

Abstract
Protein modifications, including phosphorylation, ubiquitylation, and SUMOylation, have emerged as essential components of the response to DNA double-strand breaks (DSBs). Mutations within the genes encoding effectors of these components lead to genomic instability and in selected cases, human radiosensitivity and cancer susceptibility syndromes. In this review, we highlight recent advances in the study of DSB-associated signaling events by ubiquitylation and SUMOylation and discuss how coordination among protein modification systems integrates components of the DNA damage response into a network that regulates DNA repair and transcriptional processes on contiguous stretches of chromatin.
Keywords: posttranslational modification, ubiquitylation, SUMOylation, DNA damage response
DNA damage elicits a host of coordinated cellular responses, including recognition of the genomic lesion, signal transduction to halt the cell cycle, and repair of the damage. Depending on the extent of damage and the cell of origin harboring the DNA lesions, these responses influence cell fate determination, leading to survival, death, or senescence. Cellular responses triggered by DNA damage are collectively defined as the DNA damage response (DDR).
Protein posttranslational modifications (PTMs) accommodate a vast network of functions during the DDR by altering properties of target proteins, including enzyme activity, protein stability, subcellular localization, and abilities to interact with other proteins.1 In response to double-strand breaks (DSBs), phosphoinositide-3-kinase-related protein kinase (PIKK) family members ATM, ATR, and DNA-PKcs are activated in mammalian cells, resulting in phosphorylation of more than 700 target proteins.2 Several of these phosphorylation targets reside on chromatin, including H2AX and MDC1. PIKK-dependent phosphorylation within the C-terminal tail of H2AX (γH2AX) creates a docking site for the BRCT and FHA domain–containing protein MDC1, which is in turn phosphorylated by ATM, creating a recognition site by the FHA domain of the RING (Really Interesting New Gene) domain E3 ligase RNF8. Binding of phosphorylated MDC1 by RNF8 initiates an E3 ligase cascade consisting of RNF8 and a second E3 ligase RNF168, which is biallelically mutated in the human disease syndrome known as RIDDLE syndrome (radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties). Collectively, RNF8 and RNF168 are responsible for synthesizing the majority of the conjugated ubiquitin signal at DSBs. This signaling pathway reveals a modular assembly of the DDR on chromatin, connecting DDR-dependent phosphorylation to ubiquitylation at DSBs (Figure 1).
Figure 1.
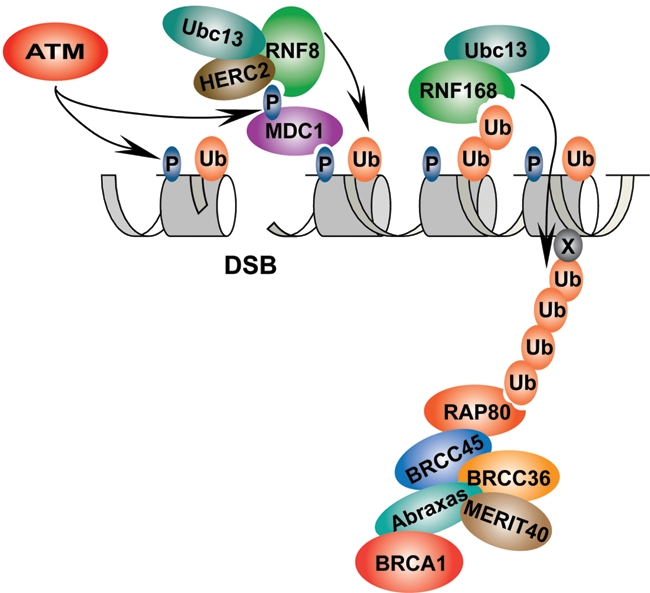
The DNA damage response (DDR) pathway. In response to DNA double-strand breaks (DSBs), ATM and related PIKKs (phosphoinositide-3-kinase-related protein kinases) phosphorylate H2AX (γH2AX), creating a platform for the assembly of DDR proteins. Localization of MDC1 to sites of damage by binding to γH2AX and its subsequent phosphorylation by ATM are required for the recruitment of the ubiquitin E3 ligase RNF8. RNF8, with the E2 ubiquitin-conjugating enzyme Ubc13 and HERC2, synthesizes K63-linked ubiquitin on histone H2A and its variants. RNF168, another RING (Really Interesting New Gene) domain ubiquitin ligase, recognizes the ubiquitin conjugates synthesized by RNF8/Ubc13, leading to its recruitment to sites of damage and to further synthesis of K63-linked ubiquitin. RAP80 (Receptor Associated Protein 80) specifically binds to K63-linked ubiquitin, recruiting a protein complex including Abraxas, MERIT40, BRCC36, BRCC45, and BRCA1 to sites of damage.
