

Abstract
BACKGROUND AND PURPOSE
Hydrogen sulphide (H2S) is a novel neuromodulator. The present study aimed to investigate the protective effect of H2S against cell injury induced by 6-hydroxydopamine (6-OHDA), a selective dopaminergic neurotoxin often used to establish a model of Parkinson's disease for studying the underlying mechanisms of this condition.
EXPERIMENTAL APPROACH
Cell viability in SH-SY5Y cells was measured using MTT assay. Western blot analysis and pharmacological manipulation were employed to study the signalling mechanisms.
KEY RESULTS
Treatment of SH-SY5Y cells with 6-OHDA (50–200 µM) for 12 h decreased cell viability. Exogenous application of NaHS (an H2S donor, 100–1000 µM) or overexpression of cystathionine β-synthase (a predominant enzyme to produce endogenous H2S in SH-SY5Y cells) protected cells against 6-OHDA-induced cell apoptosis and death. Furthermore, NaHS reversed 6-OHDA-induced loss of tyrosine hydroxylase. Western blot analysis showed that NaHS reversed the down-regulation of PKCα, ε and Akt and the up-regulation of PKCδ in 6-OHDA-treated cells. Blockade of PKCα with Gö6976 (2 µM), PKCε with EAVSLKPT (200 µM) or PI3K with LY294002 (20 µM) reduced the protective effects of H2S. However, inhibition of PKCδ with rottlerin (5 µM) failed to affect 6-OHDA-induced cell injury. These data suggest that the protective effects of NaHS mainly resulted from activation of PKCα, ε and PI3K/Akt pathway. In addition, NaHS-induced Akt phosphorylation was significantly attenuated by Gö6976 and EAVSLKPT, suggesting that the activation of Akt by NaHS is PKCα, ε-dependent.
CONCLUSIONS AND IMPLICATIONS
H2S protects SH-SY5Y cells against 6-OHDA-induced cell injury by activating the PKCα, ε/PI3K/Akt pathway.
Keywords: hydrogen sulphide, protein kinase C, apoptosis, Parkinson's disease, neuroprotection
Introduction
Hydrogen sulphide (H2S), the third endogenous gaseous mediator after nitric oxide and carbon monoxide (Wang, 2002), has been reported to be produced in the brain of rat, human, bovine and other mammals (Goodwin et al., 1989; Warenycia et al., 1989; Savage and Gould, 1990). In mammalian cells, endogenous H2S is formed from cysteine by pyridoxal-5′-phosphate-dependent enzymes (cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE)) (Stipanuk and Beck, 1982), and a pyridoxal-5′-phosphate-independent enzyme (3-mercaptopyruvate sulphurtransferase (3MST)) (Shibuya et al., 2009). CBS is mainly expressed in the hippocampus and cerebellum, where it is localized to astrocytes (Enokido et al., 2005; Ichinohe et al., 2005), whereas CSE is expressed in the ileum, portal vein and thoracic aorta (Hosoki et al., 1997). 3MST is mainly located in neurones and produces bound sulphane sulphur more efficiently than CBS in the cells (Shibuya et al., 2009). The expression of endogenous H2S generating enzymes suggests that H2S has important physiological functions.
Physically, H2S potentiates N-methyl-D-aspartate (NMDA) receptors and improves the induction of long-term potentiation (LTP) in the hippocampus, a synaptic model of learning and memory (Abe and Kimura, 1996). In addition, we and others have reported that H2S regulates Ca2+ in astrocytes and microglia, suggesting that H2S is important for the regulation of neuronal activity (Lee et al., 2006; Nagai et al., 2004). Apart from that, H2S also helps to reduce oxidative stress by inducing the production of glutathione, a kind of anti-oxidant, and suppresses oxidative stress in mitochondria (Kimura and Kimura, 2004; Kimura et al., 2010). In addition, our group has recently found that H2S protects astrocytes against oxidative stress induced by hydrogen peroxide (H2O2) by improving the glutamate transporters activities and therefore increases glutathione production as well as inhibits lipid oxidation (Lu et al., 2008; Hu et al., 2010). We also showed that H2S has an anti-inflammatory role in lipopolysaccharide (LPS)-stimulated microglial (Hu et al., 2007). In neuronal cells, we found that H2S is able to protect cells against apoptosis via preservation of mitochondrial function (Hu et al., 2009). All the above evidence suggests that H2S plays an important role in the regulation of central nervous system functions. Hence, alterations in the endogenous H2S concentration might contribute to the pathogenesis of certain neurodegenerative diseases.
Parkinson's disease (PD), the second most common neurodegenerative disease, is a condition called movement disorder characterized by muscle rigidity, tremor and slowing of physical movement. It usually affects the elderly and the age of onset is 60 years (Zhang et al., 2007). It is caused by the progressive loss of dopaminergic neurones in the substantia nigra pars compacta, accompanied by an alteration of dopamine concentration in the striatum (Henning et al., 2008). The aetiology remains unknown although oxidative stress and generation of reactive oxygen spesies (ROS) from both mitochondria impairment and dopamine metabolism might be responsible for its pathogenesis (Tian et al., 2007).
6-Hydroxydopamine (6-OHDA), a neurotoxin which selectively kills dopaminergic neurones, is widely used to induce an experimental model of PD (Fornstedt et al., 1986; Yuan et al., 2008). 6-OHDA, which has a similar molecular structure to that of a dopamine, enters the cells via a dopamine re-uptake transporter (Ljungdahl et al., 1971) and generates intracellular ROS and inhibits mitochondria to activate apoptosis cascades (Blum et al., 2001). The SH-SY5Y cell line possesses many qualities of substantia nigra neurones and is thus suitable for use as an in vitro model to study the death of dopaminergic neurones (Takahashi et al., 1994; Tian et al., 2007).
In this study, we investigated the effects of H2S on 6-OHDA-induced cell injury in SH-SY5Y cells. The involvement of various protein kinase C (PKC) isoforms and Akt signalling pathways were also examined.
