

Abstract
The NIH Chemical Genomics Center (NCGC) was the inaugural center of the Molecular Libraries and Screening Center Network (MLSCN). Along with the nine other research centers of the MLSCN, the NCGC was established with a primary goal of bringing industrial technology and experience to empower the scientific community with small molecule compounds for use in their research. We intend this review to serve as 1) an introduction to the NCGC standard operating procedures, 2) an overview of several of the lessons learned during the pilot phase and 3) a review of several of the innovative discoveries reported during the pilot phase of the MLSCN.
1. High-throughput screening at the NCGC
High-throughput screening (HTS) is one of the most established mechanisms to identify small molecules with biological activity [1]. The model of the Molecular Libraries and Screening Center Network (MLSCN) provides researchers that have established assays for either isolated targets or cellular phenotypes the resources to conduct a HTS for small molecules that affect that target or phenotype [2]. HTS has evolved to address a wide-range of biological functions by employing a multitude of formats that now includes assays for purified proteins and enzymes as well as cell-based assays for signal transduction pathways, protein complex formation, transcriptional regulation of gene function, pre-mRNA splicing events, and epigenetics [3]. The NIH Chemical Genomics Center (NCGC) has established a number of innovations aimed at reducing the burden of false positives (FP) and false negatives (FN) associated with HTS as well as providing efficient use of assay reagents and compound samples. Primary among these innovations is the establishment of multi-concentration HTS using miniaturized assay volumes (<10 μL/well) in 1536-well microtiter plates, referred to as quantitative high-throughput screening (qHTS) [4]. qHTS is capable of screening small molecules at multiple concentrations (typically between 7 and 15 specified concentrations) based upon the built-in flexibility of inter-plate titrations (Fig. 1). By miniaturizing to a 1536 well-plate format the qHTS process essentially provides cost-effective high-throughput pharmacology by delivering concentration response curves (CRCs) for each library member. The benefits of qHTS include reducing FP and capturing FN, allowing the data to be mined to determine a compound’s pharmacological profile, the ability to identify assay related artifacts, and the ability to construct structure activity relationships (SAR) from the primary screening data to drive probe optimization immediately after completion of the screen. The NCGC has applied qHTS to many different assay formats including fluorescence/luminescence biochemical and cell-based assays as well as fluorescent protein-based phenotypic assays using laser scanning microplate cytometry [5].
Fig (1).

The quantitative high-throughput screening (qHTS) paradigm. A cartoon representation of a standard inter-plate titration of small molecules and the resulting concentration response curve (CRC) generated for active library compounds.
The qHTS process has expedited our ability to accurately judge the success of a screen and move efficiently into the medicinal chemistry optimization phase of several projects. Further, in several instances, qHTS has provided the necessary data to identify probe-like compounds directly from the primary screen. qHTS has proven a great success, however, there are significant challenges associated with this screening paradigm. Primarily, an innovative workflow for compound management and the implementation of a unique informatics platform were needed. Major challenges in compound management included the need to transfer compounds received in 96/384-well plates to 1536-well plates while maintaining accurate compound registration and construction of the inter-plate serial dilutions. To accomplish this in an automated fashion, an interleaved quadrant compression of sixteen 96-well plates into four 384-well plates into one 1536 well plate was introduced followed by serial dilution using an automated protocol [6]. Even greater challenges were associated with the unprecedented data associated with the qHTS paradigm. To address this, systematic analysis processes were devised and applied to each screen. After raw data are loaded in our system, a log is processed to link assay plates with compound plates followed by an analysis template that is used in conjunction with a flexible built-in formula engine to create data layers that include corrected data for systematic artifacts due to events such as liquid handling, and a normalized data layering. Data correction is facilitated by the uniform incorporation of 1536 well-plates that serve as blanks through the screen. Once these data layers have been calculated, plate and well based data are converted to sample-oriented titration-response data. This step has built-in flexibility to support a wide variety of experimental designs and screening approaches. Novel algorithms were created to enable high-throughput unsupervised curve fitting of activity as well as other multi-channel data (Fig. 2). For a seven concentration point series the NCGC CRC algorithm requires roughly two minutes of processing time (or 13 CRCs per second). For a typical qHTS screen, our CRC fitting and classification system requires less than 0.01% manual intervention on average [7]. A CRC classification system was developed to characterize the confidence of the curve-fit data as well as the efficacy of the observed activity. Potency and Hill slope can then be considered along with the CRC class. To more efficiently view data, each compound can be sorted by CRC class and compared to other qHTS assays to define the activity signature of compounds across different assays which can be used to evaluate assay format-related artifacts or the selectivity of the probe series. This informatics assessment of activity for a given screen provides for a mineable HTS database where pharmacological parameters such as potency, efficacy of response and Hill slope can be used to fully evaluate the bioactivity of compounds. The result is a report on assay performance, active chemical series, and top singletons.
Fig (2).

The NCGC Informatics Platform. (1) Data are normalized to controls and plate background signatures used to correct for systematic patterns observed in screen. (2) Titration-response curves are created for each channel of data (automated curve fit program is available online at http://www.ncgc.nih.gov/resources/software.html). (3) Curves are classified into full curves, partial curves, single point actives, and inactives. (4) SAR reports are generated for each active chemical series.
2. High-throughput chemistry at the NCGC
Following screening and informatics analysis, the majority of lead compounds require extensive modification before they can be considered optimized probes. Further, a rigorous understanding of the SAR of a chemotype is required to appreciate the promise and/or limitations of any given probe compound. To move quickly from a confirmed screening lead to an optimized probe, the NCGC relies upon the innovations and lessons garnered by the pharmaceutical industry [8]. Both solution- and solid-phase parallel synthetic efforts are used and high-throughput methods such as microwave assisted organic chemistry, high-speed library purification methods and parallel sample handling and solution making platforms are utilized. While each chemotype is unique (eliminating a ‘one size fits all’ optimization strategy), innovations such as matrix libraries and closed-loop work flow paradigm have proven useful during the discovery of several optimized probe compounds.
3. Library Profiles at the NCGC
In any large collection of small organic molecules intended for high-throughput biochemical evaluation there are a percentage of compounds that can masquerade as inhibitors or activators of the desired target due to inherent properties that lead to compound interference of the assay signal. While the activity associated with these compounds is artifactual and unrelated to the specific target, the readout can be indistinguishable from true functional modulators. It was recognized early that reactive compounds were often noted as ‘bad actors’ in HTS [9]. Efforts to amend modern libraries eliminated compounds with overly reactive functional groups such as anhydrides, halopyrimidines and α-halo ketones has resulted in a lower burden of false positives. Further, many stochastic artifactual activities are minimized by screening in titration (i.e. qHTS). However, other mechanism by which compounds can ‘trick’ the user proved more subtle and often track in a concentration-dependent manner. Thus, we set out to establish profiles of our library to establish databases of suspected artifactual compound behavior and assist follow-up efforts.