More recently, it has been shown that DSB ubiquitylation and the recruitment of BRCA1 and other repair proteins to the sites of damage are regulated by SUMOylation, placing SUMOylation as a critical component necessary for optimal ubiquitylation at DSBs.3,4 Mounting evidence suggests that phosphorylation, ubiquitylation, and SUMOylation events are important elements of the mammalian cell DDR to DSBs and that coordination of these events is required to achieve an integrated DDR, including detection of DNA damage, DNA repair, checkpoint activation, and transcription silencing. In this review, we discuss how ubiquitylation and SUMOylation function to protect cells from DNA damage, highlighting recent advances in the crosstalk between ubiquitylation and SUMOylation in the DDR.
Principles of Protein Ubiquitylation and SUMOylation
Ubiquitin and ubiquitin-like modifiers such as SUMO and NEDD8 are small proteins (8 to 20 kDa) that cells use to alter the functions of target proteins via covalent attachment of the modifiers. Ubiquitin is a highly conserved 76–amino acid protein that shares 20% sequence identity with SUMO.5,6 Vertebrates express 3 SUMO paralogs: SUMO-1, SUMO-2, and SUMO-3. Conjugated SUMO-2 and SUMO-3 differ only in 3 N-terminal amino acid residues, and they share 50% sequence homology with SUMO-1.7,8
Conjugation of ubiquitin to a target protein via a covalent isopeptide bond is catalyzed by a highly regulated 3-step enzymatic cascade that involves sequential actions of an E1-activating enzyme, an E2-conjugating enzyme, and an E3 ligase.9 The C-terminal glycine of ubiquitin is first catalyzed by E1 to form a covalent acyl–phosphate linkage with AMP. The catalytic cysteine of the E1 then attacks the linkage between ubiquitin and AMP, subsequently forming a thioester bond with the ubiquitin C-terminal glycine and releasing AMP. Through a transthiolation reaction, ubiquitin is transferred from the E1 catalytic cysteine to the E2 catalytic cysteine. The E3 ligase functions to recruit both the target protein and E2 and catalyze the transfer of ubiquitin from the E2 catalytic cysteine to a ϵ-amino group of the target protein lysine residue. SUMOylation requires a enzyme cascade similar to that of ubiquitylation, with the notable difference being that many fewer SUMO E2 and E3 enzymes have been identified.
Substrate specificity is usually provided by the E3 ligase. Consistent with the fact that a substantial fraction of the proteome is modified by ubiquitin or SUMO, human cells are predicted by bioinformatic analysis to express more than 600 E3 ligases.10 Three major categories of E3 ligases exist: RING domain–containing E3s, HECT domain–containing E3s (i.e., Homologous to E6AP [E6-associated protein] C-Terminus domain), and E3s that are not included in the previous 2 categories.11–13 Despite sharing the ability to simultaneously recruit both E2 and the substrate, HECT and RING domain E3 ligases use distinct molecular mechanisms to transfer activated ubiquitin from E2 to the target protein. Through its conserved cysteine residue, HECT domain E3 ligases form an intermediate thioester bond with the ubiquitin C-terminus before transferring the ubiquitin to the substrate.12 Conversely, it is commonly accepted that RING E3 functions as a scaffold to bring together the E2 and the substrate to catalyze a direct transfer of ubiquitin from E2 to the substrate without the formation of an E3–ubiqutin thioester intermediate.11 In contrast to the large number of ubiquitin ligases, several SUMO E3 ligases and a single SUMO E1 (SAE1/SAE2) and SUMO E2 (Ubc9) have been identified.
All seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) and the amino terminus of ubiquitin can be conjugated to the C-terminus of another ubiquitin molecule to form ubiquitin chains of differing structures in vivo.14,15 The linkage specificity, also known as topology, of ubiquitin conjugates is essential in determining the functional outcome of ubiquitylation. Assembly of the canonical K48-linked ubiquitin chains on proteins marks them for degradation by the 26S proteasome,16,17 whereas modification by K63-linked ubiquitin polymers does not direct substrates for degradation but usually functions in protein localization and signal transduction.18 Ubiquitin chains containing heterogeneous isopeptide linkages may confer yet additional complexity. Mass spectrometry data indicate the existence of multiple types of isopeptide linkages in a single ubiquitin polymer in vivo, and branched polyubiquitin chains have been shown to form in vitro.14,15,19,20 It is also possible that ubiquitin can be attached to SUMO, leading to the formation of a hybrid structure composed of mixed ubiquitin-like modifiers.21 Nevertheless, the physiologic functions of these ubiquitin structures remain to be determined. Like ubiquitin, SUMO-2/3, but not SUMO-1, can form polymers through isopeptide linkages, yet little is known about how poly-SUMO topology relates to function.7
RING E3s and HECT E3s contribute in a manner that is not fully understood to the type of isopeptide linkages. A RING domain E3 ligase can pair with different E2s to effect ubiquitin chain topology, determined by the specificity of associated E2 enzyme.20,22 For instance, the human heterodimeric RING E3 ligase BRCA1-BARD1 pairs with an E2-conjugating enzyme Ubch5c and directs the synthesis of K6-linked ubiquitin polymers.23,24 In vitro evidence also shows that BRCA1-BARD1 can bind to Ubc13-Mms2 or Ube2k to autoubiquitylate with K63-linked or K48-linked ubiquitin chains, respectively.22 Indeed, Ubc13-Mms2 is thought to be the only E2 enzyme responsible for K63-Ub synthesis in eukaryotes. Unlike the RING E3s, HECT E3 ligases are thought to specify the chain linkage of the product.25
Consistent with the diversity of ubiquitin structures, more than 20 ubiquitin-binding domains (UBDs) have been identified to date.26 Indeed, it is thought that specific recognition by UBD-containing proteins confers functional specificity of different ubiquitin chain topologies. Such interactions would enable sorting of ubiquitylated proteins to different locales within the cell. The majority of UBDs, including ubiquitin-interacting motif (UIM), motif interacting with ubiquitin (MIU, or inverted UIM), ubiquitin-binding zinc finder, and ubiquitin-associated domain (UBA), use a single or tandem α-helices to bind a hydrophobic patch of ubiquitin that is centered around Ile44.27,28 To bind the same ubiquitin surface around Ile44, some proteins instead employ β-sheet structures that are present in the domains of ubiquitin proteases.29 Because the Ile44-containing surface is crucial for the binding of ubiquitin by UBDs, it is likely that different UBDs compete for the Ile44-containing surface, and the transduction of the ubiquitin signal may therefore be controlled by the amount and binding affinity of the UBDs available.
Because the specificity of ubiquitin chains imparts distinct roles in cellular functions, it is not surprising that an increasing number of UBD-containing proteins display specificity in their recognition of ubiquitin linkages. RAD23A, a human homologue of radiation sensitivity abnormal 23, has a UBA domain at its C-terminus that specifically binds to K48-linked, but not K63-linked, ubiquitin chains and so targets modified substrates to the 26S proteasome.30 Other UBD-containing proteins, including ubiquitin-specific peptidase 2 (USP2),31 Npl4 zinc finger domain of Tak1 binding protein 2,31 and Receptor Associated Protein 80 (RAP80),32 preferentially bind to K63-linked, instead of K48-linked, ubiquitin chains. RAP80 specifically binds to K63-linked ubiquitin chains through 2 tandem UIMs and targets a BRCA1-containing protein complex to DNA