

Abstract
A formal total synthesis of the benzothiophene selective estrogen receptor modulator (SERM) desketoraloxifene and analogs has been accomplished from alkynes bearing electron-rich aromatic rings by electrophilic cyclization using I2. This approach affords oxygen-bearing 3-iodobenzo[b]thiophenes in excellent yields, which are easily further elaborated using a two-step approach involving Suzuki-Miyaura and Mitsunobu coupling reactions.
Keywords: Iodocyclization, 3-iodobenzothiophene, benzothiophene SERM, desketoraloxifene
Early cancer drug discovery efforts focused on the design of small molecule nonsteroidal estrogen receptor (ER) ligands with antagonist properties against breast and other reproductive tissues.1 Tamoxifen (I, Figure 1) is the archetypal selective estrogen receptor modulator (SERM).2–4 It was the first marketed drug to be realized from these efforts and, while this compound and its active metabolite, 4-hydroxytamoxifen (II, Figure 1), are effective antiestrogens on estrogen receptor positive breast tissue, they subsequently were discovered to have undesirable estrogenic properties on the endometrium.5 A third triphenylethylene compound, toremifene (III, Figure 1), has also been approved for the treatment of breast cancer, although it too has been reported to have undesirable uterine stimulatory activity.6 Because more potent and safer chemotherapeutic agents are needed, due to the potential side effects of tamoxifen I, considerable attention has been paid to the development of less toxic SERMs.7 Many SERMs in clinical use and clinical development are also highly susceptible to oxidative metabolism by electrophilic, redox active quinoids simply because they are based on polyaromatic phenol scaffolds.8,9
Figure 1.
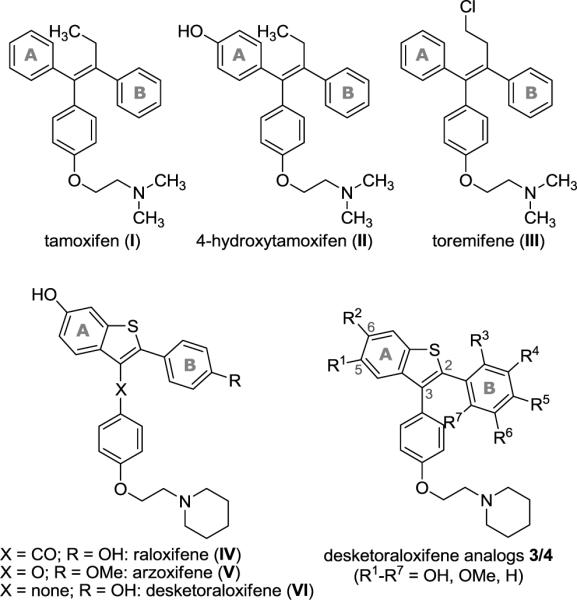
Chemical structures of tamoxifen (I), 4-hydroxytamoxifen (II), toremifene (III) and representative synthetic benzothiophene SERMs [e.g. raloxifene (IV), arzoxifene (V), and desketoraloxifene (VI)] with A and B rings corresponding to tamoxifen.
A benzothiophene SERM, raloxifene (IV, Figure 1), is in clinical use for the prevention and treatment of postmenopausal osteoporosis and is currently in clinical trials for the chemoprevention of breast cancer.4,10 Another benzothiophene SERM, arzoxifene (V, Figure 1), is a structural analog of raloxifene in which the carbonyl hinge has been replaced by an ether linkage and the 4′-hydroxy group is methylated. This SERM is currently in clinical trials for the treatment of breast cancer and, since it has similar structural characteristics to tamoxifen I, has the potential to form quinoids.7,11
Interestingly, removal of the ketone moiety in raloxifene results in a benzothiophene analog SERM desketoraloxifene (VI, Figure 1), which is more planar and conformationally more similar to 4-hydroxytamoxifen (II). Desketoraloxifene (VI) has been found to be a much stronger activator of the Activator Protein-1 (AP-1) site by ERα than ERβ, and mimics 4-hydroxytamoxifen (II) more than raloxifene (IV).10,12,13 With this information in hand, a set of desketoraloxifene analogs 3/4 was designed based on the structures of 4-hydroxytamoxifen (II) and raloxifene (IV).