Methods
All the drugs/molecular target nomenclature confirms to BJP's Guide to receptors and Channels (Alexander et al., 2008).
Cell culture
The human neuroblastoma cell line, SH-SY5Y, obtained from the American Type Culture Collection (Manassas, VA, USA), was maintained in Dulbecco's modified Eagle's Medium (DMEM) supplemented with 10% foetal bovine serum (FBS) and 0.05 U·mL−1 penicillin and 0.05 mg·mL−1 streptomycin at 37°C in a humidified atmosphere containing 5% CO2/95% air. Cultured medium was changed twice a week during cell growth.
Cell treatment
Cells were plated onto 96-well plates for viability tests or 35 mm dishes and incubated overnight as it grew into 80–90% confluency. Regular medium was replaced with low-serum medium (0.5% FBS/DMEM) immediately before treatment. Note that for LY294002 (LY) treatment, cells were deprived of serum for 4–6 h prior to treatment. After pretreatment with NaHS for 10 min or 1 h, cells were washed twice with PBS solution and incubated in fresh low-serum media with 6-OHDA. Each kinase inhibitor was added 30 min before NaHS treatment. Cells were incubated for 12 h after each exposure to toxins, and cell viability was measured using the MTT assay system.
Cell viability assay
Cell viability was measured using the MTT reduction assay as described previously (Hu et al., 2009). At the end of each treatment, MTT was added to each well at a final concentration of 0.5 mg·mL−1 and the cells were further incubated at 37°C for 4 h. Then, the insoluble formazan was dissolved in dimethyl sulphoxide (DMSO). Colorimetric determination of MTT reduction was measured at 570 nm with a reference wavelength at 630 nm.
Cell fractionation for determining PKC isoform translocation
Protein kinase C isoform translocation was detected with the cell fractionation method as described previously, with modifications (Pan et al., 2008). Treated cells were incubated and lysed at different time points (1 h, 2 h, 6 h). SH-SY5Y cells were lysed with 200 µL ice-cold lysis buffer containing 125 mM NaCl, 25 mM Tris (pH 7.5), 5 mM EDTA, 1% Nonidet P-40 and protease inhibitors and shaken on ice for 1 h. The cell lysate was centrifuged at 500× g at 4°C for 10 min to discard the nuclei-rich pellet. The supernatant was recentrifuged at 20 000× g at 4°C for 20 min. The supernatant was collected as cytosolic fraction while the pellet was resuspended in 60 µL cell lysis buffer containing 1% Triton X-100 and shaken on ice for another 1 h and then centrifuged at 20 000× g at 4°C for 20 min. The second supernatant was collected as membrane fraction. Epitopes were exposed by boiling the protein samples at 90°C for 5 min. Western blots were performed to examine the translocation of the PKC isoforms.
Preparation of cell lysates for the detection of TH and phoshorylated Akt
A cell lysate technique was adopted from the literature (Yong et al., 2008; Tamizhselvi et al., 2009). Cells were washed twice with phosphate-buffered saline (PBS) after treatment and lysed with 200 µL ice-cold lysis buffer containing 125 mM NaCl, 25 mM Tris (pH 7.5), 5 mM EDTA, 1% Nonidet P-40, 0.4% deoxycholic acid (additional 10 mM NaF and 1 mM Na3VO4 are added for detection of phosphorylated Akt) and protease inhibitor cocktail tablet (Roche Diagnostics, Penzberg, Germany) and shaken on ice for 1 h. After centrifugation at 13 000× g at 4°C for 15 min, supernatant was collected and denatured by SDS-sample buffer. Epitopes were exposed by boiling the protein samples at 100°C for 5 min.
Western blot assays
Protein concentrations were determined with NanoDrop Spectrophotometer (ND-1000, NanoDrop technology). Equal amounts of the protein samples were separated by electrophoresis using a 10% sodium dodecyl sulphate-polyacrylamide (SDS/PAGE) gel and transferred onto a nitrocellulose membrane (Whatman®, Germany). After being blocked in 10% milk with TBST buffer (10 mM Tris-HCl, 120 mM NaCl, 0.1% Tween-20, pH 7.4) at room temperature for 1 h, the membrane was incubated with various primary antibodies (1:1000) at 4°C overnight. β-Tubulin (1:1000) or β-actin (1:10 000) was used as a loading control. Membranes were washed three times in TBST buffer, followed by incubation with 1:10 000 dilutions of horseradish peroxidase-conjugated (HRP) anti-rabbit IgG or anti-mouse IgG (β-actin) at 25°C for 1 h, and washed three times in TBST. Visualization was carried out using ECL® (plus/advanced chemiluminescence) kit (GE healthcare, UK). The density of the bands on Western blots was quantified by Image J software.
Cell transfection and apoptotic detection
SH-SY5Y cells (1 × 105) were seeded onto six-well plates and transfected with CBS-PME185-HA vector (a gift from Dr Hideo Kimura) or with empty vector alone as a control using lipofectamine 2000 transfection reagent. After transfection for 24 h, cells were washed with Krebs solution twice and then treated with 6-OHDA (50 µM) for 4 h. The apoptosis was examined with an Annexin V FITC detection kit (Calbiochem, Cat. No. PF032, Darmstadt, Germany) and analysed with fluorescence microscopy under FITC and rhodamine filter sets.
To visualize nuclear morphology, cells were also stained with 2.5 µg·mL−1 DNA dye Hoechst 33342. The nuclei of healthy and viable cells are usually uniformly stained, while apoptotic cells show condensed or fragmented nuclei.