damage-induced foci.20,32,33 Structural studies of RAP80 revealed that the region linking the 2 UIMs ensures that the RAP80 UIM1 and UIM2 α-helices are arranged to specifically bind the proximal and distal ubiquitin moieties across a single K63 linkage.34,35 Contrary to RAP80, ataxin-3, an ubiquitin-specific protease associated with the development of spinocerebellar ataxia type 3, demonstrates K48 linkage specificity with 2 UIMs and a 2-residue linker region.36 Remarkably, swapping the linker between the 2 UIMs of RAP80 with the 2-residue linker of ataxin-3 results in a 6-fold increase of K48 preference of the Rap80 UIMs.35
Modification by ubiquitin or SUMO is reversible through the action of deubiquitylating enzymes (DUBs). DUBs are proteases that hydrolyze ubiquitin or ubiquitin-like proteins, and their functions include activating ubiquitin pro-proteins, reversing modifications by ubiquitylation or ubiquitin-like modifiers, and recycling ubiquitin before proteolytic degradation of an ubiquitylated substrate by the proteasome. Nearly 100 DUBs are encoded by the human genome, whereas it is estimated that there are 6 human deSumoylating enzymes.37,38 DUBs are divided into 2 categories based on the catalytic mechanism: (1) the papain-like cysteine protease family, which uses an active site cysteine for nucleophilic attack of the ubiquitin isopeptide bond, and (2) the JAB1/MPN/Mov34 metalloenzyme domain zinc-dependent metalloprotease family, which hydrolyzes the ubiquitin conjugates using Zn2+ dependent hydroxide delivery to hydrolyze the scissile ubiquitin isopeptide bond.39,40 To date, only active site cysteine-containing proteases have been implicated in deSUMOylation.41 DUB activity is tightly regulated so that cells have fine-tuned control over ubiquitin cleavage. For example, DUBs are normally constitutively inactive, and their activity is regulated by PTMs of the enzyme,42-44 the binding to substrates 45,46 or association with specific cellular organelles.47
Ubiquitylation and SUMOylation in the Protection of Genome Integrity
Emergence for Diverse Ubiquitin and SUMO Landscapes at DNA Damage-Induced Foci
DSBs are potentially cytotoxic and pose a significant threat to genome integrity. DDR proteins respond to DNA damage within minutes, resulting in rapid accumulation of signaling and repair factors at the sites of damage. The accumulation and retention of DDR factors, the number of which is estimated to be more than 1,000 molecules of a given DNA repair protein at a single DSB,48 can be visualized as repair foci by fluorescent microscopy.
Upon DSB induction, ubiquitin species with different linkage types emerge at the sites of damage, albeit with different kinetics.49 The accumulation of these ubiquitin conjugates at the sites of DNA damage depends on phosphorylation events mediated by PIKKs (Figure 1). DSBs activate ATM and related PIKKs, which phosphorylate H2AX and lead to recruitment of MDC1 to the sites of damage. The phosphorylation of MDC1 by ATM is required for the recruitment of RNF8, the first of the 3 ubiquitin E3 ligases that sequentially localize to damage-induced foci. The RING E3 ligase RNF8 binds to phosphorylated MDC1 via its forkhead-associated (FHA) domain. The FHA domain of RNF8 has recently been reported to bind to the HECT domain of HERC2 following its ionizing radiation–induced (IR-induced) phosphorylation at Thr4827. Interestingly, although RNF8 uses the same FHA domain for binding phosphorylated MDC1 and HERC2, these interactions are not mutually exclusive.50 The interaction between HERC2 and RNF8 facilitates the assembly of the E2 ubiquitin-conjugating enzyme Ubc13 with RNF8 and promotes the synthesis of DNA damage-induced K63-linked ubiquitin chains on H2A and its variants.50 Another RING domain ubiquitin E3 ligase, RNF168, recognizes and binds to K63-linked ubiquitin chains on H2A and H2AX through its 2 MIUs, amplifying the local concentration of K63-linked ubiquitin with Ubc13 and resulting in the recruitment and retention of 53BP1 and BRCA1 at the sites of lesions. Collectively, RNF8 and RNF168 are responsible for the majority of conjugated