± 0.11 pA/pF relative to the post drug washout level. There was no statistically significant difference between these induced currents (P = 0.45; t-test), indicating a near-complete reversibility of the drug effects in these cells, and that, under these conditions, leak currents had minimal effects on the drug-induced current. After washout, the initial effect was reproducible upon repeated drug application (see Figure 2D). However, ACA effects were difficult to wash out following prolonged exposure or high (>50 µM) concentrations (see Figure 3E).
Figure 3.
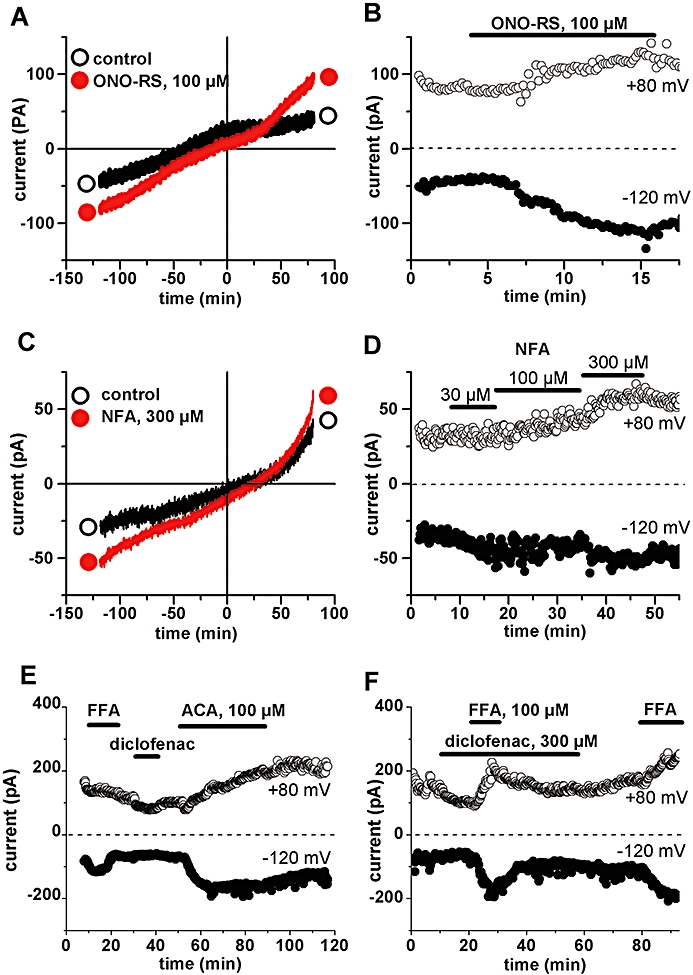
Effect of ONO-RS-082 and niflumic acid (NFA), but lack of effect of diclofenac in inducing current in ventricular myocytes. A, C: Traces of current–voltage relationships obtained using voltage ramps from +80 mV to −120 mV, under control extracellular conditions (○) and in the presence of either drug (•): 100 µM ONO-RS-082 (A) or 300 µM NFA (C) respectively. B, D: Time course of whole-cell currents measured at +80 mV and −120 mV in the same cell as in A and C respectively. The period of superfusion with drugs is indicated by horizontal bar. E, F: Failure of diclofenac to increase currents or to prevent the effect of flufenamic acid (FFA). ACA, N-(p-amylcinnamoyl)anthranilic acid.
We also tested the effects of the following related drugs: (i) ONO-RS-082, which, except for a Cl attached to the benzoic ring, is structurally identical to ACA (see Figure 1) and, like ACA, is known to inhibit secretory phospholipase A2 (PLA2) (Konrad et al., 1992); (ii) niflumic acid (NFA), which, like FFA contains a trifluoro-phenyl group but with the benzoic group replaced by a nicotinic group; and (iii) the anti-inflammatory drug diclofenac, in which the phenyl-carboxyl forming the aminobenzoic moiety has been replaced by a phenyl-acetyl (see Figure 1). Like ACA, ONO-RS-082 was able to increase membrane currents (Erev = +38 ± 4.4 mV, n = 3), but a concentration of at least 100 µM was needed for the effect (Figure 3A and 3B). Similarly NFA induced a small current (n = 4) at 300 µM (Figure 3C and 3D). In contrast, diclofenac at 100 µM (n = 6) or 300 µM (n = 4; inward current increase of −0.03 ± 0.02 pA/pF at −120 mV) was unable to induce any significant current change, even under conditions where FFA and ACA could be shown to be effective (Figure 3E and 3F). The drug also did not appear to act as an antagonist towards the other drugs as its prior application at 300 µM did not prevent the effect of 100 µM FFA applied in addition (Figure 3F).
All cells that were challenged either with FFA (≥100 µM), ACA (≥3 µM) or ONO-RS-082 (≥100 µM) responded with an increase in currents. Only two out of four cells responded to 300 µM NFA. The effects were concentration-dependent. The concentration-effect curve for ACA showed a half-maximum effective concentration (K0.5) of 24 µM, with a Hill coefficient of 1.5 (Figure 4, n = 3–23), and the relative change (ID/Imax) was similar for induced inward and outward currents (not illustrated). At −120 mV, the drug-induced current in pig cells was −0.4 ± 0.04 pA/pF for 30 µM ACA (n = 23), −0.7 ± 0.13 pA/pF for 300 µM FFA (n = 4), −0.3 ± 0.10 pA/pF for 100 µM ONO-RS-082 (n = 3) and −0.3 ± 0.07 pA/pF for 300 µM NFA (n = 4). ACA, which appeared to be the most potent of all drugs tested, was used for most subsequent experiments.