H2S measurement
The procedures are essentially described in the literature with modifications (Gilboa-Garber, 1971). In brief, aliquots (500 µL) of culture solution (Krebs' buffer) were mixed with trichloroacetic acid (10% (w/v), 250 µL), zinc acetate (1% (w/v), 250 µL), N,N-dimethyl-p-phenylenediamine sulphate (20 µM, 133 µL) in 7.2 M HCl and FeCl3 (30 µM, 133 µL) in 1.2 M HCl in parafilm-enveloped Eppendorf tubes. After 15 min, this mixture was centrifuged at 4000× g for 10 min. The supernatant was collected and its absorbance was measured in 96-well plates at a wavelength of 670 nm. All samples were assayed in duplicate and calculated against a calibration curve of NaHS dissolved in Krebs' buffer: 115 mM NaCl, 2.5 mM KCl, 2.46 mM MgSO4, 2 mM CaCl2, 5.6 mM glucose, 1.38 mM NaH2PO4 and 25 mM NaHCO3, pH 7.4.
Statistical analysis
All data are presented as mean ± SEM. Statistical significance was assessed with one-way analysis of variance (anova) followed by a post hoc (Bonferroni) test for multiple group comparison. Differences with P-values less than 0.05 were considered statistically significant.
Chemicals and reagents
Sodium hydrosulphide (NaHS), 6-OHDA, methyl thiazolyl tetrazolium (MTT) were purchased from Sigma-Aldrich (St Louis, MO, USA). Gö6976 (a PKCα inhibitor) (Zeidman et al., 1999), EAVSLKPT (a PKCε-selective peptide translocation inhibitor) (Chen et al., 2005), rottlerin (a PKCδ inhibitor) (Maher, 2001) and LY294002 (LY) (a phosphoinositol 3' kinase (PI3K) inhibitor) (Sadidi et al., 2009) were obtained from Calbiochem (Darmstadt, Germany). All chemicals were dissolved in deionized water except Gö6976, rottlerin and LY294002, which were dissolved in DMSO at a final concentration not more than 0.05%.
Primary antibody of β-tubulin and PKCε were from Santa Cruz Biotechnology while PKCα and PKCδ as well as polyclonal anti-phospho (p)-Akt rabbit IgG and polyclonal anti-total-Akt rabbit IgG were purchased from Cell Signaling Technology. Primary antibody of β-actin and anti-tyrosine hydroxylase (TH) antibody were obtained from Sigma-Aldrich (St Louis, MO, USA).
NaHS was used as an H2S donor. When H2S is dissolved in water at pH 7.4, HS- is released and forms H2S with H+. This provides a solution of H2S at a concentration that is approximately 33% of the original concentration of NaHS (Reiffenstein et al., 1992).
Results
Protective effect of H2S on 6-OHDA-induced cell injury
We first examined the toxic effect of 6-OHDA. As shown in Figure 1A, treatment with 6-OHDA (50–200 µM) for 12 h decreased the survival rate of SH-SY5Y cells. Pretreatment with NaHS (an H2S donor, 100–1000 µM) for 10 min reversed the effect of 50 µM 6-OHDA (Figure 1B). These data suggest that H2S may protect SH-SY5Y cells against 6-OHDA-induced cell injury. Figure 1C shows that the same protective effects were observed when NaHS was given 10 min or 1 h before administration of 6-OHDA.
Figure 1.
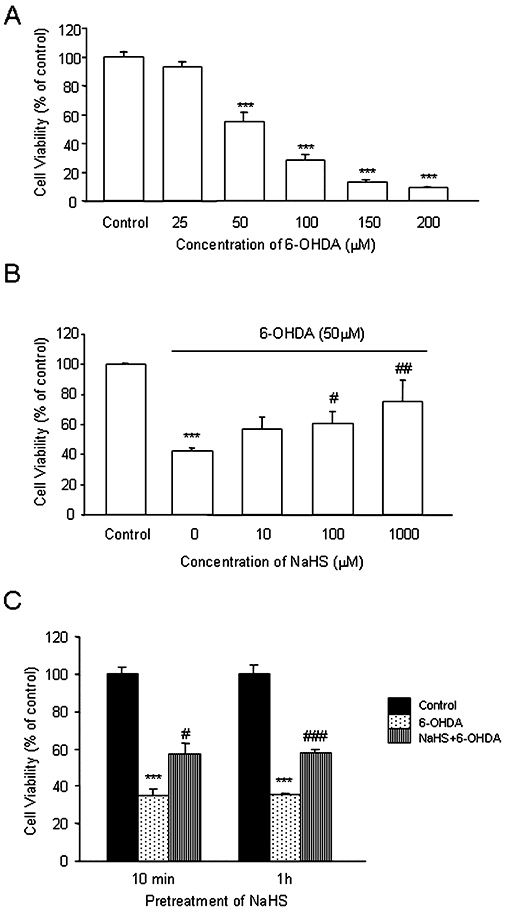
MTT assay showing the effect of NaHS and/or 6-OHDA on SH-SY5Y cell viability. (A) Effect of 6-OHDA on cell viability of SH-SY5Y cells. Cells were treated with 6-OHDA at different concentrations for 12 h. (B) Effect of NaHS on cell viability in SH-SY5Y cells treated with 6-OHDA. Cells were pretreated with various concentrations (10–1000 µM) of NaHS for 10 min before 6-OHDA (50 µM) was added for another 12 h. (C) Pretreatment of NaHS for 10 min and 1 h showed a similar protective effect against 6-OHDA-induced cell injury. Data are presented as mean ± SEM, n = 5, ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, ###P < 0.001 versus 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine.
H2S reverses 6-OHDA-induced loss of TH
Tyrosine hydroxylase (TH) is an important rate-limiting enzyme in the conversion of amino acid tyrosine to dihydroxyphenylalanine (DOPA) for the production of dopamine (Pardridge, 2005). To confirm the neuroprotective effect of H2S, we examined the effect of H2S (100 µM) on TH protein level. As shown in Figure 2, treatment with 6-OHDA for 12 h significantly decreased TH level in SH-SY5Y cells. This effect was reversed by H2S, suggesting that H2S treatment produces protective effects against 6-OHDA-induced dopaminergic neurone injury.
Figure 2.