Profiles for aggregation
Shoichet and coworkers were among the first to report on the phenomenon of nonspecific enzyme inhibition through promiscuous small molecule aggregation [10]. Following these early reports, it became standard practice to perform a post-screen analysis of potential leads as potential aggregators before exploring them further. While subsequent analyses aided researchers in avoiding false leads, these collective follow-up efforts yielded little information about the phenomenon of aggregation itself and provided no data for a large-scale library profile. In an effort to 1) evaluate the feasibility of conducting a high-throughput analysis of aggregation, 2) examine how widespread the phenomenon of aggregation is and 3) evaluate our own libraries’ propensity for aggregation, we conducted a comprehensive profile of our compound collection. In collaboration with Dr. Brian Shoichet (Dept. of Pharmaceutical Chemistry, University of California at San Francisco), we performed a qHTS screen of Amp C β-lactamase in the absence and presence of Triton X-100 (a detergent with known ability to disrupt aggregate formation) [11]. The results provided the first large-scale assessment of the divergence of activities associated with aggregation (detergent sensitive: 1.7% hit rate) and true enzyme inhibition (detergent insensitive: 0.1% hit rate). Numerous leads were followed up in more conventional, low-throughput assays to confirm these results. This study was enabled through the qHTS process and established the ability to screen in the presence of detergents as well as the feasibility of determining a proactive database of likely aggregators within a large compound collection. Further, the results from this experiment provided a wider correlative association between steep dose response curves (Hill slope > 2) and inhibition through aggregation [12]. Beyond this study, we have recently reported a thorough analysis of the mechanism by which each active from the screen (and a related virtual screen) was affecting enzyme inhibition (i.e. aggregation, inhibition through covalent modification or inhibition through binding) [13]. To date, the aggregation profile of our library has aided numerous follow-up efforts.
Profiles of cytotoxicity
There are numerous advantages to utilizing cell-based screening technologies and qHTS has proven a major success in cell-based screens. Application of qHTS in profiling cytotoxicity in cell based assays has yielded highly reproducible data and greatly reduced the level of false-positive and false-negative rates relative to tradition HTS. In any cell-based assays it is important to consider the affects of cellular toxicity when assessing the results from a high-throughput screen. Cell death (be it necrosis, apoptosis or autophagic) will often result in a decrease in signal output that typically tracks in a concentration dependent manner and is often independent of the assay format and platform. These signals can mimic antagonism or inhibition (though signals associated with cell death are sometimes associated with steep Hill slopes). There is also a typical decrease of intracellular ATP content following cell death that can have associated affects on assay outputs. It is important to note that cytotoxic effects of library compounds are dependent on the cell line being utilized, and therefore profiles of cytotoxicity cannot typically be generally applied. To date, we have explored cytotoxicity profiling of 9 human cell lines, 2 rat cell lines and 2 mouse cell lines in an effort to 1) better understand the underlying mechanism of cellular cytotoxicity, 2) understand how cytotoxicity affects the output of cell-based assays and 3) produce general method for prioritizing compounds for advanced toxicologic evaluation [14]. These studies include an assay of membrane integrity as marker of cytotoxicity and an in vitro correlation between cellular viability and caspase activity [15, 16]. Data associated with these studies are not only furthering our understanding of how cell viability and HTS are intertwined, but also establishing novel paradigms for advanced toxicology [17].
Profiles of autofluorescence
The basic readout for the various assays performed at NCGC involves the generation of light energy (fluorescence or luminescence). These assays rely heavily on common fluorophores ranging from the blue region of the spectrum (i.e. coumarin-based fluorophores with λex and λem in the region of 450 nm) to dyes in the red region of the spectrum (i.e. xanthene-based fluorophores with λex and λem in the region of 650 nm). While the properties of known fluorophores are generally well understood, the overall generalizations that govern fluorescence are not fully known. As such, there is little ability to predict which small organic molecules are capable of a fluorescent output in these common spectral regions. As the degree of fluorescence will typically track as a measure of concentration, any molecule capable of autofluorescence can mimic the typical CRC’s associated with any given qHTS campaign. Molecular entities with these properties can greatly complicate the analysis of a screen. To nullify this, we profiled the fluorescent properties of the entire small molecule collection at wavelengths associated with eight common fluorophores ranging from the blue to the red regions of the visible spectrum (4-methylumbelliferone, Rhodamine, Alexafluor 350, Resorufin, Fluorescein, Texas Red, Alexafluor 488 and AlexaFluor 647)[18]. Several lessons were learned from this experiment. Foremost, the percentage of library compounds that possessed autofluorescence was dramatically greater when excited in the blue spectral region in comparison to the red spectral region; nearly 5% of the collection showed autofluorescence at excitation and emission wavelengths associated with 4-MU compared to 0.01 % at excitation and emission wavelengths associated with Texas Red. Additionally, we found through direct analysis of well samples for library members with autofluorescence that the spectral output was occasionally associated with impurities rather than the small molecule sample. This profile also prompted a reassessment of common assumptions about how the general assay environment affects a molecule’s fluorescent properties. It should be noted that several recent manuscripts have highlighted the use of HTS as a means for seeking out novel fluorphores [19]. This profile has afforded us the ability to further annotate our follow-up efforts based upon the probability that a given ‘active’ is the result of autofluorescence. From these lessons learned we have made a marked effort to incorporate red-shifted dyes into our screening efforts whenever possible to avoid such effects outright.
Profiles for firefly luciferase inhibition
Numerous advances in biotechnology have been translated into the realm of HTS. Of particular importance is the adaptation of bioluminescent enzymes such as luciferase that are oftentimes used as a reporter for cell-based assays. Luciferase has found vast utility as a reporter in gene expression assays and as an ATP sensor in both cell-based or biochemical assay formats, as well as other enzymatic reactions or cell-based events. Firefly luciferase is one of the most commonly used reporters for constructing assays. However, this enzyme has two small molecules as substrates (D-luciferin and ATP) and inhibition of firefly luciferase enzyme activity can occur when screening typical small molecules libraries. An inhibitor of luciferase will track in a concentration-dependent manner and show SAR reflective of the mode of inhibition. Therefore, luciferase inhibitors are an important source of false leads in bioluminescent assays. As HTS-based assays extensively utilize this reporter including many undertaken by the MLSCN, a profile aimed at identifying the small molecules from our collection that inhibit luciferase was warranted. As such, a qHTS profile of the library was assayed for activity versus the ATP-dependent Photinus pyralis luciferase [20]. Nearly 1% of the library was noted to inhibit this luciferase variant at-or-below 10 μM. Several chemotypes identified were further profiled versus other commonly used firefly luciferase detection conditions and luciferase variants. Consistent with specific inhibition of P. pyralis luciferase enzyme, the inhibitors were found to be largely inactive at the unrelated Renilla reniformis luciferase as well as commercial reagents that employ a different variant of firefly luciferase (Ultra-Glo™, Promega Corp). Several of these compounds were examined in advanced SAR and modeling analysis to better explain the divergence of activities across different luciferase variants. The results from this profile have prompted a comprehensive review of screening results generated in house and elsewhere. In several instances, the plurality of activities reported from a screen has been the result of luciferase inhibition rather than direct modulation of the pathway of interest. Further, we have endeavored to use the Renilla luciferase as a means to mitigate these effects.