Figure 4.
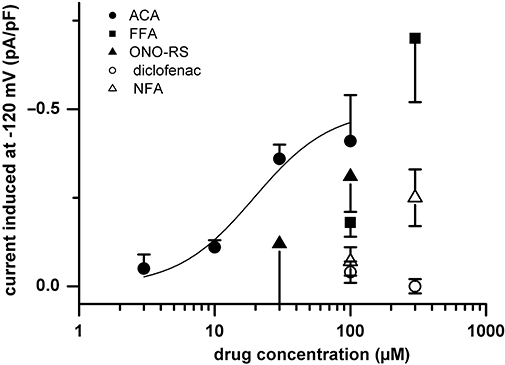
Concentration dependence of the effect of N-(p-amylcinnamoyl)anthranilic acid (ACA). Pooled data (n = 3–23) on the effects of different concentrations of the drugs tested. Data points for ACA are fitted by the Hill equation (see Methods), with Vmax = −0.50 pA/pF, K0.5 = 24 µM; and nHill = 1.5 (continuous curve). Data for the other drugs were not fitted because only two concentrations of each were tested. FFA, flufenamic acid; NFA, niflumic acid.
Whereas the above and subsequent data describe the results obtained in pig cells, similar effects could be obtained in sheep ventricular and atrial myocytes. Figure 5 shows an example from a sheep left ventricular myocyte, and further illustrates that the ACA-induced current was time-independent during a voltage step.
Figure 5.
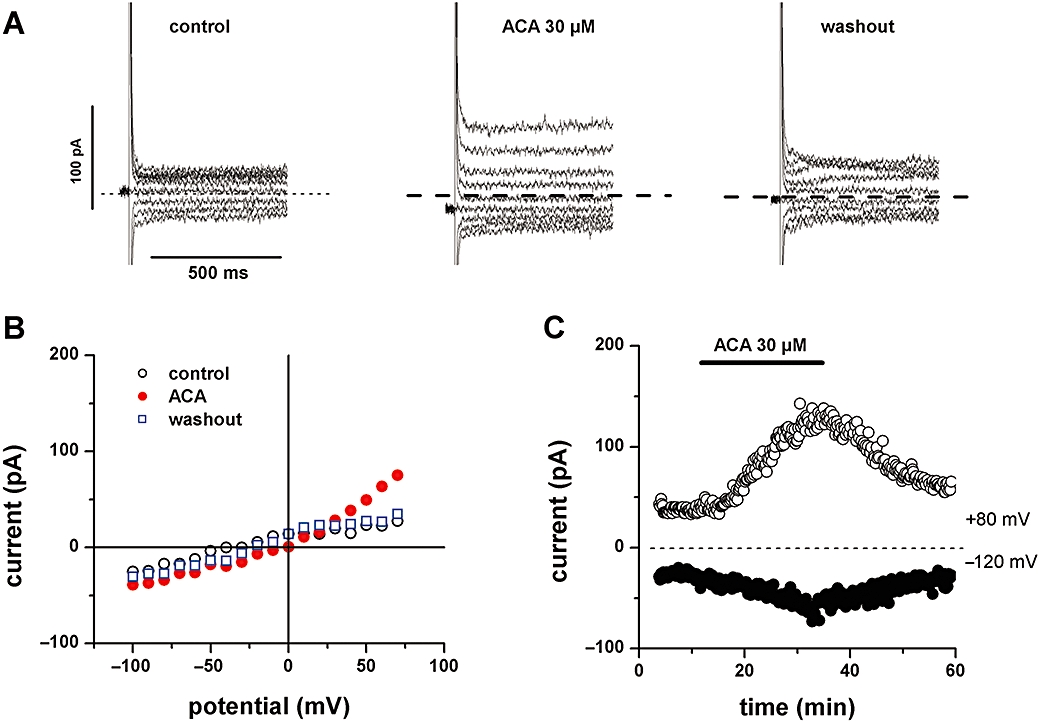
N-(p-amylcinnamoyl)anthranilic acid (ACA)-induced current in sheep ventricular myocytes. A: Traces of currents obtained using voltage steps between −100 mV and +70 mV, with 10 mV increments (but displayed with 20 mV increments), under control conditions (left), in the presence of 30 µM ACA (middle), and following drug washout (right). B: Current–voltage relationships obtained using end-of-pulse currents in A: under control conditions (○), in the presence of ACA (•) and after drug washout (□). C: Time course of currents at +80 mV and −120 mV obtained using voltage ramps in the same cell as in the other panels. The period of superfusion with ACA is indicated by horizontal bar.
Na+ as charge carrier of the inward current
The magnitude of the ACA-induced current or its Erev were not changed upon lowering extracellular Cl- concentrations by substituting the anion with methylsulfate (not illustrated). Given our experimental conditions, where Na+ is the ion with equilibrium potential (ENa: +67 mV) closest to the above Erev values, it is very likely that Na+ carries the current induced by ACA. Alternatively, a combination of divalent and monovalent cations could be involved. We therefore tested the effect of ACA in cells superfused with Na+-free solutions. In the absence of extracellular Na+, the inward component but not the outward component of the ACA-induced current was markedly decreased (Figure 6A and 6B) and could be restored with Na+ re-admission (Figure 6A and 6C; see also Figure 2D and summary data in Figure 6F). In addition, during drug application the inward current in the absence of extracellular Na+ and Ca2+ was not modified by re-admitting 1.8 mM Ca2+ (Figure 6D). Similarly, the ACA effect in the presence of extracellular Na+ was not modified by removing external both Ca2+ and Mg2+ (n = 3, not illustrated). Inward current could still be induced in the absence of extracellular Na+, Ca2+ and Mg2+ (n = 6, Figure 6F), indicating permeability to either Cs+ or NMDG+. These results suggest that the inward component of the ACA-induced current was largely carried by Na+ with no or little contribution of the divalent cations at their physiological concentrations. The outward component of the ACA-induced current could be due to intracellular Na+ or Cs+. In cells dialysed with Na+-free intracellular solutions (n = 4), outward currents could still be induced by ACA (Figure 6E), making it likely that it was carried by Cs+, the only remaining major intracellular cation under our experimental conditions. We noticed that the outward component of the ACA-induced current was very small or absent in cells where 25 mM TEA-Cl was included in the pipette solution, suggesting an apparent block by internal TEA+, but this was not further investigated.