Effect of NaHS on TH expression in SH-SY5Y cells treated with 6-OHDA. Cells were pretreated with NaHS for 10 min before addition of 6-OHDA (50 µM) for another 12 h. Proteins were extracted and subjected to Western blot analysis using the anti-TH and β-tubulin (as a loading control) antibodies. Data are presented as mean ± SEM, n = 5, ***P < 0.001 versus control; ##P < 0.01 versus 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine; TH, tyrosine hydroxylase.
Effect of NaHS on translocation of PKC isoforms in 6-OHDA-treated SH-SY5Y cells
It has been reported that 6-OHDA-induced cell death is via regulating PKC activity (Tian et al., 2007). In the present study, we examined the effect of 6-OHDA on the translocation of PKC isoforms. The time-courses were shown in Figure 3A. Treatment with 6-OHDA for 1–6 h inhibited the translocation of PKCα and PKCε from cytosol to membrane, but stimulated the activity of PKCδ. The maximum responses were found at 2 h for PKCδ, and 6 h for PKCα and ε. NaHS treatment significantly reversed the effect of 6-OHDA on the activities of the three PKC isoforms, suggesting that the protective effect of H2S may involve different PKC isoforms (Figure 3B).
Figure 3.

Role of PKC isoforms in the neuroprotective effects of NaHS in SH-SY5Y cells treated with 6-OHDA. (A–B) a: PKCδ, b: PKCα, c: PKCε. (A) Time course for the effect of 6-OHDA on the translocation of PKC isoforms. (B) Effect of NaHS (100 µM) on the translocation of PKC isoforms caused by 6-OHDA (50 µM, 2 h). Right panel shows the effect of various PKC isoform inhibitors on the translocation of PKC isoforms. The ratios of membrane/cytosol fraction are normalized to that of control group. β-Actin was used as a loading control. Data are presented as mean ± SEM, n = 5–8, *P < 0.05, **P < 0.01, ***P < 0.001 versus control; ###P < 0.001 versus 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine; PKC, protein kinase C.
Effect of NaHS on cell viability in 6-OHDA-treated SH-SY5Y cells in the presence and absence of inhibitors of PKC isoforms
To further confirm the involvement of PKC, we pretreated the cells with rottlerin (5 µM, a PKCδ inhibitor), Gö6976 (2 µM, a PKCα inhibitor) or EAVSLKPT (200 µM, a specific PKCε inhibitor) for 30 min prior to administration of NaHS (100 µM). The MTT assay showed that the protective effect of H2S was significantly diminished by Gö6976 and EAVSLKPT (Figure 4A). However, inhibition of PKCδ with rottlerin did not reverse cell injury caused by 6-OHDA (Figure 4B). This finding implies that the protective effect of H2S may mainly originates from PKCα and PKCε, not PKCδ.
Figure 4.

The protective effect of NaHS on cell viability in the presence and absence of various PKC isoform inhibitors. (A) Blockade of PKCα with Gö6976 (2 µM, 30 min pretreatment) or PKCε with EAVSLKPT (200 µM, 30 min pretreatment) attenuated the protective effect of NaHS (100 µM, 12 h) on 6-OHDA (50 µM, 12 h)-induced cell injury. (B) Blockade of PKCδ with rottlerin did not reverse cell injury caused by 6-OHDA. Data are presented as mean ± SEM, n = 5, ***P < 0.001 versus control; #P < 0.05, ###P < 0.001 versus 6-OHDA-treated cells; +P < 0.05, ++P < 0.01 versus NaHS + 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine; PKC, protein kinase C.
Involvement of PI3K/Akt in H2S-induced neuroprotection
We also investigated the contribution of the PI3K/Akt pathway in the neuroprotective effects of NaHS. We first examined the time-course for the effect of 6-OHDA on Akt activity. As shown in Figure 5A, 6-OHDA significantly suppressed Akt activity after treatment for 6 h. Figure 5B shows the time-course for the effect of NaHS on Akt activity. There were two peaks (20–40 min and 4–12 h) for NaHS-induced Akt activation. Bearing the above data in mind, we examined the effect of NaHS on Akt activity in cells treated with 6-OHDA for 6 h and 12 h. As shown in Figure 5C, treatment with NaHS reversed the inhibitory effect of 6-OHDA on Akt activity at both time points.
Figure 5.

Involvement of PI3K/Akt pathway in the neuroprotective effects of NaHS. (A–B) Time-courses for the effect of 6-OHDA (50 µM, A) and NaHS (100 µM, B) on Akt activity. (C) NaHS reversed the inhibitory effect of 6-OHDA (6 h and 12 h) on Akt phosphorylation. The histograms represent the ratio of phosphorylated protein over total Akt. Data are presented as mean ± SEM, n = 5, *P < 0.05, ***P < 0.001 versus control; ###P < 0.001 versus 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine.
The involvement of the PI3K/Akt pathway was further confirmed with cell viability data using MTT assay. As shown in Figure 6, pretreatment with LY294002 (a PI3K inhibitor) for 30 min, which alone had no significant effect, markedly reversed the protective effects of NaHS on cell viability. Taken together, these data clearly suggest that the protective effect of NaHS is via stimulation of the PI3K/Akt pathway.
Figure 6.

Blockade of PI3K with LY294002 (20 µM, 30 min pretreatment) abolished the protective effect of NaHS (100 µM, 12 h) on cell injury induced by 6-OHDA (50 µM, 12 h). Data are presented as mean ± SEM, n = 5, ***P < 0.001 versus control; ##P < 0.01 versus 6-OHDA-treated cells; +++P < 0.001 versus NaHS + 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine.
Correlation between PKC and Akt
We further examined whether PI3K/Akt is downstream to PKC activation. As shown in Figure 7, the effect of NaHS on Akt activity was abolished by pretreatment of the cells with Gö6976 (2 µM, an inhibitor of PKCα) or EAVSLKPT (200 µM, an inhibitor of PKCε) for 30 min. These results suggest that activation of Akt by H2S is dependent on PKCα and PKCε.
Figure 7.