A further lesson from this profile is the paradox of cell-based luminescent signal activation due to inhibition of firefly luciferase within the cellular milieu. Following the identification of the chemotypes associated with luciferase inhibition from our profiling experiment, we noted that these luciferase inhibitors were frequently reported as agonists/activators in cell-based assays using firefly luciferase as a reporter within the PubChem database [21]. Enzyme activity, overall, is the result of not only expression levels but also of the protein half-life (due to stability and resistance to degradation) (Fig. 3). Protein stabilization via ligand binding is a well known phenomenon and the resilience of luciferase to proteolysis when bound to small molecules had been previously reported [22]. To expand upon and explore this suspicion, we conducted several experiments that demonstrated luciferase signal activation in cells tracked in accordance with luciferase inhibition determined in a purified enzyme assay [23]. Representative inhibitor classes identified in the profile were found to increase luciferase activity in a manner independent of transcriptional/translational events and prolonged the half-life of the luciferase enzyme in the cells. Further, we found that the presence of excess substrates that are typically present in the bioluminescent detection reagents used in HTS can compete with small molecule inhibitors of firefly luciferase, thereby allowing free luciferase enzyme to be readily measured during the detection step of the assay. Thus, an inhibitor of luciferase has the paradoxical effect of extending the half-life of the enzyme and thus providing exaggerated enzyme levels generating the appearance of agonism/activation. As bioluminescent assays enable a large range of experiments within the domain of HTS, the luciferase inhibitor profile should provide for more efficient interpretation of the results reported within this important class of assays.
Fig (3).

The reporter inhibition-nonspecific activation paradox. A) The typical fate of a protein reporter being depleted over time via proteolysis and B) the stabilization and extension of reporter half-life through inhibitor binding and the false impression of activation.
4. Small Molecule Chaperones of Glucocerebrosidase
Protein misfolding leads to numerous cellular disorders and several characterized genetic diseases. Gaucher disease is one such genetic disorder caused by mutations of the sugar hydrolase glucocerebrosidase (GC). Many of these mutations result in a misfolding of the protein that hinders the ability of GC to traffic from the endoplasmic reticulum to the lysosome [24]. The deficit of this important sugar hydrolase causes the improper accumulation of glycosphingolipids in the lysosomes and results in a disease phenotype. While enzyme replacement therapy has proven efficacious for systemic treatment of the disease, it is widely hoped that chemical chaperone therapy will be more successful for treatment of the CNS associated symptoms of Gaucher disease [25]. The concept of chemical chaperone therapy revolves around the ability of small molecules to tightly bind to a misfolded protein and produce a conformational change that allows proper trafficking. Several successes have been reported utilizing this concept including a chemical chaperone for the V2 vasopressin receptor [26].
In collaboration with Dr. Ellen Sidransky (National Human Genome Research Institute, NIH) we set out to identify novel chemical chaperones of GC. Initial efforts surrounded the development and execution of a qHTS utilizing purified GC and a caged fluorogenic substrate (4-methylumbelliferone-β-D-glucopyranoside) (PubChem AIDs 348, 360). GC is capable of cleaving the sugar-fluorophore conjugate resulting in the ‘uncaged’ 4-methylumbelliferone that is fluorescence (Fig. 4). The presence of small molecule inhibitors would reduce this output in a quantifiable manner. From this screen, three general small molecule scaffolds were identified as inhibitors of GC, including a series of substituted (1,3,4-thiadiazol-2-yl)-4-phenylsulfonamides, a series of substituted (4-methylquinolin-6-yl)carboxamides and a series of 1,3,5-triazines [27]. This effort underscored the utility of the qHTS paradigm as several leads from the primary screen demonstrated potencies in the low nanomolar range and selected compounds were entered into follow-up efforts directly. In order to optimize several of the other leads, we relied upon the immense pool of commercially available analogues that were noted to maintain acceptable similarity to leads found in the primary screen. By direct compound acquisition, we identified several analogues with improved potencies. Additionally, a synthetic effort was engaged within the optimization of the 1,3,5-triazine series resulting in several analogues with improved potency [28].
Fig (4).

Novel glucocerebrosidase (GC) Probes. A) The fluorescence GC enzyme assay developed using the pro-fluorescent substrate 4-methylumbelliferone-β-D-glucopyranoside as a substrate for the GC enzyme. B) The entire qHTS data set. Samples where the CRCs could be fit to the data are shown in light blue (high-quality CRCs) or dark blue (CRCs showing fits of lower confidence), while data that did not show any CRC is shown in orange. C) The chemical structure of NCGC00092410 (1) D) the selectivity profile from 1 versus GC, α-glucosidase, α-galactosidase or β-N-acetylflucosaminidase. E) Overlay image from the dual labeling [(GC Ab, red) and (Lysotracker, green)] of cells expressing the clinically relevant N370S GC mutant in the presence of 40 μM of NCGC00092410. Colocalization of N370S GC to the lysosome presents as yellow.
The most potent analogues from each compound series were analyzed for inhibitory potential versus several related sugar hydrolases (including α-glucosidase, α-galactosidase and β-N-acetylflucosaminidase) to gauge the selectivity of these novel reagents. Additionally, as the goal was to find chemical chaperones, selected analogues were analyzed for enhancement of GC activity and evidence of lysosomal localization in Gaucher fibroblasts utilizing a clinically relevant N370S mutant. GC activity was measured utilizing a pulse-chase assay and localization of the mutant GC was accomplished utilizing a known GC polyclonal antibody and a fluorescent lysosome marker (Lysotracker DND-99).
While several compounds were noted as active, one member of the (4-methylquinolin-6-yl)carboxamides series possessed several attractive qualities. N-(4-methyl-2-morpholinoquinolin-6-yl)cyclohexanecarboxamide (NCGC00092410, 1) was noted to inhibit native GC with a Ki of 21 nM while possessing no inhibitory capacity for α-glucosidase, α-galactosidase or β-N-acetylflucosaminidase (Fig. 4). Further, treatment of N370S mutant GC fibroblasts with NCGC00092410 at 40 μM demonstrated both increased GC activity (40% to 90%) and co-localization of this mutant form of GC to the lysosome (Fig. 4). These results matched or exceeded the ability of a known GC chaperone (the iminosugar nonyl-DNJ) to effect similar GC activity and chaperone potential in these experiments. Given the promising results for NCGC00092410, we determined various pharmacokinetic properties for this compound. NCGC00092410 was noted to have acceptable aqueous solubility, microsomal stability (rat), Caco-2 permeability and no relevant Cyp inhibition. However, NCGC00092410 was noted to possess a high level of plasma protein binding (low free fraction). It is likely that this property will be limiting for this reagent and efforts to improve its protein binding profile will be necessary.