Figure 6.
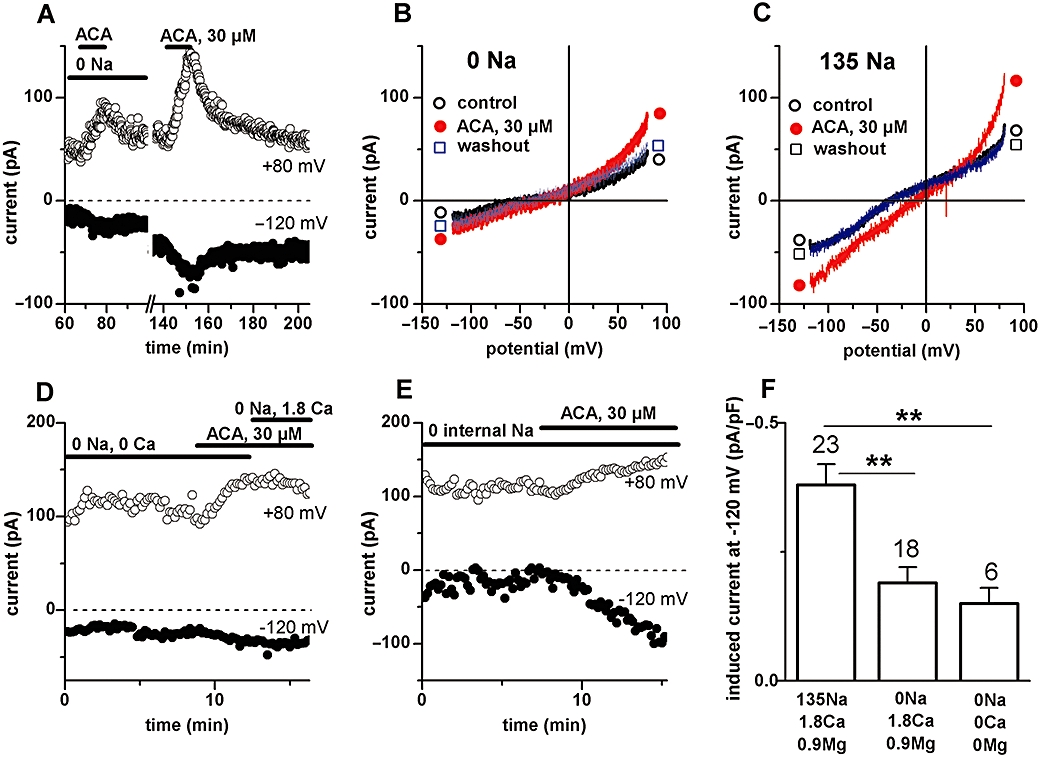
Cation permeability of the N-(p-amylcinnamoyl)anthranilic acid (ACA)-induced conductance. A, B, C: Effects of ACA (30 µM) during superfusion with Na+-free (Na+o replaced by NMDG+o) or Na+-containing extracellular solutions. Data from one same single cell. A: Time course of current at +80 mV and −120 mV. B, C: Current–voltage relationships under basal condition (○), in the presence of ACA (•) and after drug washout (□), during superfusion with Na+-free extracellular solution (B) or during superfusion with standard (Na+-containing) extracellular solution (C). D: Failure of Ca2+ to increase inward current in the presence of ACA (30 µM) in a cell superfused with Na+-free extracellular solution. E: Failure of removal of intracellular Na+ to suppress the outward current induced by ACA (30 µM) at positive potentials. F: Pooled data of ACA-induced currents measured at −120 mV in the presence of different types of extracellular solutions. **P < 0.01 versus control; anova with post hoc test.
Failure of diacylglycerol to activate the current
Fenamates have been shown to potentiate α-adrenoceptor-mediated induction of a cation current in smooth muscle (Yamada et al., 1996). In this preparation, the fenamate-induced currents are carried by divalent cations and are likely to be due to the activation of TRPC6 channels by diacylglycerol (DAG) (Jung et al., 2002). Despite the difference in charge carrier compared with the currents described here in cardiac cells, we examined the possibility that DAG-sensitive channels underlie the ACA-induced current in our preparation. On testing whether exogenous application of the DAG analogue 1-oleoyl-2-acetyl-sn-glycerol (OAG) could produce effects like those of ACA, we found that 100 µM OAG failed to induce any current, in four cells (Figure 7A). In addition, we examined whether endogenous DAG putatively released upon receptor activation by muscarinic and adrenoceptor agonists could enhance the current induced by ACA. No effect was obtained in seven cells with carbachol (100 µM) or phenylephrine (40 µM) given either separately or together in addition to ACA (Figure 7B).
Figure 7.