Previously, we have developed a general synthesis of 2,3-disubstituted benzo[b]thiophenes by the palladium/copper-catalyzed cross-coupling of various o-iodothioanisoles and terminal alkynes, followed by electrophilic cyclization under mild reaction conditions (Scheme 1).14 Very recently, a simple and efficient method for the parallel synthesis of multi-substituted benzo[b]thiophenes has also been described via known palladium-catalyzed couplings for generation of a diverse set of building blocks starting from 3-iodobenzo[b]thiophenes.15,16
Scheme 1.
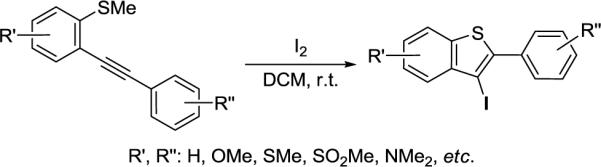
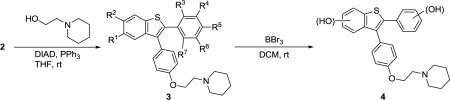
We wish to report here a new efficient method for the preparation of oxygen-functionalized 3-iodobenzo[b]thiophenes 1 by electrophilic cyclization using I2 and their further elaboration to desketoraloxifene analogs 3/4 (Scheme 2 and Table 1). The 3-iodobenzo[b]thiophenes 1, having oxygen substituents at the C-5 and/or C-6 benzothiophene positions, are promising precursors to a wide variety of desketoraloxifene analogs 3/4.
Scheme 2.

Table 1.
Synthesis of desketoraloxifene analogs 3 and 4a
| Entry | 2 | R | Product | 3/4 | Yield (%)d |
|---|---|---|---|---|---|
| 1 | 2a | R1,R5 = OMe |
|
3a | 89 |
| 2 | 2a | R1,R5 = OH | 4a b | 58 | |
| 3 | 2b | R1,R4 = OMe |
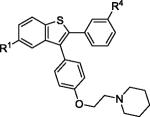
|
3b | 86 |
| 4 | 2b | R1,R4 = OH | 4b b | 61 | |
| 5 | 2c | R1,R3 = OMe |
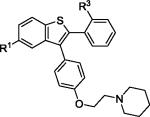
|
3c | 87 |
| 6 | 2c | R1,R3 = OH | 4c b | 52 | |
| 7 | 2d | R1,R4,R6 = OMe |
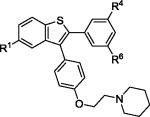
|
3d | 83 |
| 8 | 2d | R1,R4,R6 = OH | 4d c | 39 | |
| 9 | 2e | R2,R5 = OMe |
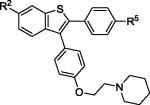
|
3e | 83 |
| 10 | 2e | R2,R5 = OH | 4e (VI) b | 78 | |
| 11 | 2f | R1,R2,R5 = OMe |
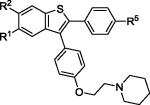
|
3f | 76 |
| 12 | 2f | R1,R2,R5 = OH | 4f c | 41 |
Reagents and conditions: i. Mitsunobu coupling: 2 (0.2 mmol), alkylaminoethanol (1.5 equiv), DIAD (1.5 equiv), PPh3 (2.0 equiv), THF (2.0 mL), rt, 24–36 h. ii. Demethylation: 3 (0.1 mmol), BBr3, CH2C12 (1.0 mL), rt, N2, 3 h.
4.0 Equiv of BBr3 used.
6.0 Equiv of BBr3 used.
Isolated yields after column chromatography. All isolated products were characterized by 1H and 13C NMR spectroscopy (see the Supporting Information).