Effect of NaHS on Akt activation was dependent on PKC activity. Blockade of PKCα with Gö6976 (2 µM, A) or PKCε with EAVSLKPT (200 µM, B) attenuated NaHS-up-regulated Akt phosphorylation in SH-SY5Y cells treated with 6-OHDA for 6 h. Whole cell lysates were prepared for Western blot analysis of total Akt and phosphorylated Akt level. The histograms represent the ratio of phosphorylated protein to total Akt. Results shown are the mean ± SEM, n = 5, **P < 0.01 versus control; ###P < 0.001 versus 6-OHDA-treated cells; ++P < 0.01, +++P < 0.001 versus NaHS + 6-OHDA-treated cells. 6-OHDA, 6-hydroxydopamine; PKC, protein kinase C.
CBS overexpression attenuates 6-OHDA-induced apoptosis in SH-SY5Y cells
To confirm the role of endogenous H2S, SH-SY5Y cells were transfected with the cDNA of CBS, the main enzyme that produces H2S in these cells. As shown in Figure 8A, CBS protein expression in SH-SY5Y cells was obviously increased 24 h after transfection. Accordingly, H2S concentration in CBS-overexpressed cellular culture medium was significantly elevated compared with empty vector–transfected group (Figure 8B). The H2S level increased to 13.13 ± 1.33 µM 24 h after CBS transfection compared with that in the control group of 9.93 ± 0.23 µM. After application of NaHS (100 µM) for 10 min, the H2S level was 42.73 ± 1.92 µM in medium. Thus, the elevated H2S level caused by overexpression of CBS is much lower than that caused by exogenous application of NaHS.
Figure 8.

Effect of endogenous H2S on 6-OHDA-induced cell apoptosis in SH-SY5Y cells. (A) Transfection of CBS cDNA into SH-SY5Y cells increased the protein expression of CBS. β-Tubulin was used as a loading control. (B) Effect of CBS overexpression and exogenous application of NaHS on endogenous H2S level. Mean ± SEM, n = 8, *P < 0.05, ***P < 0.001 versus GFP; ###P < 0.001 versus CBS. (C) CBS overexpression alleviated 6-OHDA-induced apoptosis. Green: early apoptosis indicated by FITC fluorescence; red: late phase apoptosis stained by propidium iodide; blue: nuclei stained by Hoechst 33342. Photos were taken at ×20 magnification. Scale bar: 200 nm. 6-OHDA, 6-hydroxydopamine; CBS, cystathionine β-synthase; GFP, green fluorescent protein.
Phosphatidylserine (PS) is located on the cytoplasmic surface of the cell membrane during normal conditions. Early apoptosis leads to exposure of PS on the cell surface. The extracellular PS therefore binds with Annexin V-FITC. Late apoptotic cells not only bind with Annexin V-FITC but can also be stained with propidium iodide. As shown in Figure 8C, 6-OHDA increased the number of cells with green (AnnexinV stained by FITC) and red (stained by propidium iodide) fluorescence in the vehicle group (empty vector-transfected cells). This effect was significantly attenuated by the overexpression of CBS.
The beneficial effects of endogenous H2S against 6-OHDA-induced apoptosis were also confirmed by Hoechst 33342 staining assay. Representative photomicrographs of nuclei morphology of SH-SY5Y cells are shown in the right panel of Figure 8C. The effect of 6-OHDA led to the condensation and fragmentation of nuclei (a characteristic of apoptosis). CBS overexpression significantly attenuated this effect. Taken together, these data suggest that 6-OHDA-induced cell apoptosis was attenuated by endogenous H2S, which is generated by CBS overexpression.
Discussion
The cornerstone of signs and symptoms of PD are the loss of dopaminergic neurones and subsequent dopamine deficit in the brain (Zigmond et al., 2002; Tian et al., 2007; Henning et al., 2008; Yuan et al., 2008). Dopamine loss in PD is unable to be replaced directly by dopamine replacement therapy, as this monoamine does not cross the brain capillary endothelial wall, which forms the blood brain- barrier (BBB). However, the precursor to dopamine, dihydroxyphenylalanine (DOPA), is able to cross the BBB owing to its transport via the BBB large neutral amino acid transporter. DOPA is then decarboxylated to dopamine by aromatic amino acid decarboxylase (AAAD). The rate-limiting step in cerebral production of dopamine is the conversion of tyrosine to DOPA via tyrosine hydroxylase (TH) (Pardridge, 2005). Therefore, at present, levodopa (L-dopa) is widely used as a treatment to restore dopamine concentration in PD patients. However, studies have shown that long-term usage of L-dopa leads to pro-oxidant damage (Tian et al., 2007); hence new therapy is needed. In the present study, we provide evidence that H2S, which can easily penetrate biological membranes (Kimura et al., 2005), is able to protect against 6-OHDA-induced cell injury. In addition, H2S significantly increased the level of TH, the rate-limiting enzyme in dopamine production. Our in vitro findings suggest that H2S protects dopaminergic neurones against 6-OHDA-induced injury.
We also examined the protective effect of endogenous H2S on 6-OHDA-induced cell damage. We found that overexpression of CBS only moderately increased the H2S level, whereas exogenous application of NaHS markedly elevated the H2S level. However, CBS overexpression still produced obvious protective effects against cell apoptosis. This is probably because overexpression of CBS may increase H2S level stably and persistently, whereas exogenous application of NaHS causes a transient increase in H2S level in the buffer (Hu et al., 2009). This finding reveals that endogenously produced H2S is important to protect brain against oxidative stress-induced neurodegenerative diseases. Our observation is consistent with Kimura's finding that transfection with 3MST also showed significant resistant to oxidative stress in Neuro2a cells (Kimura et al., 2010).
We further investigated the possible signalling mechanisms underlying the protective effect of H2S on 6-OHDA-induced cell injury. We focused on PKC and Akt, because both of them are well-known prosurvival protein kinases and are hence thought to be involved in anti-oxidation effects that induce neuronal protection (Louis et al., 1988; Tian et al., 2007).