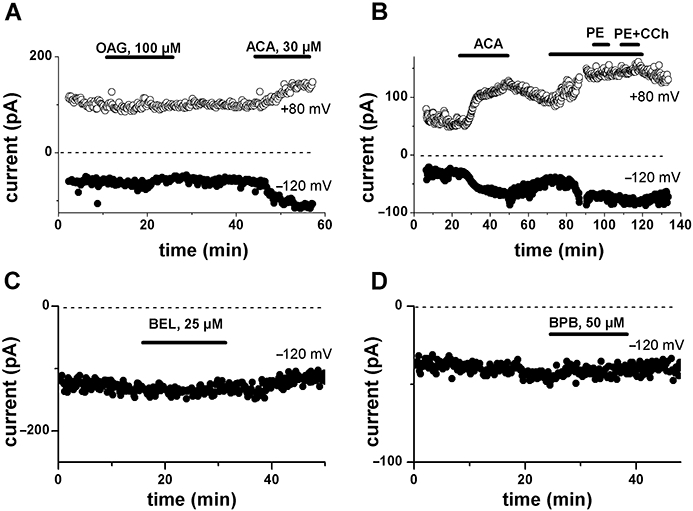
Lack of involvement of diacylglycerol or phospholipase A2. A: Lack of effect of extracellularly applied oleoyl acetyl glycerol (OAG; 100 µM) on membrane currents measured at +80 mV and −120 mV using voltage ramps. B: Lack of effect of α-adrenoceptor (phenylephrine, PE; 40 µM) and muscarinic (carbachol, CCh; 100 µM) agonists on the current induced by N-(p-amylcinnamoyl)anthranilic acid (ACA). C, D: Lack of effect of extracellular application of bromoenol lactone (BEL; 25 µM; C) or bromophenacyl bromide (BPB; 50 µM; D) on membrane currents measured at −120 mV. Periods of drug application are indicated by horizontal bars.
As various aminobenzoic derivatives, including ACA and ONO-RS-082, inhibit PLA2, we also examined whether their effect could eventually be due to the modification of some tonic action of PLA2 catalysis products. We therefore tested the effect of two other but chemically unrelated PLA2 inhibitors. Figure 7C and 7D show that neither the selective inhibitor of Ca2+-independent PLA2, BEL (also known as haloenol lactone suicide substrate or HELSS; 25 µM; n = 5) nor the non-specific PLA2 inhibitor bromophenacyl bromide (50 µM; n = 5) had any effect on membrane currents. These data exclude a mechanism operating via PLA2 as the basis for the change in membrane currents by ACA.
No influence of intracellular Ca2+-buffering. Modulation by ion channel blockers
Highly buffering intracellular Ca2+ and Mg2+ (10 mM EDTA, n = 9) did not prevent ACA from inducing an inward current on top of the TRPM7 current present under these conditions (not illustrated). Finally, the current induced by ACA was completely blocked by 100 µM Gd3+ (n = 3), but was found to be insensitive to 50–100 µM 2-aminoethoxydiphenyl borate (2-APB; n = 3) and 10 µM amiloride (n = 4) (not illustrated).
In preliminary experiments examining whether the ACA effect was modified by pH, lowering pH to 6.6 did not decrease, but was rather associated with an increase of the ACA-induced current compared with the effect at normal pH (ACA-induced current at −120 mV: −0.78 ± 0.24 pA/pF at pH 6.6 vs. −0.34 ± 0.12 pA/pF at pH 7.4 in the same cells; P = 0.11; paired t-test; n = 3). No current change was obtained in the absence of ACA (n = 4).
Effect on action potential
An isolated effect of ACA, FFA and related drugs on membrane potentials, due to the induced current described above, is difficult to investigate because the drugs affect other ion channels that normally contribute to the action potential, including voltage-dependent channels, Ca2+-activated Cl- currents and delayed rectifier K+ currents (see Introduction). Figure 8A shows that a prominent effect of ACA was a decrease in the peak amplitude of the action potential, measured under ruptured-patch conditions, due to a lowering of the overshoot potential, an effect likely due to a block of voltage-dependent Na+ channels (our unpublished results). At low (≤30 µM) ACA concentration, this effect occurred without a depolarization of the resting potential and was associated with a decrease of the action potential duration. As the ACA concentration was increased (≥30 µM), there was a further decrease in the overshoot potential, and in addition the resting membrane was depolarized and the action potential lengthened relative to its duration with lower concentrations (Figure 8A; see summary data in Figure 8B and 8C). In some cells, this later lengthening could bring the action potential to a longer duration compared with control. Because diclofenac was unable to induce the novel current while sharing with ACA the effects on many other channels underlying the action potential (our unpublished data), its effects served as reference for comparison with those of ACA. Like ACA, diclofenac decreased the overshoot potential and the total duration of the action potential (Figure 8D). However, in contrast to ACA, the drug was unable to cause the secondary action potential lengthening (Figure 8D; see summary data in Figure 8E) or depolarize the cells (Figure 8D; see Figure 8F). Similar results were obtained when applying the drugs while measuring under perforated-patch conditions (not illustrated). These results indicate that the latter two effects can, to some extent, be attributable to the novel current induced by ACA.
Figure 8.