5. Inhibitors of AmpC β-Lactamase
AmpC β-lactamases have emerged as important targets of study based upon their clinical relevance to antibiotic resistant bacteria. The hydrolytic properties of AmpC β-lactamases are similar to other cephalosporinases and, importantly, these enzymes have demonstrated drug resistant properties for the third generation cephalosporins. The study of AmpC further provided an interesting case study as it is an enzymatic target that crystallizes readily and is well characterized enzymologically.
In collaboration with Dr. Brian Shoichet (Dept. of Pharmaceutical Chemistry, University of California at San Francisco), we set out to identify novel chemical inhibitors of AmpC. Additionally, this collaboration provided us with an opportunity to analyze each ‘hit’ compound from a mechanistic standpoint and perform a comparison between the screening experiment and a parallel virtual screen run using the same compound collection. An assay of β-lactamase activity was accomplished through the monitoring of nitrocefin hydrolysis in the presence and absence of Triton X-100 detergent. The qHTS effort [AID: 584 (with detergent) 585 (without detergent)] proved successful and several novel inhibitors were identified [11, 13]. Additionally, a number of cephalosporins (the library contains more than 50) were uncovered providing a new, comprehensive SAR analysis for this classical inhibitor versus AmpC. Several novel inhibitors were noted from the primary screen including numerous substituted 2-sulfonyl-1,3,4-oxadiazoles that maintained potencies ranging from 16 nM to 8 μM. Selected compounds from this class were tested against a panel of well-characterized enzymes. Inhibition was noted in several cases including selected serine and cysteine proteases. Further, it was found that inhibition was time dependent, suggestive of a covalent modification of these enzymes. The most potent of these agents was (S)-tert-butyl 1-(5-(2-chloro-6-fluorobenzylsulfonyl)-1,3,4-oxadiazol-2-yl)-2-methylpropylcarbamate (NCGC00067197, 2) that possessed an IC50 value of 16 nM (Fig 5). Mass analysis of AmpC following a preincubation with 2 resulted in an increase in molecular weight equivalent to the (1,3,4-oxadiazol-2-yl)propylcarbamate moiety of this structure (mass addition of 239 a.m.u.). The covalent adduct was confirmed via crystallographic analysis of AmpC following incubation with 2 that clearly showed the oxadiazole moiety bound to Ser64 (Fig 5).
Fig (5).

Novel AmpC Probes. A) The chemical structure of NCGC00067197 (2) and NCGC00161790 (3) B) Structure of AmpC- NCGC00067197 adduct at a 2 Å resolution. C) Structure of a representative phthalimide bound to AmpC at 1.8 Å resolution.
Another prominent chemotype discovered during this study was a series of phthalimides that were not detected during the primary screen (the screening cutoff for activity was 30 μM). However, docking analysis suggested that analogues resembling (R)-2-(1-carboxy-2-(naphthalen-1-yl)ethyl)-1,3-dioxoisoindoline-5-carboxylic acid (NCGC00161790, 3) had binding potential to AmpC. The initial follow-up studies explored racemic versions of these molecules which maintained only modest potencies (the best registered Ki was 140 μM). Despite this poor affinity, a crystal structure was obtained that provided a roadmap for optimization of this chemotype (Fig 5). From this data, the foremost adjustment was to focus on chiral versions of this compound. Additional SAR was also generated at both the phthalimide moiety and the amino acid derived portion of the molecule. These efforts resulted in an analogue (NCGC00161790, 3) with improved potency (registered Ki was 5 μM). NCGC00161790 represents one of the most potent, non-covalent modifying inhibitors of AmpC recorded to date.
The comprehensive analysis of the experimental and virtual screen yielded several additional lessons, including the apparent lack of ‘hits’ derived from compounds that maintain reactive functionalities (including Michael acceptors, alkyl halides and isocyanates). This result highly suggests that there is still much more to learn about the acknowledged ‘best practices’ regarding the makeup of library collections for screening.
6. Antagonists and Agonists of the Thyroid Stimulating Hormone Receptor
Thyroid stimulating hormone receptor (TSHR), luteinizing hormone receptor (LHR) and follicle-stimulating hormone receptor (FSHR) belong to the glycoprotein hormone receptor (GPHR) subfamily of G-protein coupled receptors. Disruption and malfunction of these related receptors has been implicated in a number of human diseases. Of particular importance are the role of TSHR in Graves disease and the use of recombinant thyroid stimulating hormone (TSH) in post-operative thyroid cancer patients. Small molecule agonists may replace recombinant TSH in this role, while small molecule antagonists of TSHR may have therapeutic potential for TSHR-mediated hyperthyroidism caused by constitutively activating mutations or stimulating autoantibodies associated with Graves’ disease.
In collaboration with Dr. Marvin Gershengorn (National Institute of Diabetes and Digestive and Kidney Diseases, NIH), we set out to identify novel small molecule agonists and antagonists of TSHR. A cell-based assay of TSHR activation was established based upon a cyclic nucleotide gated ion channel approach [29]. Differing amounts of cAMP produced by TSHR activation alter the membrane potential through the cyclic nucleotide gated ion channel and can be measured by a membrane potential dye. Through careful control of reagents, this approach allows the identification of both agonists and antagonists. Additionally, the qHTS effort was subsequently run against the parental cell line (no TSHR) as a control. The qHTS screen was performed (AID: 926) and several novel agonists were discovered through this effort. Among these were a series of 4-oxo-1,2,3,4-tetrahydroquinazolin-2-yl based scaffolds including N-(4-(5-(3-benzyl-4-oxo-1,2,3,4-tetrahydroquinazolin-2-yl)-2-methoxybenzyloxy)phenyl)acetamide (NCGC00161876, 4) (Fig 6). An optimization effort was pursued for these agents and will be presented in due course.
Fig (6).

Novel TSHR Probes. A) The chemical structure of NCGC00161876 (4) and NCGC00166332 (5). B) Molecular model of a potential docking orientation of NCGC00166332 (5) bound to the transmembrane core of TSHR. C) Intracellular cAMP accumulation in HEK-EM 293 cells stably expressing TSHR in the presence of sera from patients with Graves’ disease (GD) or the EC50 concentration of TSH (1.8 nM) in the presence or absence of NCGC00166332 (5).
While the qHTS effort succeeded in discovering several agonists, an antagonist of TSHR was not identified. Therefore, we sought out alternate methods to create a novel small molecule antagonist for TSHR. A recent report detailed the success of a series of substituted thieno[2,3-d]pyrimidines as LHR agonists that culminated with the description of the highly potent Org 41841 (Scheme 1) [30]. Our interest in this small molecule was based upon a hypothesis that the agonism displayed by Org 41841 was mediated through binding to the transmembrane helix of LHR.