Protein kinase C is a family of well-studied serine-threonine kinases. It is involved in many cell functions including cell proliferation, differentiation, apoptosis and gene expression. The hallmark for PKC activation is a process called translocation, whereby PKC isoforms translocate from the cytosol to subcellular membrane regions (Mackay and Mochly-Rosen, 2001; Pan et al., 2008). The PKC family consists of at least 12 isoforms, among which PKCδ, α and ε are expressed in SH-SY5Y neuroblastoma cells (Zeidman et al., 1999; Mackay and Mochly-Rosen, 2001; Pan et al., 2008). Various studies have shown that different isoforms play different roles in cell functions. PKCα and PKCε have been associated with cell proliferation (Dlugosz et al., 1994; Kampfer et al., 1998), while PKCδ activation contributes to apoptosis (Ohba et al., 1998; Li et al., 1999a; Maher, 2001). Weinreb et al. (2004) reported that PKCα phosphorylates Bcl-2 in a site which increases its anti-apoptotic function, while overexpression of PKCε elevates the expression of Bcl-2 which inhibits apoptosis (Itano et al., 1996; Akao et al., 2002; Weinreb et al., 2004). In view of this, we examined the effect of NaHS on translocation of these isoforms in SH-SY5Y cells treated with 6-OHDA. We found that 6-OHDA transiently (∼2 h) stimulated translocation of PKCδ and sustainably (>6 h) inhibited the activities of PKCα and PKCε. These effects were inhibited by NaHS. Blockade of PKCα and PKCε with their inhibitors abolished the neuroprotection caused by H2S. However, inhibition of PKCδ with rottlerin did not reverse the cell injury caused by 6-OHDA in SH-SY5Y cells. These findings suggest that the toxic effect of 6-OHDA may predominantly originate from sustained inhibition (>6 h) of PKCα and PKCε, but not from the transient (∼2 h) activation of PKCδ. In a similar way, the neuroprotection offered by NaHS may mainly result from stimulation of PKCα and PKCε, instead of inhibition of PKCδ.
Akt, also known as protein kinase B (PKB), is a key molecule in growth factor signalling pathways mediating neuronal survival in both development and disease in multiple paradigms, including resistance against oxidative insults in the brain (Rodriguez-Blanco et al., 2008). Stimulation of the PI3K pathway is necessary for Akt activation in most instances (Li et al., 1999b). Once activated, Akt, in turn, inactivates several pro-apoptotic proteins including BAD and caspase-9 (Li et al., 2006) and therefore promotes cell survival. In the present study, we found that blockade of PI3K with its selective inhibitor, LY294002, abolished the protective effects of NaHS on cell viability. More importantly, NaHS reversed the down-regulated Akt activity caused by 6-OHDA. Our data clearly suggest that the protective effect of NaHS on 6-OHDA-induced cell injury is mediated by stimulation of the PI3K/Akt pathway.
A physical interaction between PKC and Akt in human vascular endothelial cells results in induction of Bcl-2 and enhancement of protection against apoptotic cell death via caspase-3 cleavage inhibition (Yonekawa and Akita, 2008). The activation sequence between PKC and PI3K/Akt has not been fully elucidated and several different mechanisms have been reported. It has been reported that activation of PI3K/Akt controls PKC activity (Le Good et al., 1998). However, up-regulated Akt activity was also observed when PKCα was overexpressed in 32D myeloid progenitor cells (Li et al., 1999b; Rodriguez-Blanco et al., 2008). In the present study, we found that inhibition of PKCα and PKCε isoforms down-regulated Akt expression level, which led to increased cell apoptosis. Our findings suggest that Akt could be the downstream effector of PKCα and PKCε.
The results in the present study suggest that the neuroprotection offered by H2S is a preconditioning-like effect. Our previous study showed that H2S quickly decays to an undetectable level within 30 min after addition of NaHS into cell culture buffer (Hu et al., 2009). This suggests that H2S rapidly stimulates the PKC/PI3K/Akt pathway and triggers a series of persistent intracellular responses and therefore protects the cells. This preconditioning effect is similar to the mechanisms for the cardioprotection conferred by H2S preconditioning (Bian et al., 2006; Pan et al., 2006; Hu et al., 2008; Pan et al., 2008; 2009). Our time-course study shows that the preconditioning period of NaHS lasts for at least 1 h. At this time point, the H2S concentration has already decayed to an undetectable level (Hu et al., 2009). This finding excludes the possibility that the neuroprotection of H2S is caused by direct inhibition of extracellular auto-oxidation of 6-OHDA, another important mechanism for 6-OHDA-induced dopaminergic cell death (Abad et al., 1995; Blum et al., 2000).
In summary, the present observations identify the potential of H2S in protecting SH-SY5Y cells against 6-OHDA-induced cell injury. The neuroprotective effect of H2S involves the PKC-dependent PI3K/Akt pathway.
Acknowledgments
The authors gratefully thank Tan Choon Ping for the technical assistance. This work is supported by research grants from Singapore National Medical Research Council (1183/2008).
Glossary
Abbreviations
- 3MST
3-mercaptopyruvate sulphurtransferase
- 6-OHDA
6-hydroxydopamine
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- H2S
hydrogen sulphide
- L-dopa
levodopa
- LTP
long-term potentiation
- NaHS
sodium hydrosulphide
- PD
Parkinson's disease
- PI3K
phosphoinositol 3′ kinase
- PKB
protein kinase B
- PKC
protein kinase C
- PS
phosphatidylserine
- ROS
reactive oxygen spesies
- TH
tyrosine hydroxylase
Conflicts of interest
None.