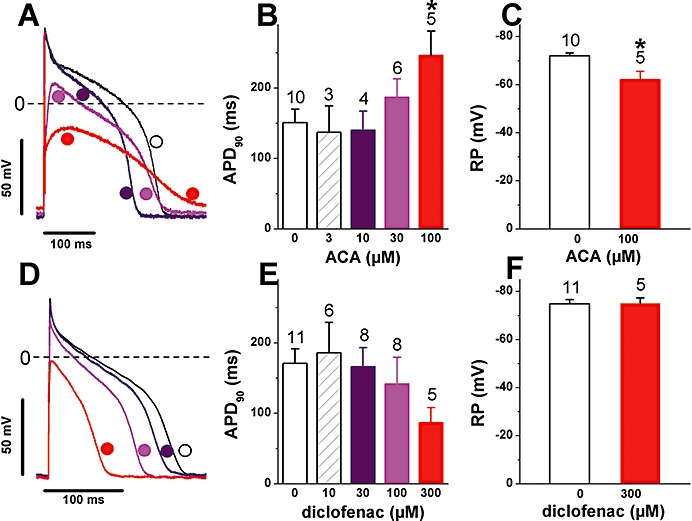
N-(p-amylcinnamoyl)anthranilic acid (ACA) and diclofenac effects on the action potential. A, D: Superimposed action potentials recorded before and during application of increasing concentrations of ACA (A) or of diclofenac (D). The symbol and trace colours in A and D correspond to the column colours for the same concentrations in B and E respectively. The horizontal dashed lines indicate the 0 mV level. B, E: Action potential duration measured at 90% repolarization (APD90) in presence of various concentrations of ACA (B) or of diclofenac (E). (*P < 0.05 for ACA vs. control; anova with post-test) C, F: Effect of 100 µM ACA (C) or 300 µM diclofenac (F) on the resting membrane potential (*P < 0.05 for ACA vs. control). Notice the depolarization of the resting membrane and the relative lengthening of the action potential as the concentration of ACA but not that of diclofenac is raised. Number of cells for each drug concentration indicated above the columns. Stimulation at 1 Hz.
Discussion
Non-selective cation channels in cardiac cells
A large variety of non-selective cation channels have been detected in cardiac cells (e.g. Colquhoun et al., 1981; Matsumoto and Pappano, 1989; Kim and Fu, 1993; Ruknudin et al., 1993; Tarr et al., 1994; Parker and Scarpa, 1995; Zhang and Hancox, 2003; Gwanyanya et al., 2004; 2006; Guinamard et al., 2006a; Youm et al., 2006). One important issue concerning these channels is to identify the underlying proteins. For some of them, the underlying proteins are now known: HCN proteins for the pacemaker channel (Moroni et al., 2001; Robinson and Siegelbaum, 2003), TRPM4 or TRPM5 proteins for the Ca2+-activated Na+-conducting channels (Guinamard et al., 2006b; Guinamard and Bois, 2007) and connexins for the gap junction channels (John et al., 1999; Kondo et al., 2000). Recent data suggest that TRP proteins may also underlie many of the heretofore unidentified non-selective cation channels. For example, cardiac stretch-activated channels are proposed to be formed by TRPC1 (Maroto et al., 2005; Watanabe et al., 2008; Dyachenko et al., 2009), purinergic receptor-activated channels by TRPC6 (Alvarez et al., 2008) and the cardiac Mg2+-sensitive, PIP2-dependent channel by TRPM7 (Gwanyanya et al., 2006). However, for many other channels, the constituent proteins remain unknown.
The other issue concerns the role of the various non-selective channels. Except for an involvement of If (HCN) and eventually Ca2+-activated TRPM4/5 channels (Demion et al., 2007) in pacemaking, the other channels are supposed to induce electrophysiological or ion concentration changes associated with pathophysiological processes such as arrhythmogenesis, myocardial hypertrophy or heart failure.
Novel channel activated by FFA and related drugs
In the present study we describe a current, induced by FFA, ACA and related drugs, that displays various characteristics, of which some are in many respects similar to those of already known channels, while others are different. One first and major characteristic is the activation by FFA that distinguishes it from other cation channels, generally inhibited by fenamates. Other exceptions to this inhibition by fenamates are HCN channels, purinergic receptor-activated channels and insulin-activated channels. The second major characteristic of the conductance pathway identified in the present study is its permeability profile. The pathway displays high permeability to Na+ (PNa), as indicated by the value of the reversal potential (Erev), close to ENa, and by the large suppression of the inward component of the induced-current following substitution of external Na+ by NMDG+. Cs+ may also permeate the conductance, as indicated by the incomplete suppression of inward currents in cells superfused with Na+-free solutions, and the persistence of outward currents in cells dialysed with Na+-free internal solution. However, in order to account for the positive Erev, PNa has to be larger than that of the other monovalent cations present (PNa > PCs). Based on the Erev value of +35 mV obtained in cells superfused with Cs+-free extracellular solution and dialysed with Na+-free pipette solution, and assuming no permeation of NMDG+, we estimate PNa/PCs to be about 3.6. In cardiac cells, except for HCN and TRPM4/5 channels, other non-selective channels are usually relatively less permeable to Na+. HCN channels, which display substantial Na+ permeability, are voltage-dependent and are blocked by Cs+ in contrast to the current described in the present study. TRPM4/5 channels are highly Na+-selective (Guinamard et al., 2006a); however, they are activated by intracellular Ca2+, in contrast to the conductance described in the present study, which was present in cells dialysed with high EDTA concentrations. The possibility cannot be totally excluded that drugs could modify Ca2+-dependent channels and remove the requirement for Ca2+i. The involvement of TRPM4/5 channels is made more unlikely by the fact that, instead of being stimulated, they are known to be blocked by FFA (Guinamard et al., 2006b).