Scheme 1.

a Reagents and conditions: (i) K2CO3, EtOH, 60 °C, 5 h; (ii) POCl3, dioxane, reflux, 2 h; (iii) NaOEt, EtOH, 50 °C 3 h; (iv) LiOH, dioxane/H2O; (v) PyBop, DIPEA, DMF followed by addition of amine.
Interestingly, the transmembrane domains for each of the glycoprotein hormone receptors possess a remarkably high degree of homology and we reasoned that Org 41841 would likely display activity at the related TSHR and FSHR. We showed that Org 41841 does produce an agonistic response at TSHR, albeit at a lower potency relative to LHR [31, 32]. Docking studies using homology models of LHR and TSHR suggested a binding pocket for Org 41841 between transmembrane helixes 3, 4, 5 and 6 and the extracellular loop 2 in both receptors. With these docking poses as a guide, we systematically altered the core structure of Org 41841 in order to find derivatives that would antagonize the native signal of TSHR. One analogue, containing a 2-methoxyvinyl moiety at the para position of the 4-phenyl ring of the thieno[2,3-d]pyrimidine core heterocycle, was noted as an antagonist and was designated as NCGC00166332 (5) (Fig 6) [33]. Direct evidence that NCGC00166332 interacts with the transmembrane helixes was established through interactions between NCGC00166332 and selected TSHR mutants with specified alterations determined as critical for binding based upon docking models. These computational models suggested a pose that is similar to that of Org41841 (Fig 6).
Having arrived at a novel inhibitor of TSHR, we placed additional effort into examining its potential. NCGC00166332 was shown to be capable of blocking thyroperoxidase mRNA expression in human thyrocytes at concentrations of 10 μM and 30 μM. Finally, a cAMP accumulation assay showed that NCGC00166332 was capable of blocking the activity of thyroid-stimulating antibodies collected directly from 4 Graves’ disease patients (GD5, GD19, GD29 and DG30) (Fig 6). Currently, we our examining the core structure of NCGC00166332 to improve its potency, water solubility and selected ADME/DMPK properties.
7. Modulators of the Cyclic AMP Response Element Binding Protein
The cyclic AMP Responsive Elements (CRE) are common nucleotide sequences found in the promoter regions of numerous genes [34]. There are several characterized cyclic AMP response element binding (CREB) proteins that serve as transcription factors within multi-component signaling pathways associated with neuronal development and activity. Genetic analyses have linked the CREB pathway to memory and have evolved to mouse models where deficient CREB signaling has resulting in long-term memory impairment [35]. These results suggest that pharmacological modulation of CRE may result in the means to ameliorate memory disorders.
In collaboration with Dr. Marshall Nirenberg (National Heart, Lung and Blood Institute, NIH), we set out to identify novel small molecule modulators of the CRE pathway. A cell-based reporter gene assay was developed as a means to identify such compounds (AID: 662). The basis of this assay relied upon a β-lactamase reporter gene under the control of cAMP response element (CRE) in the presence of the chimeric fluorophore CCF4 (Fig 7). Excitation of the CCF4 fluorophore, while intact, elicits a FRET transition and fluorescent emission in the green region of the visible spectrum (530 nm). In the cellular system, small molecule induced activation of CREB proteins results in the expression of β-lactamase, a cleavage of the cephalosporin moiety linking the coumarin and fluorescene fluorphores and results in emission in the blue spectral region. A total of 95 structural series were identified and based upon selected metrics, a total of 38 separate chemotypes were selected for advanced study.
Fig (7).

Novel CREB pathway Probes. A) The fluorescence CREB pathway assay developed using an inducible β-lactamase reporter gene assay and the chimeric CCF4 fluorophore. B) The entire qHTS data set. Samples where the CRCs could be fit to the data are shown in blue while data that did not show any CRC is shown in orange.
Cell-based (or pathway) assays present unique opportunities in screening paradigms and offer great potential in terms of target choice and phenotype exploration. However, the deconvolution effort needed to discover the exact interactions between protein targets and ‘hit’ compounds is often a large barrier to advancing such studies (vide supra). Within the CRE pathway, there are numerous receptors and enzymes that play a role in the eventual activation or inhibition of transcription. These include a variety of GPCR’s, adenylate cyclases, phosphdiesterases and protein kinase A. In order to pair each lead chemotype with a target protein class, several purified protein assays associated with these targets were established and representatives from the 38 lead chemotypes were evaluated therein. Among these target proteins is phosphodiesterase 4 (PDE4). PDE4 is capable of degrading cyclic AMP, resulting in a suppression of CRE signaling. Inhibitors of PDE4 have been shown to potentiate CRE signaling via the disruption of this process [36].
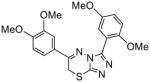
During the deconvolution phase of this work, one chemotype based upon a core [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine heterocycle was noted to be a potent inhibitor of PDE4A in a purified protein assay. The original leads from the primary screen were noted to have potencies as low at 40 nM. To expand upon this chemotype we prepared numerous analogues based upon existing literature precedence (Scheme 2) [37–39].
Scheme 2.

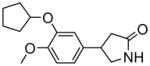
a Reagents and conditions: (i) hydrazine monohydrate, H2O, reflux, 3 h, then conc. HCl; (ii) Br2, CHCl3, rt 5 min. then reflux 30 min to 4 h; (iii) EtOH, 105 °C, 4 h.
Our initial SAR evaluation involved the creation of a matrix library based upon specific substitutions on the adjunct 3- and 6- phenyl rings (the screening leads maintained numerous methoxy substitutions and the bulk of our SAR explorations expanded upon this). From this library, we identified the 3,4-dimethoxy substitution pattern on the 6-phenyl ring system as the core pharmacophore. One analogue, 3-(2,5-dimethoxyphenyl)-6-(3,4-dimethoxyphenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (NCGC00165289, 6) was noted to maintain an 8 nM IC50 value versus PDE4A (Table 1). There are numerous PDE isoforms expressed in the various tissue of the human body including 11 primary PDE families (designated PDE1 through PDE11). Most isoforms have been characterized in terms of tissue distribution and therapeutic potential. The PDE4 family further contains 4 principle gene products designated PDE4A, PDE4B, PDE4C and PDE4D. Selectivity is an important aspect of many small molecule modulators of protein function and it was of interest to profile our novel PDE4A inhibitor across a panel of PDE isozymes. Therefore, we subjected NCGC00165289 and the classical PDE4 inhibitor rolipram to a commercially available panel of 18 PDE assays including five PDE4 variants. We were gratified to note that NCGC00165289 maintained activity primarily versus the PDE4 family of enzymes. Modest activities were additionally noticed against PDE10A1 and PDE3B. Currently, NCGC00165289 and several advanced derivatives of this chemotype are being evaluated in advanced models of neurogenesis and memory enhancement.