Supplemental material
Supporting Information: Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Abad F, Maroto R, Lopez MG, Sanchez-Garcia P, Garcia AG. Pharmacological protection against the cytotoxicity induced by 6-hydroxydopamine and H2O2 in chromaffin cells. Eur J Pharmacol. 1995;293:55–64. doi: 10.1016/0926-6917(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akao Y, Maruyama W, Yi H, Shamoto-Nagai M, Youdim MB, Naoi M. An anti-Parkinson's disease drug, N-propargyl-1(R)-aminoindan (rasagiline), enhances expression of anti-apoptotic bcl-2 in human dopaminergic SH-SY5Y cells. Neurosci Lett. 2002;326:105–108. doi: 10.1016/s0304-3940(02)00332-4. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY, Zhou S, et al. Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther. 2006;316:670–678. doi: 10.1124/jpet.105.092023. [DOI] [PubMed] [Google Scholar]
- Blum D, Torch S, Nissou MF, Benabid AL, Verna JM. Extracellular toxicity of 6-hydroxydopamine on PC12 cells. Neurosci Lett. 2000;283:193–196. doi: 10.1016/s0304-3940(00)00948-4. [DOI] [PubMed] [Google Scholar]
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA. Specific modulation of Na+ channels in hippocampal neurons by protein kinase C epsilon. J Neurosci. 2005;25:507–513. doi: 10.1523/JNEUROSCI.4089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosz AA, Cheng C, Williams EK, Dharia AG, Denning MF, Yuspa SH. Alterations in murine keratinocyte differentiation induced by activated rasHa genes are mediated by protein kinase C-alpha. Cancer Res. 1994;54:6413–6420. [PubMed] [Google Scholar]
- Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005;19:1854–1856. doi: 10.1096/fj.05-3724fje. [DOI] [PubMed] [Google Scholar]
- Fornstedt B, Rosengren E, Carlsson A. Occurrence and distribution of 5-S-cysteinyl derivatives of dopamine, dopa and dopac in the brains of eight mammalian species. Neuropharmacology. 1986;25:451–454. doi: 10.1016/0028-3908(86)90242-x. [DOI] [PubMed] [Google Scholar]
- Gilboa-Garber N. Direct spectrophotometric determination of inorganic sulfide in biological materials and in other complex mixtures. Anal Biochem. 1971;43:129–133. doi: 10.1016/0003-2697(71)90116-3. [DOI] [PubMed] [Google Scholar]
- Goodwin LR, Francom D, Dieken FP, Taylor JD, Warenycia MW, Reiffenstein RJ, et al. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: postmortem studies and two case reports. J Anal Toxicol. 1989;13:105–109. doi: 10.1093/jat/13.2.105. [DOI] [PubMed] [Google Scholar]
- Henning J, Strauss U, Wree A, Gimsa J, Rolfs A, Benecke R, et al. Differential astroglial activation in 6-hydroxydopamine models of Parkinson's disease. Neurosci Res. 2008;62:246–253. doi: 10.1016/j.neures.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Hu LF, Wong PT, Moore PK, Bian JS. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J Neurochem. 2007;100:1121–1128. doi: 10.1111/j.1471-4159.2006.04283.x. [DOI] [PubMed] [Google Scholar]
- Hu LF, Lu M, Wu ZY, Wong PT, Bian JS. Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol Pharmacol. 2009;75:27–34. doi: 10.1124/mol.108.047985. [DOI] [PubMed] [Google Scholar]
- Hu LF, Lu M, Tiong CX, Dawe GS, Hu G, Bian JS. Neuroprotective effects of hydrogen sulfide on Parkinson's disease rat models. Aging Cell. 2010;9:135–146. doi: 10.1111/j.1474-9726.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, et al. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 2008;455:607–616. doi: 10.1007/s00424-007-0321-4. [DOI] [PubMed] [Google Scholar]
- Ichinohe A, Kanaumi T, Takashima S, Enokido Y, Nagai Y, Kimura H. Cystathionine beta-synthase is enriched in the brains of Down's patients. Biochem Biophys Res Commun. 2005;338:1547–1550. doi: 10.1016/j.bbrc.2005.10.118. [DOI] [PubMed] [Google Scholar]
- Itano Y, Ito A, Uehara T, Nomura Y. Regulation of Bcl-2 protein expression in human neuroblastoma SH-SY5Y cells: positive and negative effects of protein kinases C and A, respectively. J Neurochem. 1996;67:131–137. doi: 10.1046/j.1471-4159.1996.67010131.x. [DOI] [PubMed] [Google Scholar]
- Kampfer S, Uberall F, Giselbrecht S, Hellbert K, Baier G, Grunicke HH. Characterization of PKC isozyme specific functions in cellular signaling. Adv Enzyme Regul. 1998;38:35–48. doi: 10.1016/s0065-2571(97)00005-8. [DOI] [PubMed] [Google Scholar]
- Kimura H, Nagai Y, Umemura K, Kimura Y. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid Redox Signal. 2005;7:795–803. doi: 10.1089/ars.2005.7.795. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Lee SW, Hu YS, Hu LF, Lu Q, Dawe GS, Moore PK, et al. Hydrogen sulphide regulates calcium homeostasis in microglial cells. Glia. 2006;54:116–124. doi: 10.1002/glia.20362. [DOI] [PubMed] [Google Scholar]
- Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol Cell Biol. 1999a;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Sampat K, Hu N, Zakari J, Yuspa SH. Protein kinase C negatively regulates Akt activity and modifies UVC-induced apoptosis in mouse keratinocytes. J Biol Chem. 2006;281:3237–3243. doi: 10.1074/jbc.M512167200. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang J, Flechner L, Hyun T, Yam A, Franke TF, et al. Protein kinase C-alpha overexpression stimulates Akt activity and suppresses apoptosis induced by interleukin 3 withdrawal. Oncogene. 1999b;18:6564–6572. doi: 10.1038/sj.onc.1203065. [DOI] [PubMed] [Google Scholar]
- Ljungdahl A, Hokfelt T, Jonsson G, Sachs C. Autoradiographic demonstration of uptake and accumulation of 3H-6-hydroxydopamine in adrenergic nerves. Experientia. 1971;27:297–299. doi: 10.1007/BF02138157. [DOI] [PubMed] [Google Scholar]