The ACA-induced channel does not appear to display marked permeability for divalent cations at physiological concentrations as the current was not affected by Ca2+ concentration changes, even when effected in Na+-free solution. This lack of significant permeability to Ca2+ makes it unlikely that the current is due to channels with substantial PCa relative to PNa, including purinergic receptor-activated channels (Alvarez et al., 2008) and insulin-activated channels (Zhang and Hancox, 2003). The latter conductance also shows equal PNa and PCs and can be activated by OAG, in contrast to the conductance described in the present study, which shows less PCs and is not activated by OAG. TRPC6 and TRPA1, which are activated by fenamates, as well as TRPC3 and TRPC7, which are also not inhibited and may eventually display some activation by FFA (Jung et al., 2002; Foster et al., 2009), are all candidates for the current described in the present study. However, their reported permeability profile, PCa ≫ PNa, is in apparent contrast with our findings in cardiac cells. In addition, TRPC6 is activated by DAG and, so far, it is uncertain whether TRPA1 is expressed in cardiac myocytes.
The fact that the current was induced in cells dialysed with Mg2+i concentrations known to inhibit TRPM7 channels, suggests that these proteins were not involved in the fenamate action. Additional features allowing the differentiation from TRPM7 channels (Mubagwa et al., 1997; Zakharov et al., 1999; 2003; Gwanyanya et al., 2004) include: the substantial ACA-induced inward current in the presence of extracellular divalent cations, the insensitivity of the induced current to the removal of extracellular divalent cations and the fact that the current amplitude was not decreased by acidification of the perfusing solution as observed for TRPM7 channels (Gwanyanya et al., 2004; Jiang et al., 2005).
Although we did not test the sensitivity of the current to tetrodotoxin it is unlikely that the effect of FFA and related drugs was due to a persistent current involving voltage-dependent Na+ (NaV) channels (Saint et al., 1992), given the linear current–voltage relationship and the fact that current could be obtained at potentials as negative as −120 mV without prior activation by depolarization.
Finally, the relationship to the background inward Na+ current induced by muscarinic receptor activation in cardiac myocytes, in an apparently G-protein independent way (Matsumoto and Pappano, 1989; Shirayama et al., 1993), is unknown. Similar Na+-permeable conductances are activated by neurotransmitters or peptides in a G-protein-independent way in other cell types, including neuronal (e.g. Guerineau et al., 1995) and pancreatic cells (e.g. Rolland et al., 2002). These latter conductances are due to a tetrodotoxin-insensitive, voltage-independent Na+ leak (also called NALCN or NaVi) channel (Lu et al., 2007; Swayne et al., 2009). NALCN is expressed in cardiac cells (Swayne et al., 2009) and is therefore another candidate channel protein underlying the current described here. In our experiments, we failed to induce any current by carbachol, and the agonist did not change the magnitude of the current induced by ACA. Clearly, more studies, eventually using cells from knock out animals, are needed to identify the protein underlying the current described here.
Role of the novel current
Besides the molecular identity, the physiological activators and the role of this channel remain unknown. When activated, the channel will cause changes in resting and action potentials. Our data show that a depolarization of the resting membrane and a lengthening of the action potential duration are caused by ACA, which is able to induce the new current, but not by diclofenac, which was unable to activate the current. Using an action potential simulation model (Clancy and Rudy, 2002), the addition of a current similar to the one measured in the myocytes (with Erev of +35 mV) was also able to cause resting membrane depolarization and action potential duration lengthening (not illustrated). Increasing the magnitude of the current many-fold compared with the value recorded in our measurements was able to cause early delayed afterdepolarizations. In addition to the electrophysiological effects, the current may be implicated in intracellular Na+ and subsequent Ca2+ overloads associated with disease processes.
When used clinically, FFA is reported to have a plasma concentration of 4–12 µg·mL−1 (Aly et al., 2000), that is, about 42 µM. Although we did not test the effect of FFA concentrations lower than 100 µM, based on the concentration–effect curve, the clinical concentration may be expected to cause a small activation of the novel current. Part of the plasma FFA may be bound to proteins, but the fraction of the bound versus unbound drug is not known. The pathophysiological relevance of the current described here will clearly depend on whether there are alternative mechanisms of activation via receptors or kinases.
In conclusion, the present study shows that the fenamate FFA and related drugs induce a current with novel characteristics in cardiac cells. The action mechanism of these drugs remains unknown. The novel conductance pathway is largely permeable to Na+, but additional studies are needed to identify the underlying channel proteins and to explore its physiological or pathophysiological role.
Acknowledgments
We thank Dr V. Bito, Patricia Holemans and Christel Huysmans for assistance with cell dissociation. This study was supported by Grant G.0634.07 from FWO, the Flemish Foundation for Science. RM was supported by grant SF/08/021 from the Research Council of the University of Leuven. AG was supported by the Belgian Technical Cooperation.
Glossary
Abbreviations
- 2-APB
aminoethoxydiphenylborate
- ACA
N-(p-amylcinnamoyl)anthranilic acid
- APD
action potential duration
- BEL
bromoenol lactone
- BPB
bromophenacyl bromide
- CCh
carbachol
- DAG
diacylglycerol
- Erev
reversal potential
- FFA
flufenamic acid
- HCN
hyperpolarization-activated channel
- NALCN
sodium leak channel
- NFA
niflumic acid
- NMDG
N-methyl-D-glucamine
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- PE
phenylephrine
- PLA2
phospholipase A2
- TEA
tetraethylammonium
- TRP
transient receptor potential
- TRPA
transient receptor potential, ankyrin
- TRPC
transient receptor potential, canonical
- TRPM
transient receptor potential, melastatin
- TRPP
transient receptor potential, polycystin
Conflicts of interest
All authors declare that they have no conflict of interest.