Results and Discussion
Our first goal was the efficient preparation of a variety of oxygen-bearing 3-iodobenzo[b]thiophenes 1 (Scheme 2). In this series, we proposed to initially change the substituents at the C-2, C-3, C-5, and C-6 positions of the benzothiophene ring system. This decision was based on the structure of desketoraloxifene (VI), which has a para-phenol at the 2-position, a basic aliphatic amine chain at the 3-position and an hydroxyl group at the 6-position of the benzothiophene ring system.
The cyclization proceeds smoothly when the substituent on the distal end of the alkyne is an electron-rich methoxy-aryl group. These 3-iodobenzo[b]thiophenes 1 are easily further elaborated using a two-step approach involving Suzuki-Miyaura and Mitsunobu coupling reactions to give desketoraloxifene analogs 3. The first step, the palladium-catalyzed Suzuki-Miyaura coupling of the 3-iodobenzo[b]thiophenes 1 with a tetrahydropyranyl (THP) ether-protected boronic acid, e.g.p-THPOC6H4B(OH)2, for 6–8 h, followed by aqueous HCl deprotection, afforded the desired phenolic oxygen products 215 in high yield (Scheme 2).
In the second step, amine-coupled SERM precursors have been produced by reaction of the phenolic oxygen species 2 with 1-(2-hydroxyethyl)piperidine under Mitsunobu reaction conditions,17 using Ph3P and diethyl azodicarboxylate (DEAD), to afford the desketoraloxifene analogs 3 in good yields. The use of multimethoxy-substituted desketoraloxifene analogs 3 affords considerable diversity. The final step in our synthesis delivers hydroxy-substituted desketoraloxifene analogs 4 using BBr3. The results are summarized in Table 1.
As illustrated in Table 1, entry 10, desketoraloxifene (VI) itself has been synthesized using the approach outlined. The desired dimethoxy-substituted desketoraloxifene analog 3e was obtained from compound 2e using 1-(2-hydroxyethyl)piperidine under the general Mitsunobu coupling conditions in 83% yield. Compound 3e was then converted by demethylation using BBr3 to the corresponding desketoraloxifene 4e (VI) in 78% yield. In a similar manner a variety of desketoraloxifene analogs 3 and 4 have been prepared in good yields and a minimum of steps.
In summary, a number of benzothiophene SERM analogs and the desketoraloxifene analogs 3/418 have been successfully synthesized starting from various oxygen-bearing 3-iodobenzo[b]thiophenes 1 by a two-step approach involving sequential Suzuki-Miyaura and Mitsunobu couplings. We believe that this approach to oxygen-bearing 3-iodobenzo[b]thiophenes 1 should readily afford many other functionalized desketoraloxifene analogs 3/4 using known chemistry and parallel synthesis strategies.
Supplementary Material
Acknowledgments
We thank the National Institute of General Medical Sciences (GM070620 and GM079593) and the National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (GM069663) for support of this research; Johnson Matthey, Inc. for donation of tetrakis(triphenylphosphine)palladium(0) and Frontier Scientific, Inc. for donation of 4-[(tetrahydro-2H-pyran-2-yl)oxy]benzeneboronic acid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:########.