ubiquitin signal at DSBs.51,52 BRCA1 is yet another ubiquitin E3 ligase that localizes to damage-induced foci. Unlike RNF8 or RNF168, BRCA1 localization requires its interaction with a complex composed of BRCC36, BRCC45, Abraxas, MERIT40, and RAP80.53-55 RAP80 specifically recognizes K63-linked ubiquitin conjugates through its 2 UIMs and recruits BRCA1 and the other members of the BRCA1–RAP80 complex to DNA damage sites.32,33,56 BRCA1 functions as a heterodimer with the RING domain protein BARD1 and has been proposed to synthesize K6-linked ubiquitin at sites of DNA damage. Indeed, it has been shown to be autoubiquitylated by K6-linked ubiquitin in the presence of Ubch5c in vitro, and the accumulation of K6-linked ubiquitin at damage-induced foci depends on both BRCA1 and Ubch5c.23,24,57 In response to DNA damage, BRCA1 has also been reported to ubiquitylate BRCA1 carboxy–terminal interacting partner CtBP-interacting protein (CtIP) in a manner that depends on a phosphorylation-mediated interaction between CtIP and BRCA1 BRCT domains.58 The E3 ligase activity of BRCA1 through ubiquitylation of CtIP was suggested to be required for its control over a transient G2/M checkpoint.58,59 To address the role of BRCA1 E3 ligase activity in a clean genetic system, a hypomorphic variant of BRCA1 with respect to E3 ligase activity was knocked into murine embryonic stem cells. This rationally designed mutation reduces BRCA1 RING domain association with Ubch5c, limiting BRCA1 in vitro E3 ligase activity.60 To the surprise of many, BRCA1I26A did not show obvious deficits in homologous recombination-mediated repair of a nuclease-induced DSB, suggesting that either compensatory mechanisms exist within the cell or that BRCA1 E3 ligase activity plays a heretofore unappreciated role in a process, other than the repair of DSBs by homologous recombination.61 An alternative possibility is that additional regulatory factors enable the BRCA1I26A mutant to be an active E3 ligase in vivo. Recent evidence that BRCA1 E3 activity is increased 10- to 20-fold in vitro by SUMOylation4 raises the possibility that BRCA1 SUMOylation could surmount the reduced affinity of BRCA1I26A for E2 enzymes, thereby maintaining BRCA1-dependent DSB ubiquitylation. A detailed analysis of BRCA1I26A knockin cells will be necessary to address these possibilities.
With regard to the role of SUMOylation in the DDR, it has been shown to participate in base excision repair, regulation of translesion DNA synthesis, and both homologous recombination and nonhomologous end-joining repair of DSBs.3,4,62,63 A base excision repair protein, thymine-DNA glycosylase (TDG), plays a central role in the cellular defense against mutations caused by the spontaneous deamination of 5-methylcytosine and cytosine. TDG removes the resultant uracil or thymine that mispairs with guanine, leaving an abasic site for the ensuing actions of base excision repair.64,65 DNA-bound TDG can be SUMOylated, which alters the conformation of TDG and dissociates the glycosylase from the abasic site.66 Another example with regard to roles of SUMOylation in DNA repair is RAD52, which plays a central role in homologous recombination. Rad52 promotes strand annealing by exchanging RPA with Rad51 on single-stranded DNA. SUMOylation of Rad52 sustains its activity and protects Rad52 from proteaosomal degradation, implicating a role of SUMOylation in the homologous recombination pathway for the protection of genome integrity.67 SUMOylation also participates in nonhomologous end joining. Repair proteins involved in nonhomologous end joining, including Ku70-Ku80 and XRCC4, and 53BP1 have also been found to be modified by SUMOylation, and failure to execute SUMOylation reactions at DSBs reduces nonhomologous end-joining mechanisms of repair.3,4,68-70
Circumstantial evidence exists for a far broader role of SUMO in the DDR. Sequence alignments showed that out of the 700 proteins that are potential phosphorylation targets of ATM/ATR,2 618 of them contain at least one SUMO modification motif, ψKxE (our analysis), suggesting that DNA damage may target PIKK substrates for SUMOylation.