Supplemental material
Supporting Information: Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez J, Coulombe A, Cazorla OF, Ugur M, Rauzier JM, Magyar J, et al. ATP/UTP activate cation-permeable channels with TRPC3/7 properties in rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;285:H21–H28. doi: 10.1152/ajpheart.00135.2008. [DOI] [PubMed] [Google Scholar]
- Aly FA, Al-Tamimi SA, Alwarthan AA. Determination of flufenamic acid and mefenamic acid in pharmaceutical preparations and biological fluids using flow injection analysis with tris(2,2′-bipyridyl)ruthenium(II) chemiluminescence detection. Anal Chim Acta. 2000;416:87–96. [Google Scholar]
- Clancy CE, Rudy Y. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation. 2002;105:1208–1213. doi: 10.1161/hc1002.105183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Neher E, Reuter H, Stevens CF. Inward current channels activated by intracellular Ca in cultured cardiac cells. Nature. 1981;294:752–754. doi: 10.1038/294752a0. [DOI] [PubMed] [Google Scholar]
- Demeuse P, Penner R, Fleig A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127:421–434. doi: 10.1085/jgp.200509410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc Res. 2007;73:531–538. doi: 10.1016/j.cardiores.2006.11.023. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D. Characterization of single pacemaker channels in cardiac sino-atrial node cells. Nature. 1986;324:470–473. doi: 10.1038/324470a0. [DOI] [PubMed] [Google Scholar]
- Dyachenko V, Husse B, Rueckschloss U, Isenberg G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium. 2009;45:38–54. doi: 10.1016/j.ceca.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Ehara T, Noma A, Ono K. Calcium-activated non-selective cation channel in ventricular cells isolated from adult guinea-pig hearts. J Physiol. 1988;403:117–133. doi: 10.1113/jphysiol.1988.sp017242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster RR, Zadeh MA, Welsh GI, Satchell SC, Ye Y, Mathieson PW, et al. Flufenamic acid is a tool for investigating TRPC6-mediated calcium signalling in human conditionally immortalised podocytes and HEK293 cells. Cell Calcium. 2009;45:384–390. doi: 10.1016/j.ceca.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Guerineau NC, Bossu JL, Gahwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R, Bois P. Involvement of transient receptor potential proteins in cardiac hypertrophy. Biochim Biophys Acta. 2007;1772:885–894. doi: 10.1016/j.bbadis.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Chatelier A, Lenfant J, Bois P. Activation of the Ca-activated nonselective cation channel by diacylglycerol analogues in rat cardiomyocytes. J Cardiovasc Electrophysiol. 2004;15:342–348. doi: 10.1046/j.1540-8167.2004.03477.x. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Demion M, Chatelier A, Bois P. Calcium-activated nonselective cation channels in mammalian cardiomyocytes. Trends Cardiovasc Med. 2006a;16:245–250. doi: 10.1016/j.tcm.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Demion M, Magaud C, Potreau D, Bois P. Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension. 2006b;48:587–594. doi: 10.1161/01.HYP.0000237864.65019.a5. [DOI] [PubMed] [Google Scholar]
- Gwanyanya A, Amuzescu B, Zakharov SI, Macianskiene R, Sipido KR, Bolotina VM, et al. Magnesium-inhibited, TRPM6/7-like channel in cardiac myocytes: permeation of divalent cations and pH-mediated regulation. J Physiol. 2004;559:761–776. doi: 10.1113/jphysiol.2004.067637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwanyanya A, Sipido K, Vereecke J, Mubagwa K. ATP- and PIP2-dependence of the magnesium-inhibited, TRPM7-like cation channel in cardiac myocytes. Am J Physiol Cell Physiol. 2006;291:C627–C635. doi: 10.1152/ajpcell.00074.2006. [DOI] [PubMed] [Google Scholar]
- Harteneck C, Frenzel H, Kraft R. N-(p-amylcinnamoyl)anthranilic acid (ACA): a phospholipase A2 inhibitor and TRP channel blocker. Cardiovasc Drug Rev. 2007;25:61–75. doi: 10.1111/j.1527-3466.2007.00005.x. [DOI] [PubMed] [Google Scholar]
- Hu H, Tian J, Zhu Y, Wang C, Xiao R, Herz JM, et al. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Arch. 2010;459:579–592. doi: 10.1007/s00424-009-0749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Li M, Yue L. Potentiation of TRPM7 inward currents by protons. J Gen Physiol. 2005;126:137–150. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SA, Kondo R, Wang SY, Goldhaber JI, Weiss JN. Connexin-43 hemichannels opened by metabolic inhibition. J Biol Chem. 1999;274:236–240. doi: 10.1074/jbc.274.1.236. [DOI] [PubMed] [Google Scholar]
- Jung S, Strotmann R, Schultz G, Plant TD. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- Kim D, Fu C. Activation of a nonselective cation channel by swelling in atrial cells. J Membr Biol. 1993;135:27–37. doi: 10.1007/BF00234649. [DOI] [PubMed] [Google Scholar]
- Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol. 2000;32:1859–1872. doi: 10.1006/jmcc.2000.1220. [DOI] [PubMed] [Google Scholar]
- Konrad RJ, Jolly YC, Major C, Wolf BA. Inhibition of phospholipase A2 and insulin secretion in pancreatic islets. Biochim Biophys Acta. 1992;1135:215–220. doi: 10.1016/0167-4889(92)90139-3. [DOI] [PubMed] [Google Scholar]
- Kraft R, Harteneck C. The mammalian melastatin-related transient receptor potential cation channels: an overview. Pflugers Arch. 2005;451:204–211. doi: 10.1007/s00424-005-1428-0. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LY, Fei XW, Li ZM, Zhang ZH, Mei YA. Diclofenac, a nonsteroidal anti-inflammatory drug, activates the transient outward K+ current in rat cerebellar granule cells. Neuropharmacology. 2005;48:918–926. doi: 10.1016/j.neuropharm.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129:371–383. doi: 10.1016/j.cell.2007.02.041. [DOI] [PubMed] [Google Scholar]