- Louis JC, Magal E, Yavin E. Protein kinase C alterations in the fetal rat brain after global ischemia. J Biol Chem. 1988;263:19282–19285. [PubMed] [Google Scholar]
- Lu M, Hu LF, Hu G, Bian JS. Hydrogen sulfide protects astrocytes against H(2)O(2)-induced neural injury via enhancing glutamate uptake. Free Radic Biol Med. 2008;45:1705–1713. doi: 10.1016/j.freeradbiomed.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Mackay K, Mochly-Rosen D. Localization, anchoring, and functions of protein kinase C isozymes in the heart. J Mol Cell Cardiol. 2001;33:1301–1307. doi: 10.1006/jmcc.2001.1400. [DOI] [PubMed] [Google Scholar]
- Maher P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J Neurosci. 2001;21:2929–2938. doi: 10.1523/JNEUROSCI.21-09-02929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Tsugane M, Oka J, Kimura H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- Ohba M, Ishino K, Kashiwagi M, Kawabe S, Chida K, Huh NH, et al. Induction of differentiation in normal human keratinocytes by adenovirus-mediated introduction of the eta and delta isoforms of protein kinase C. Mol Cell Biol. 1998;18:5199–5207. doi: 10.1128/mcb.18.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan TT, Feng ZN, Lee SW, Moore PK, Bian JS. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. J Mol Cell Cardiol. 2006;40:119–130. doi: 10.1016/j.yjmcc.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Pan TT, Neo KL, Hu LF, Yong QC, Bian JS. H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am J Physiol Cell Physiol. 2008;294:C169–C177. doi: 10.1152/ajpcell.00282.2007. [DOI] [PubMed] [Google Scholar]
- Pan TT, Yong QC, Bain JS. All in the timing: a comparison between the cardioprotection induced by H2S preconditioning and post-infarction treatment. Eur J Pharm. 2009;616:160–165. doi: 10.1016/j.ejphar.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Tyrosine hydroxylase replacement in experimental Parkinson's disease with transvascular gene therapy. NeuroRx. 2005;2:129–138. doi: 10.1602/neurorx.2.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Blanco J, Martin V, Herrera F, Garcia-Santos G, Antolin I, Rodriguez C. Intracellular signaling pathways involved in post-mitotic dopaminergic PC12 cell death induced by 6-hydroxydopamine. J Neurochem. 2008;107:127–140. doi: 10.1111/j.1471-4159.2008.05588.x. [DOI] [PubMed] [Google Scholar]
- Sadidi M, Lentz SI, Feldman EL. Hydrogen peroxide-induced Akt phosphorylation regulates Bax activation. Biochimie. 2009;91:577–585. doi: 10.1016/j.biochi.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage JC, Gould DH. Determination of sulfide in brain tissue and rumen fluid by ion- interaction reversed-phase high-performance liquid chromatography. J Chromatogr. 1990;526:540–545. doi: 10.1016/s0378-4347(00)82537-2. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Deng Y, Maruyama W, Dostert P, Kawai M, Naoi M. Uptake of a neurotoxin-candidate, (R)-1,2-dimethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline into human dopaminergic neuroblastoma SH-SY5Y cells by dopamine transport system. J Neural Transm Gen Sect. 1994;98:107–118. doi: 10.1007/BF01277014. [DOI] [PubMed] [Google Scholar]
- Tamizhselvi R, Sun J, Koh YH, Bhatia M. Effect of hydrogen sulfide on the phosphatidylinositol 3-kinase-protein kinase B pathway and on caerulein-induced cytokine production in isolated mouse pancreatic acinar cells. J Pharmacol Exp Ther. 2009;329:1166–1177. doi: 10.1124/jpet.109.150532. [DOI] [PubMed] [Google Scholar]
- Tian LL, Zhou Z, Zhang Q, Sun YN, Li CR, Cheng CH, et al. Protective effect of (+/-) isoborneol against 6-OHDA-induced apoptosis in SH-SY5Y cells. Cell Physiol Biochem. 2007;20:1019–1032. doi: 10.1159/000110682. [DOI] [PubMed] [Google Scholar]
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- Warenycia MW, Goodwin LR, Benishin CG, Reiffenstein RJ, Francom DM, Taylor JD, et al. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem Pharmacol. 1989;38:973–981. doi: 10.1016/0006-2952(89)90288-8. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Bar-Am O, Amit T, Chillag-Talmor O, Youdim MB. Neuroprotection via pro-survival protein kinase C isoforms associated with Bcl-2 family members. FASEB J. 2004;18:1471–1473. doi: 10.1096/fj.04-1916fje. [DOI] [PubMed] [Google Scholar]
- Yonekawa H, Akita Y. Protein kinase Cepsilon: the mitochondria-mediated signaling pathway. FEBS J. 2008;275:4005–4013. doi: 10.1111/j.1742-4658.2008.06558.x. [DOI] [PubMed] [Google Scholar]
- Yong QC, Lee SW, Foo CS, Neo KL, Chen X, Bian JS. Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am J Physiol Heart Circ Physiol. 2008;295:H1330–H1340. doi: 10.1152/ajpheart.00244.2008. [DOI] [PubMed] [Google Scholar]
- Yuan WJ, Yasuhara T, Shingo T, Muraoka K, Agari T, Kameda M, et al. Neuroprotective effects of edaravone-administration on 6-OHDA-treated dopaminergic neurons. BMC Neurosci. 2008;9:75. doi: 10.1186/1471-2202-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidman R, Pettersson L, Sailaja PR, Truedsson E, Fagerstrom S, Pahlman S, et al. Novel and classical protein kinase C isoforms have different functions in proliferation, survival and differentiation of neuroblastoma cells. Int J Cancer. 1999;81:494–501. doi: 10.1002/(sici)1097-0215(19990505)81:3<494::aid-ijc26>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson's disease. J Pharmacol Exp Ther. 2007;322:913–922. doi: 10.1124/jpet.107.124669. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Hastings TG, Perez RG. Increased dopamine turnover after partial loss of dopaminergic neurons: compensation or toxicity? Parkinsonism Relat Disord. 2002;8:389–393. doi: 10.1016/s1353-8020(02)00019-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.