Table 1.
PDE isoform selectivity data for rolipram and NCGC116
| PDE isoform* | rolipram IC50/% inh. | 6 IC50/% inh. | |
|---|---|---|---|
 rolipram |
PDE1A | inactive | inactive |
| PDE1B | inactive | inactive | |
| PDE1C | inactive | 26% | |
| PDE2A | inactive | 41% | |
 6 (NCGC00165289) |
PDE3A | inactive | 1.7 μM |
| PDE3B | inactive | 720 nM | |
| PDE4A1A | 102 nM | 12.9 nM | |
| PDE4B1 | 901 nM | 48.2 nM | |
| PDE4B2 | 534 nM | 37.2 nM | |
| PDE4C1 | 40% | 452 nM | |
| PDE4D2 | 403 nM | 49.2 nM | |
| PDE5A1 | inactive | 60% | |
| PDE7A | inactive | 73% | |
| PDE7B | inactive | 33% | |
| PDE8A1 | inactive | 57% | |
| PDE9A2 | inactive | inactive | |
| PDE10A1 | inactive | 823 nM | |
| PDE11A4 | inactive | inactive |
data generated by BPS Biosciences (CA) [http://www.bpsbioscience.com] and represents either the IC50 value or the % inhibition at 10 uM of compound.
8. Inhibitors of Thioredoxin Glutatione Reducatase
Schistosomiasis is a chronic inflammatory disease that is estimated to afflict more than 200 million people causing more than 280,000 deaths annually [40]. Schistosomiasis is caused by trematode flatworms of the genus Schistosoma, a parasite with a complex lifecycle with both definitive (human) and intermediate (snail) hosts [41]. There is currently no vaccine for schistosomiasis and control of the disease is mediated exclusively through the use of praziquantel (PZQ). The widespread reliance on PZQ has raised concern over the development of drug-resistant parasites and recently PZQ resistant isolates of S. mansoni and S. haematobium have been identified [42]. It is of great interest to identify novel chemotherapeutics for the control of this widespread affliction.
The complex lifecycle of the parasite includes migrations throughout the human body. Following skin penetration, the cercariae localize in the liver where they mature and mate. Once paired, the now adult parasites migrate to the urogenital system where eggs are produced. Thus, the parasites primarily reside in highly aerobic environments and must deal with numerous attacks by reactive oxygen species (from both aerobic process and the host’s innate immune system). It has been shown that S. mansoni is highly reliant on the NADPH-dependent flavoenzyme thioredoxin-glutathione reductase (TGR) to maintain a proper redox environment [43]. Treatment of larval schistosome parasites with interfering RNA targeting TGR produced high levels of parasite death (>90%) over a four day span. Therefore, Williams and coworkers reasoned that TGR would represent a good prospective target for novel therapeutics for schistosomiasis.
In collaboration with Dr. David Williams (Rush University Medical Center), we set out to identify novel small molecules capable of inhibiting TGR. A coupled assay was developed utilizing TGR and peroxiredoxin (Prx), enzymes responsible for the resolution of peroxide species [44]. This assay utilized TGR and Prx in stoichiometrically equal quantities and relied upon the quantitative reduction of fluorescence that occurred during NADPH consumption (Fig 8). Recently we reported a colorimetric assay of TGR based upon the reduction of 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman’s reagent) [45]. These assays successfully identified numerous leads and several chemotypes were selected for follow-up studies in orthogonal assays, including a direct measurement of ex-vivo worm killing.
Fig (8).

Novel thioredoxin-glutathione reductase (TGR) Probes. A) The fluorescence TGR/Prx coupled enzyme assay developed using the reduction of fluorescence that occurs during NADPH consumption. B) The entire qHTS data set. Samples where the CRCs could be fit to the data are shown in blue (activating CRCs) or red (inhibitory CRCs), while data that did not show any CRC is shown in orange. C) The chemical structure of furoxan (7) D) The survival of ex-vivo S. mansoni adult worms at selected concentrations of furoxan. E) In vivo analysis of worm burdens following furoxan (IP administration) treatment in subpopulations of S. mansoni infected mice. Treatment 1 represents dosing during the larvae stage of parasite infection. Treatment 2 represents dosing during the juvenile stage of parasite infection. Treatment 3 represents dosing during the adult stage of parasite infection.
Among the most compelling chemotypes were a series of oxadiazole-2-oxides, including 3-cyano-4-phenyl-1,2,5-oxadiazole 2-oxide (commonly referred to as furoxan) (NCGC00015800, 7) (Fig 8) [46]. Furoxan was shown to possess modest inhibition potential (between 3 μM and 8 μM based upon assay format) in the primary screen and in follow-up efforts directed to ascertain TGR inhibition directly. Interestingly, furoxan effected between 90% and 100% parasite death when tested at a concentration of 10 μM. Often, it is expected that potency will drop significantly when transitioning from a purified protein assay to more complex in vivo assays. Based upon the success of this agent in both the primary screen and follow-up parasite studies, we advanced this chemotype for additional studies.
Furoxan (7) is a known nitric oxide (NO) donor compound introduced by Gasco and coworkers in the mid 1990’s [47]. NO donation by furoxan is presumed to be mediated by a reactive/nucleophilic thiol attack on the compound followed by a molecular rearrangement that ultimately releases a single equivalent of NO (several mechanisms have been postulated). With the current understanding of the biochemical effects of NO, this chemotype has entered into studies of β2-adrenoceptor agonism, within aspirin conjugates for platelet aggregation inhibition and as H2-receptor antagonists [48–50]. Based upon this known activity, we examined the release of NO from furoxan following a single 10 μM exposure to 15 nM S. mansoni TGR in the presence of NADPH. Under these conditions furoxan was noted to produce an appreciable amount of NO [46]. Additionally, we found that the worm killing potential of furoxan was largely mitigated by administering a 100 μM concentration of carboxy-PTIO (a known NO scavenger) during the worm killing assay. Accordingly, we are currently exploring the role that NO release has on TGR inhibition and parasite death.
Based upon the promising ex-vivo worm data, we next explored the capability of furoxan to act as a therapeutic cure for S. mansoni infected mice. In vivo treatment (IP administration) at 10 mg/kg was examined in three mouse populations based upon the stage of the infection [deemed treatment 1 (larvae stage: treatment at 1 day post infection), treatment 2 (juvenile stage: treatment at 23 days post infection) and treatment 3 (adult stage: treatment at 37 days post infection)] [46]. Significant reductions in worm burdens were noted for all treatment regiments compared to controls (Fig 8). The effects demonstrated by furoxan exceed the activity criteria set by the World Health Organization for development of novel leads for the control of schistosomiasis. We are currently exploring novel derivatives of this chemotype, the mechanism of action associated with worm killing and generating ADME/DMPK properties for selected compounds for consideration as clinical leads.