References and notes
- (1).Lerner LJ, Holthaus JF, Thompson CR. Endocrinology. 1958;63:295. doi: 10.1210/endo-63-3-295. [DOI] [PubMed] [Google Scholar]
- (2).Harper MJ, Walpole AL. Nature. 1966;212:87. doi: 10.1038/212087a0. [DOI] [PubMed] [Google Scholar]
- (3).Jordan VC. Nat. Rev. Drug Discovery. 2003;2:205. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- (4).Jordan VC, Phelps E, Lindgren JU. Breast Cancer Res. Tr. 1987;10:31. doi: 10.1007/BF01806132. [DOI] [PubMed] [Google Scholar]
- (5).Killackey MA, Hakes TB, Pierce VK. Cancer Treat. Rep. 1985;69:237. [PubMed] [Google Scholar]
- (6).Robinson SP, Goldstein D, Witt PL, Borden EC, Jordan VC. Breast Cancer Res. Tr. 1990;15:95. doi: 10.1007/BF01810781. [DOI] [PubMed] [Google Scholar]
- (7).Liu H, Liu J, van Breemen RB, Thatcher GRJ, Bolton JL. Chem. Res. Toxicol. 2005;18:162. doi: 10.1021/tx049776u. [DOI] [PubMed] [Google Scholar]
- (8).Macgregor JI, Jordan VC. Pharmacol. Rev. 1998;50:151. [PubMed] [Google Scholar]
- (9).Bolton JL, Yu L, Thatcher GRJ. Methods Enzymol. 2004;378:110. doi: 10.1016/S0076-6879(04)78006-4. [DOI] [PubMed] [Google Scholar]
- (10).Weatherman RV, Carroll DC, Scanlan TS. Bioorg. Med. Chem. Lett. 2001;11:3129. doi: 10.1016/s0960-894x(01)00646-1. [DOI] [PubMed] [Google Scholar]
- (11).Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, Sporn MB. Cancer Res. 2001;61:8412. [PubMed] [Google Scholar]
- (12).Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, Matsumoto K, Palkowitz AD, Sato M, Termine JD, Winter MA, Yang NN, Dodge JA. Proc. Nat. Acad. Sci. U.S.A. 1997;94:14105. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Carta G, Knox AJS, Lloyd DG. J. Chem. Inf. Model. 2007;47:1564. doi: 10.1021/ci600471m. [DOI] [PubMed] [Google Scholar]
- (14).Yue D, Larock RC. J. Org. Chem. 2002;67:1905. doi: 10.1021/jo011016q. [DOI] [PubMed] [Google Scholar]
- (15).Cho C-H, Neuenswander B, Lushington GH, Larock RC. J. Comb. Chem. 2009;11:900. doi: 10.1021/cc9000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cho C-H, Neuenswander B, Larock RC. J. Comb. Chem. 2010;12:278. doi: 10.1021/cc900172u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mitsunobu O, Yamada Y. Bull. Chem. Soc. Japan. 1967;40:2380. [Google Scholar]
- (18).General procedure for iodocyclization using I2 to form compounds 1. To a solution of 5.0 mmol of the alkyne and 20 mL of CH2Cl2 was added gradually 1.2 equiv of I2 dissolved in 30 mL of CH2Cl2. The reaction mixture was allowed to stir at room temperature for up to 10 min. The reaction was monitored by TLC to establish completion. The remaining I2 was removed by washing with satd aq Na2S2O3. The mixture was then extracted by EtOAc (2 × 100 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by flash chromatography using EtOAc/hexanes as the eluent to afford the corresponding products 1.3-Iodo-5-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (1a). The product was obtained as a pale yellow solid (94% yield): mp 114–115 °C (uncorrected); 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 3.90 (s, 3H), 6.95–7.00 (m, 3H), 7.24 (d, J = 2.4 Hz, 1H), 7.58–7.60 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 55.5, 55.8, 78.8, 108.4, 114.0 (×2), 115.7, 123.0, 127.1, 131.1 (×2), 131.3, 143.2, 143.5, 158.6, 160.2; HRMS calcd for C16H13IO2S [M+], 395.9681, found 395.9684.General procedure for Suzuki-Miyaura coupling to form compounds 2. To a solution of 1 (1.0 mmol) and 5 mol % Pd(PPh3)4 in toluene (10 mL) was added K2CO3 (2.5 mmol) under an Ar atmosphere. To the resulting mixture was added p-THPOC6H4B(OH)2 (1.5 mmol) dissolved in ethanol (2 mL) and water (0.5 mL) and the reaction mixture heated to 80 °C for 6–8 h with vigorous