DNA Damage-Associated Ubiquitylation and SUMOylation in the Protection of Genome Integrity
Disruption of the formation, recognition, and clearance of ubiquitin or SUMO conjugates at DSBs lead to genomic instability and in selected cases, human radiosensitivity and cancer susceptibility syndromes. Mutations in BRCA1 and BRCA2 occur in about 20% of the families with inherited breast cancers.71,72 The RING domain of BRCA1 is the catalytic domain for its ubiquitin E3 ligase activity. Mutations in the RING domain, which account for approximately 20% of the clinical BRCA1 missense mutations, disrupt the E3 ligase activity of BRCA1 by altering its interaction with ubiquitin-conjugating enzymes.73,74 In addition, RNF168, an ubiquitin E3 ligase, was found mutated in the recently discovered RIDDLE syndrome, which manifested as immunodeficiency and radiosensitivity. Disruption of RNF168-dependent conjugation of K63-linked ubiquitin results in impaired localization of 53BP1 and BRCA1 to the sites of DSBs.51,52
Disruption of the SUMO pathway also results in decreased repair and sensitizes cells to DNA damaging agents. Saccharomyces cerevisiae cells expressing a temperature-sensitive SUMO E2 mutant, Ubc9, display hypersensitivity to the alkylating agent MMS and to ultraviolet radiation at a semipermissive temperature.75 Furthermore, overexpression of a dominant-negative mutant of Ubc9 in the human breast cancer cell line, MCF-7, resulted in increased sensitivity to anticancer agents requiring DSB repair by homologous recombination.76 Furthermore, studies in yeast and human cells revealed that deficiency of the SUMO ligase MMS21/hMMS21 resulted in decreased capacity to repair DNA lesions.77,78
Besides the synthesis of ubiquitin conjugates, the specific recognition of ubiquitin structures at the sites of damage is important for preserving genome integrity and preventing the onset of familial breast cancer. Through its tandem UIMs, RAP80 specifically recognizes K63-linked ubiquitin chains synthesized at the sites of DNA damage and mediates the recruitment of BRCA1 to DSBs. Sequencing for mutations in the RAP80 gene in BRCA1/BRCA2 mutation-negative Finnish breast cancer families identified a single–amino acid deletion (delE81) in one of the highly conserved UIMs of RAP80.79 This mutant protein showed severely impaired ability to bind to ubiquitin and to localize itself and BRCA1 to DSBs. In addition, expression of the mutant protein significantly increased the level of chromosomal aberrations.79 Interestingly, a screen for mutations in MERIT40 and Abraxas, core components of the BRCA1–RAP80 complex, in breast and ovarian cancer families did not find disease-related sequence variations, suggesting that loss of function mutations within the BRCA1–RAP80 complex will not be a common genetic cause of inherited breast cancer.80,81 However, given that multiple recognition events for linkage-specific ubiquitin species are required for the proper execution of DDR signaling and tumor suppression,82-85 it is possible that mutations in genes central to these pathways will contribute to the onset of familial cancer syndromes. RNF8, RNF168, and other components of the ubiquitin and SUMO DDR pathways are new candidate genes worthy of investigation in breast cancer and in other malignancies.
In addition to the synthesis and recognition of ubiquitin, evidence has emerged in the last several years that clearance of the ubiquitin by DUBs is an indispensible element of the DDR. Similarly, clearance of γH2AX phosphorylation is required for a full recovery from the DDR.86,87 Following IR, USP3-depleted cells showed persistent γH2AX and conjugated ubiquitin foci and a prolonged G2/M checkpoint.88 Yet, DUBs may protect the integrity of the genetic information of an organism by eliminating the cells with extensive DNA damage through apoptosis. For example, USP28 has been shown to be required for DNA damage-induced apoptosis mediated by the Chk2-p53-PUMA pathway through stabilizing Chk2 and 53BP1.89 The temporal control of DUBs activity and the functions of target protein altered by deubiquitylation are also critical determinants of the role of DUBs in preserving genome stability. For instance, the Fanconi anemia syndrome proteins FANCD2 and FANCI are each monoubiquitylated, and this modification is critical for their localization to DSBs.90,91 USP1 binds to FANCD2, and the inhibition of USP1 leads to the accumulation of monoubiquitylated FANCD2 and mitomycin C hypersensitivity, suggesting that failure to deubiquitylate FANCD2, like impaired ubiquitylation, inhibits its function in repairing DNA interstrand cross-links.92,93 As with deubiquitylation, regulated cleavage of SUMO from target proteins is important for the proper execution of DDR. Inhibition of Ulp2, SUMO homolog Smt3-deconjugating enzyme, in S. cerevisiae as well as mutations in Ulp1 in Schizosaccharomyces pombe led to hypersensitivity to hydroxyurea and UV radiation, respectively.94,95
SUMOylation-Dependent Ubiquitylation