8. Conclusions and Perspectives
The NIH Chemical Genomics Center is a comprehensive research center incorporating HTS, informatics and chemistry. To date, scientists at the NCGC have developed novel paradigms in screening and informatics, described innovative methods to profile large chemical libraries and discovered and improved numerous chemical probes of wide interest to the biomedical and chemical biology communities. Chemical probes serving as chaperones for glucocerebrosidase, inhibitors of AmpC β–lactamases, agonists and antagonists of the thyroid stimulating hormone receptor, small molecules capable of potentiating of the cyclic AMP response element and inhibitors of thioredoxin-glutathione reductase represent several of the most promising findings at the NCGC during the pilot phase of the Molecular Libraries program. The NCGC enters the production phase of the program as a comprehensive center and is currently pursuing small molecule modulators of numerous biochemical targets, pathways and phenotypes.
Acknowledgments
The authors would foremost like to acknowledge the talent, dedication and hard work of the entire staff of the NIH Chemical Genomics Center. We thank Ms. Allison Peck for critical reading of this manuscript. Special thanks to Adam Yasgar and Paul Shin. We further thank the Sidransky, Shoichet, Gershengorn, Nirenberg and Williams’s laboratories for their exceptional expertise and efforts. The research reviewed here was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
References
- 1.Inglese J, Auld DS. High Throughput Screening Techniques: Overview of Applications in Chemical Biology in Wiley Encyclopedia of Chemical Biology. John Wiley & Sons, Inc; Hoboken, NJ: 2008. [Google Scholar]
- 2.Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 3.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 4.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auld DS, Johnson RL, Zhang YQ, Veith H, Jadhav A, Yasgar A, Simeonov A, Zheng W, Martinez ED, Westwick JK, Austin CP, Inglese J. Fluorescent protein-based cellular assays analyzed by laser-scanning microplate cytometry in 1536-well plate format. Methods Enzymol. 2006;414:566–589. doi: 10.1016/S0076-6879(06)14029-X. [DOI] [PubMed] [Google Scholar]
- 6.Yasgar A, Shinn P, Jadhav A, Auld D, Michael S, Zheng W, Austin CP, Inglese J, Simeonov A. JALA. 2008:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.http://ncgc.nih.gov/docs/ has a link to “Enabling the Large Scale Analysis of Quantitative High Throughput Screening Data”
- 8.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. Application of Combinatorial Chemistry Science on Modern Drug Discovery. J Comb Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 9.Rishton GM. Reactive compounds and in vitro false positives in HTS. Drug Discov Today. 1997;2:382–384. [Google Scholar]
- 10.McGovern SL, Helfand BT, Feng B, Shoichet BK. A Specific Mechanism of Nonspecific Inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 11.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A High-Throughput Screen for Aggregation-Based Inhibition in a Large Compound Library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 12.Shoichet BK. Interpreting Steep Dose-Response Curves in Early Inhibitor Discovery. J Med Chem. 2006;49:7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 13.Babaoglu K, Simeonov A, Irwin JJ, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive Mechanistic Analysis of Hits from High-throughput and Docking Screens against β-Lactamase. J Med Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia M, Huang R, Witt KL, Southall N, Fostel J, Cho MH, Jadhav A, Smith CS, Inglese J, Portier CJ, Tice RR, Austin CP. Compound Cytotoxicity Profiling Using Quantitative High-Throughput Screening. Environ Health Perspect. 2008;3:284–291. doi: 10.1289/ehp.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho MH, Niles A, Huang R, Inglese J, Austin CP, Riss T, Xia M. A bioluminescent cytotoxicity assay for assessment of membrane integrity using a proteolytic biomarker. Toxicol In Vitro. 2008;22:1099–1106. doi: 10.1016/j.tiv.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang R, Southall N, Cho M-H, Xia M, Inglese J, Austin CP. Characterization of Diversity in Toxicity Mechanism Using in Vitro Cytotoxicity Assays in Quantitative High Throughput Screening. Chem Res Toxicol. 2008;21:659–667. doi: 10.1021/tx700365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins FS, Gray GM, Bucher JR. Tranforming Environmental Health Protection. Science. 2008;319:906–907. doi: 10.1126/science.1154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simeonov A, Jadhav A, Thomas CJ, Wang Y, Huang R, Southall NT, Shinn P, Smith J, Austin CP, Auld DS, Inglese J. Fluorescence Spectroscopic Profiling of Compound Libraries. J Med Chem. 2008;51:2363–2371. doi: 10.1021/jm701301m. [DOI] [PubMed] [Google Scholar]
- 19.Ahn YH, Lee JS, Chang YT. J Am Chem Soc. 2007;129:4510–4511. doi: 10.1021/ja068230m. [DOI] [PubMed] [Google Scholar]
- 20.Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, Austin CP, Inglese J. Characterization of Chemical Libraries for Luciferase Inhibitory Activity. J Med Chem. 2008;51:2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- 21.http://pubchem.ncbi.nlm.nih.gov/
- 22.Thompson JF, Hayes LS, Lloyd DB. Modulation of firefly luciferase stability and impact on studies of gene regulation. Gene. 1991;103:171–177. doi: 10.1016/0378-1119(91)90270-l. [DOI] [PubMed] [Google Scholar]
- 23.Auld DS, Thorne N, Nguyen DT, Inglese J. A Specific Mechanism for Nonspecific Activation in Reporter-Gene Assays. ACS Chem Biol. 2008;3:463–470. doi: 10.1021/cb8000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hruska KS, LaMarca ME, Sidransky E. In: Gaucher Disease. Futerman AH, Zimran A, editors. CRC Press; Boca Raton FL: 2006. pp. 13–48. [Google Scholar]
- 25.Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scoot CR, Wappner RS, Zimran A. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 1991;113:112–119. doi: 10.1016/s0002-9343(02)01150-6. [DOI] [PubMed] [Google Scholar]