- Macianskiene R, Moccia F, Sipido KR, Flameng W, Mubagwa K. Channels involved in transient currents unmasked by removal of extracellular calcium in cardiac cells. Am J Physiol Heart Circ Physiol. 2002;282:H1879–H1888. doi: 10.1152/ajpheart.00952.2001. [DOI] [PubMed] [Google Scholar]
- Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol. 2005;7:179–185. doi: 10.1038/ncb1218. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Pappano AJ. Sodium-dependent membrane current induced by carbachol in single guinea-pig ventricular myocytes. J Physiol. 1989;415:487–502. doi: 10.1113/jphysiol.1989.sp017733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni A, Gorza L, Beltrame M, Gravante B, Vaccari T, Bianchi ME, et al. Hyperpolarization-activated cyclic nucleotide-gated channel 1 is a molecular determinant of the cardiac pacemaker current If. J Biol Chem. 2001;276:29233–29241. doi: 10.1074/jbc.M100830200. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Stengl M, Flameng W. Extracellular divalent cations block a cation non-selective conductance unrelated to calcium channels in rat cardiac muscle. J Physiol (Lond) 1997;502:235–247. doi: 10.1111/j.1469-7793.1997.235bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B. TRP channels in disease. Biochim Biophys Acta. 2007;1772:805–812. doi: 10.1016/j.bbadis.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, et al. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor AM, Randolph KM. Inhibition of the Na+/dicarboxylate cotransporter by anthranilic acid derivatives. Mol Pharmacol. 2007;72:1330–1336. doi: 10.1124/mol.107.035352. [DOI] [PubMed] [Google Scholar]
- Parker KE, Scarpa A. An ATP-activated nonselective cation channel in guinea pig ventricular myocytes. Am J Physiol. 1995;269:H789–H797. doi: 10.1152/ajpheart.1995.269.3.H789. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Rolland JF, Henquin JC, Gilon P. G protein-independent activation of an inward Na+ current by muscarinic receptors in mouse pancreatic beta-cells. J Biol Chem. 2002;277:38373–38380. doi: 10.1074/jbc.M203888200. [DOI] [PubMed] [Google Scholar]
- Ruknudin A, Sachs F, Bustamante JO. Stretch-activated ion channels in tissue-cultured chick heart. Am J Physiol. 1993;264:H960–H972. doi: 10.1152/ajpheart.1993.264.3.H960. [DOI] [PubMed] [Google Scholar]
- Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am J Physiol Cell Physiol. 2008;294:C833–C841. doi: 10.1152/ajpcell.00313.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama T, Matsumoto K, Pappano AJ. Carbachol-induced sodium current in guinea pig ventricular myocytes is not regulated by guanine nucleotides. J Pharmacol Exp Ther. 1993;265:641–648. [PubMed] [Google Scholar]
- Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J Biol Chem. 2003;278:11909–11915. doi: 10.1074/jbc.M210878200. [DOI] [PubMed] [Google Scholar]
- Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S, et al. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep. 2009;10:873–880. doi: 10.1038/embor.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr M, Arriaga E, Goertz KK, Valenzeno DP. Properties of cardiac Ileak induced by photosensitizer-generated reactive oxygen. Free Radic Biol Med. 1994;16:477–484. doi: 10.1016/0891-5849(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Volk T, Schwoerer AP, Thiessen S, Schultz JH, Ehmke H. A polycystin-2-like large conductance cation channel in rat left ventricular myocytes. Cardiovasc Res. 2003;58:76–88. doi: 10.1016/s0008-6363(02)00858-1. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol Ther. 2008;118:337–351. doi: 10.1016/j.pharmthera.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Murakami M, Ohba T, Ono K, Ito H. The pathological role of transient receptor potential channels in heart disease. Circ J. 2009;73:419–427. doi: 10.1253/circj.cj-08-1153. [DOI] [PubMed] [Google Scholar]
- Yamada K, Waniishi Y, Inoue R, Ito Y. Fenamates potentiate the alpha 1-adrenoceptor-activated nonselective cation channels in rabbit portal vein smooth muscle. Jpn J Pharmacol. 1996;70:81–84. doi: 10.1254/jjp.70.81. [DOI] [PubMed] [Google Scholar]
- Youm JB, Han J, Kim N, Zhang YH, Kim E, Joo H, et al. Role of stretch-activated channels on the stretch-induced changes of rat atrial myocytes. Prog Biophys Mol Biol. 2006;90:186–206. doi: 10.1016/j.pbiomolbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Zakharov SI, Mongayt DA, Cohen RA, Bolotina VM. Monovalent cation and L-type Ca2+ channels participate in calcium paradox-like phenomenon in rabbit aortic smooth muscle cells. J Physiol (Lond) 1999;514:71–81. doi: 10.1111/j.1469-7793.1999.071af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov SI, Smani T, Leno E, Macianskiene R, Mubagwa K, Bolotina VM. Monovalent cation (MC) current in cardiac and smooth muscle cells: regulation by intracellular Mg2+ and inhibition by polycations. Br J Pharmacol. 2003;138:234–244. doi: 10.1038/sj.bjp.0705074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Hancox JC. A novel, voltage-dependent nonselective cation current activated by insulin in guinea pig isolated ventricular myocytes. Circ Res. 2003;92:765–768. doi: 10.1161/01.RES.0000065920.64121.FC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.