Because a specific lysine residue in a protein can be subject to either ubiquitylation or SUMOylation, it is not surprising that a single protein can perform distinct functions based on the PTM mark it bears. For example, proliferating cell nuclear antigen (PCNA) can be modified either by monoubiquitin, K63-linked ubiquitin chains, or SUMO on the same lysine residue (K164).96 During S phase, in response to stalled replication forks, monoubiquitylation of PCNA on K164 directs cells for translesion synthesis, whereas polyubiquitylation leads to recombination-related error-free repair, of which the detailed mechanism remains unclear.96-98 SUMOylation of PCNA on K164 in yeast specifically recruits Srs2 to prevent unwanted recombination between newly formed sister chromatids following DNA replication.99,100
DSBs trigger a DDR that is characterized by the synthesis of ubiquitin conjugates at the sites of damage-induced repair foci. Sequential recognition of the K63-linked ubiquitin structures eventually leads to the recruitment of BRCA1 and 53BP1. Two recently published studies report that the synthesis of ubiquitin conjugates mediated by BRCA1 depends on 2 SUMO E3 ligases, PIAS1 and PIAS4.3,4 Depletion of PIAS1 or PIAS4 significantly reduced the level of FK-2-recognized ubiquitin conjugates at the sites of IR-induced foci or microirradiation stripes.3,4 Although it has been shown that the disruption of PIAS1 or PIAS4 activity leads to impaired recruitment of BRCA1,3,4 it remains unclear how the SUMO E3 ligases participate in the recruitment of BRCA1. In these studies, depletion of PIAS4 has been shown to affect the sequential recruitment of DNA damage-responsive proteins up to RNF168, whereas depletion of PIAS1 affects only the accumulation of RAP80 and BRCA1, suggesting that the 2 SUMO E3 ligases affect the recruitment of BRCA1 through distinct mechanisms.3,4 BRCA1 has 2 SUMO target motifs (ψKxE) immediately following its N-terminal RING domain, and mutation of the lysine residue (K119R) of one of these motifs is associated with decreased ubiquitin accumulation at the site of BRCA1 foci following IR.4 In vitro experiments showed that SUMOylated BRCA1–BARD heterodimer has approximately 10- to 20-fold increased ubiquitin ligase activity, compared to the unmodified heterodimer.4 Together, these results suggest that PIAS1- or PIAS4-dependent synthesis of ubiquitin conjugates at the sites of DNA damage might be mediated through the regulation of BRCA1 ligase activity via SUMO modification.
Consistent with this interpretation, BRCA1 has been shown to be modified by SUMO in response to DNA damage,3,4 yet it is not clear whether it is modified by SUMO-1 or SUMO-2/3 or both. PIAS1 has been demonstrated to be responsible for SUMO-2/3 conjugation, whereas PIAS4 is mainly responsible for the synthesis of SUMO-1 and, to a lesser extent, the synthesis of SUMO- 2/3.3 However, it has also been shown that depletion of either PIAS1 or PIAS4 impaired the pulling down of BRCA1 by both anti-SUMO-1 and anti-SUMO-2/3 antibodies.4
Consistent with the roles of PIAS1 and PIAS4 in the DDR, depleting either of them resulted in decreased DSB repair by homologous recombination and nonhomologous end joining.3,4 Together, these studies established SUMOylation as a key element in the DDR.
DSB-Induced Transcription Silencing: Functions of DSB-Associated Ubiquitin beyond DNA Repair Protein Recruitment
The ultimate goal of the DDR is to protect the integrity of genetic information and its faithful transmission either to DNA by replication or to mRNA by transcription. Although physiological DSBs have been shown to elicit global transcriptional activation and chromatin relaxation in an ATM-dependent manner,101,102 little is known about how DDR influences the transcription of genes local to the sites of DNA damage. A common modification displayed at both DSBs and transcriptionally silent chromatin is ubiquitylated histone H2A (uH2A).49,103 However, until recently, it has been unclear if uH2A could mediate cross talk between these processes. Elucidation of the relationship between the DDR and transcriptional silencing local to the sites of DSBs has recently been reported (Shanbhag et al.104). By modifying a system previously used for visualizing gene transcriptional in a single living cell,105 this study investigated how the DDR influences the transcription of genes on the same stretch of chromatin distal to DSBs. The system was developed by integrating transgene array integrated into a single location in the genome of human U2OS cells on chromosome 1p3.6.105 Each unit of the transgene array is composed of multiple DNA sequence elements, including 256 copies of the lac operator, 96 copies of tetracycline response elements (TRE), a minimal CMV promoter, CFP fused to the peroxisomal targeting signal SKL, and 24 copies of the MS2 RNA stem loop sequence (Figure 2A). FokI endonuclease is fused to the lac repressor–mCherry fusion protein to introduce DSBs at least 4 kb away from the CMV promoter. Doxycycline can be used to activate the transcription of the genes downstream of the TRE. Nascent transcription can be visualized by YFP-MS2 that binds to MS2 stem loops, and translation can be indicated by the accumulation of CFP-SKL in the cytoplasm.