stirring. After concentration of the solvent under reduced pressure, 10% aq HCl was added to the crude product in THF (0.1 M) at room temperature and stirred for 1 h. The mixture was then extracted by EtOAc (2 × 20 mL), and the aqueous phase was also extracted with EtOAc or CH2Cl2. The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by flash chromatography using EtOAc/hexanes as the eluent to afford the corresponding products 2.3-(4-Hydroxyphenyl)-5-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (2a). The product was obtained as a pale yellow oil (89% yield): 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 3.78 (s, 3H), 5.12 (br s, 1H), 6.78 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 6.96–7.03 (m, 2H), 7.20 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 55.5, 55.8, 105.8, 114.0 (×2), 114.3, 115.9 (×2), 122.9, 127.1, 128.3, 130.8 (×2), 131.1, 131.85 (×2), 131.89, 140.7, 142.4, 155.0, 157.8, 159.2; HRMS calcd for C22H18O3S [M+], 362.0977, found 362.0983.General procedure for the Mitsunobu reaction to form compounds 3. To a solution of 2 (0.2 mmol), triphenylphosphine (PPh3) (0.4 mmol), and alkylaminoethanol (0.3 mmol) in anhydrous THF (2 mL) was added diisopropyl azodicarboxylate (DIAD) (0.3 mmol) with stirring at 0–5 °C. The resulting solution was stirred at room temperature for 24–32 h (monitored by TLC until completion) and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using methanol/ethyl acetate/hexanes as the eluent to afford the corresponding products 3.6-Methoxy-2-(4-methoxyphenyl)-3-{4-[2-(1-piperidinyl)ethoxy]phenyl}benzo[b]thiophene (3e). The product was obtained as a pale yellow oil (83% yield): 1H NMR (400 MHz, CDCl3) δ 1.40–1.50 (m, 2H), 1.58–1.66 (m, 4H), 2.55 (br s, 4H), 2.81 (t, J = 6.0 Hz, 2H), 3.79 (s, 3H), 3.89 (s, 3H), 4.15 (t, J = 6.0 Hz, 2H), 6.78 (d, J = 8.9 Hz, 2H), 6.90–6.97 (m, 1H), 6.92 (d, J = 8.9 Hz, 2H), 7.22 (d, J = 8.8 Hz, 4H), 7.32 (d, J = 2.3 Hz, 1H), 7.44 (d, J = 8.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 24.4, 26.1 (×2), 55.3 (×2), 55.4, 55.9, 58.2, 66.1, 104.8, 114.0 (×2), 114.3, 114.9 (×2), 124.0, 127.2, 128.2, 130.7 (×2), 131.6 (×2), 131.7, 135.5, 136.4, 139.9, 157.5, 158.2, 159.0; HRMS calcd for C29H31NO3S [M+], 473.2025, found 473.2029.General procedure for the demethylation of 3e to form 6-hydroxy-2-(4-hydroxyphenyl)-3-{4-[2-(1-piperidinyl)ethoxy]phenyl}benzo[b]thiophene (desketoraloxifene, 4e). To a solution of compound 3e (0.095 mmol, 45 mg) in anhydrous CH2Cl2 (2 mL) cooled in an ice water bath under N2 was added BBr3 (0.38 mL, 0.38 mmol) while stirring. The solution turned orange in color. This solution was stirred for 3 h after slowly warming to room temperature. The reaction was quenched with satd aq NaHCO3 (2 × 2 mL) and the product was extracted with 5% CH3OH/CHCl3 (3 × 5 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by column chromatography using 5–10% CH3OH/CHCl3 as the eluent to provide 33 mg (78%) of desketoraloxifene (4e) as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 1.34–1.43 (m, 2H), 1.48–1.57 (m, 4H), 2.72 (br s, 2H), 3.35 (br s, 4H), 4.10 (t, J = 5.7 Hz, 2H), 6.67 (d, J = 8.7 Hz, 2H), 6.84 (dd, J = 2.2, 8.7 Hz, 1H), 6.99 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.17 (d, J = 8.7 Hz, 2H), 7.23 (d, J = 8.7 Hz, 1H), 7.28 (d, J = 2.2 Hz, 1H), 9.62 (s, 1H), 9.65 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 23.7, 25.3 (×2), 54.3 (×2), 57.2, 65.3, 107.0, 114.6, 114.7 (×2), 115.3 (×2), 123.2, 124.6, 127.4, 130.1 (×2), 130.7, 131.0 (×2), 133.5, 134.8, 138.8, 155.1, 156.9, 157.6; HRMS calcd for C27H27NO3S [M+], 445.1712, found 445.1725.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.