- 26.Robben JH, Sze M, Knoers NVAM, Deen PMT. Rescue of Vasopressin V2 Receptor Mutants by Chemical Chaperones: Specificity and Mechanism. Mol Biol Cell. 2005;17:379–386. doi: 10.1091/mbc.E05-06-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpann O, Simeonov A, Goldin E, Auld D, LeMarca ME, Inglese J, Austin CP, Sidransky E. Proc Natl Acad Sci USA. 2007;104:13192–13197. doi: 10.1073/pnas.0705637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang W, Zheng W, Urban DJ, Inglese J, Sidransky E, Austin CP, Thomas CJ. Bioorg Med Chem Lett. 2007;17:5783–5789. doi: 10.1016/j.bmcl.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Titus S, Neumann S, Zheng W, Southall N, Michael S, Klumpp C, Yasgar A, Shinn P, Thomas CJ, Inglese J, Gershengorn MC, Austin CP. Quantitative High-Throughput Screening Using a Live-Cell cAMP Assay Identifies Small-Molecule Agonists of the TSH Receptor. J Biomol Screening. 2008;13:120–127. doi: 10.1177/1087057107313786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Straten NCR, Schoonus-Gerritsma GG, van Someren RG, Draaijer J, Adang AEP, Timmers CM, Hanseen RGJM, van Boeckel CAA. The First Orally Active Low Molecular Weight Agonists for the LH Receptor: Thienopyr(im)idines with Therapeutic Potential for Ovulation Induction. Chem Bio Chem. 2002;3:1023–1026. doi: 10.1002/1439-7633(20021004)3:10<1023::AID-CBIC1023>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Jäschke H, Neumann S, Moore S, Thomas CJ, Colson AO, Costanzi S, Kleinau G, Jiang J-K, Paschke R, Raaka BM, Krause G, Gershengorn MC. A Low Molecular Weight Agonist Signals by Binding to the Transmembrane Domain of Thyroid-stimulating Hormone Receptor (TSHR) and Luteinizing Hormone/Chorionic Gonadotropin Receptor (LHCGR) J Biol Chem. 2006;281:9841–9844. doi: 10.1074/jbc.C600014200. [DOI] [PubMed] [Google Scholar]
- 32.Moore S, Jaeschke H, Kleinau G, Neumann S, Costanzi S, Jiang JK, Childress J, Raaka BM, Colson A, Paschke R, Krause G, Thomas CJ, Gershengorn MC. Evaluation of Small-Molecule Modulators of the Luteinizing Hormone/Choriogonadotropin and Thyroid Stimulating Hormone Receptors: Structure-Activity Relationships and Selective Binding Patterns. J Med Chem. 2006;49:3888–3896. doi: 10.1021/jm060247s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann S, Kleinau G, Costanzi S, Moore S, Jiang J-k, Raaka BM, Thomas CJ, Krause G, Gershengorn MC. A Low Molecular Weight Antagonist for the Human Thyrotropin Receptor with Therapeutic Potential for Hyperthyroidism. Endocrinology. 2008 doi: 10.1210/en.2008-0836. ASAP article online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Yin JC, Del Vecchio M, Zhou H, Tully T. CREB as a Memory Modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in drosophila. Cell. 1995;81:107–115. doi: 10.1016/0092-8674(95)90375-5. [DOI] [PubMed] [Google Scholar]
- 36.Zhang KYJ, Ibrahim PN, Gillette S, Bollag G. Phosphodiesterase-4 as a potential drug target. Expert Opin Ther Targets. 2005;9:1283–1303. doi: 10.1517/14728222.9.6.1283. [DOI] [PubMed] [Google Scholar]
- 37.Jacob JN, Nichols DE. Dopamine agonist properties of N-alkyl-4-(3,4-dihydroxyphenyl)-1,2,3,4-tetrahydroisoquinolines. J Med Chem. 1981;24:1013. doi: 10.1021/jm00140a021. [DOI] [PubMed] [Google Scholar]
- 38.Moreno I, Tellitu I, Dominguez E, SanMartin R. A Simple Route to New Phenanthro- and Phenanthroid-Fused Thiazoles by a PIFA-Mediated (Hetero)biaryl Coupling Reaction. Eur J Org Chem. 2002;13:2126. [Google Scholar]
- 39.Skoumbourdis AP, Huang R, Southall N, Leister W, Guo V, Cho MH, Inglese J, Nirenberg M, Austin CP, Xia M, Thomas CJ. Identification of a potent new chemotype for the selective inhibition of PDE4. Bioorg Med Chem Lett. 2008;18:1297–1303. doi: 10.1016/j.bmcl.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hotez PJ, Brindley PJ, Bethony JM, King CH, Pearce EJ, Jacobson J. Helminth infections: the great neglected tropical diseases. J Clin Invest. 2008;118:1311–1321. doi: 10.1172/JCI34261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vermund SH, Bradley DJ, Ruiz-Tiben E. Survival of Schistosoma mansoni in the human host: estimates from a community-based prospective study in Puerto Rico. Am J Tropical Med & Hyg. 1983;32:1040–1048. doi: 10.4269/ajtmh.1983.32.1040. [DOI] [PubMed] [Google Scholar]
- 42.Fallon PG, Doenhoff MJ. Drug-resistant schistosomiasis: resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drugs specific. Am J Tropical Med & Hyg. 1994;51:83–88. doi: 10.4269/ajtmh.1994.51.83. [DOI] [PubMed] [Google Scholar]
- 43.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ES, Williams DL. Thioredoxin glutathione reductase from Schistosoma mansoini: an essential parasite enzyme and a key drug target. PLoS Med. 2007;4:e260. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simeonov A, Jadhav A, Sayed AA, Wang Y, Nelson ME, Thomas CJ, Inglese J, Williams DL, Austin CP. Quantitative High-Throughput Screen Identifies Inhibitors of the Schistosoma mansoni Redox Cascade. PLoS Neg Trop Dis. 2008;2:e127. doi: 10.1371/journal.pntd.0000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lea WA, Jadhav A, Rai G, Sayed AA, Cass CL, Inglese J, Williams DL, Austin CP, Simeonov A. A 1,536-Well-Based Kinetic HTS Assay for Inhibitors of Schistosoma mansoni Thioredoxin Glutathione Reductase. Assay and Drug Dev Tech. 2008;6:551–555. doi: 10.1089/adt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nature Med. 2008;14:407–412. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medina C, Ermondi G, Fruttero R, Di Stilo A, Ferretti C, Gasco A. Furoxans as Nitric Oxide Donors. 4-Phenyl-3-furoxancarbonitrile: Thiol-Mediated Nitric Oxide Release and Biological Evaluation. J Med Chem. 1994;37:4412–4416. doi: 10.1021/jm00051a020. [DOI] [PubMed] [Google Scholar]
- 48.Buonsanti MF, Bertinaria M, Di Stilo A, Cena C, Fruttero R, Gasco A. Nitric Oxide Donor β2-Agonists: Furoxan Derivatives Containing the Fenterol Moiety and Related Furazans. J Med Chem. 2007;50:5003–5011. doi: 10.1021/jm0704595. [DOI] [PubMed] [Google Scholar]
- 49.Turnbull CM, Cena C, Fruttero R, Gasco A, Rossi AG, Megson IL. Mechanism of action of NO-releasing furoxan derivatives of aspirin in human platelets. Br J Pharm. 2006;148:517–526. doi: 10.1038/sj.bjp.0706743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bertinaria M, Sorba G, Medana C, Cena C, Adami M, Morini G, Pozzoli C, Coruzzi G, Gasco A. Synthesis and Pharmacological Characterization of New H2-Antagonists Containing NO-Donor Moieties, Endowed with Mixed Antisecretory and Gastroprotective Activities. Helv Chem Acta. 2000;83:287–299. [Google Scholar]