Figure 2.
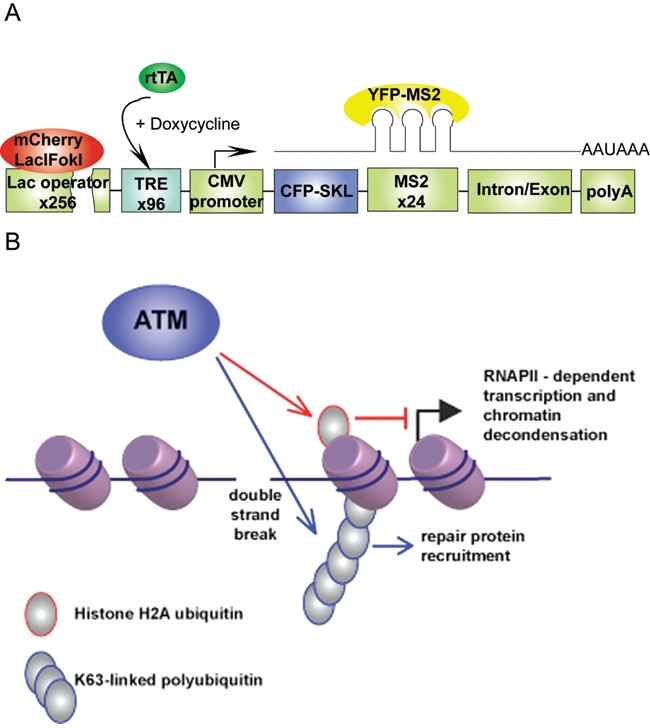
Double-strand break–induced (DSB-induced) transcriptional silencing. (A) Schematic of the transcription reporter locus. The lac operator (256 copies) is followed by the tetracycline response element (96 repeats) and a CMV promoter-driven reporter gene that codes for the CFP-SKL protein and the MS2 RNA stem loop (24 repeats). Expression of the mCherry-Lac repressor-FokI nuclease domain fusion protein induces DSBs within the lac operator region. Transcription of the report gene can be induced by doxycycline and visualized by YFP-MS2 that binds to the MS2 stem loops. (B) DSB-induced local transcriptional silencing. DSBs induce the synthesis of histone H2A ubiquitin and K63-linked polyubiquitin chains at sites of damage. Although histone H2A ubiquitin is responsible for DSB-induced ATM-dependent transcriptional silencing, K63-linked polyubiquitin chains are responsible for the recruitment of repair factors.
Using the single-cell reporter system, a novel DDR-associated transcriptional silencing program, DSB-induced silencing cis (DISC), has been identified (Shanbhag et al.104). FokI endonuclease-induced DSBs resulted in transcription inhibition of distal genes on the contiguous stretch of DNA, as demonstrated by decreased YFP-MS2 signal close to the sites of damage as well as decreased MS2 transcript levels measured by qRT-PCR. DISC has been shown to be a local cellular response to the damage and is dependent on the activity of ATM, given that ATM inhibition reverses the damage-induced transcriptional silencing. Similar to polycomb repressive complex 1–related (PRC1-related) transcription repression,106-108 DSBs-associated transcription silencing is correlated with the ubiquitylation of H2A but not with DSB-associated K63-Ub levels (Figure 2B). Mechanistically, DISC may be achieved through hypophosphorylation of RNAPII at the Serine 2 residue of the c-terminal domain repeat region, thereby preventing RNAPII-dependent transcriptional elongation. ATM-dependent inhibition of RNAPII elongation during DISC prevented transcription-associated chromatin decondensation, a particularly interesting observation in light of the strong connection between transcription and aberrant chromosomal translocations found in malignancy.109,110 Perhaps ATM-dependent silencing prevents large-scale chromatin movements that could disrupt synapsis of chromosomal termini at a DSB. Indeed, ATM deficiency is highly correlated with transcription-associated chromosome translocations.109,111,112
Similarities may exist between DISC and polycomb-dependent transcriptional silencing.113 Polycomb-repressive complex 2–related (PRC2-related) transcription silencing is centered at trimethylation of H3K27, which has been reported to be enriched at DSBs.114 PRC1 complexes cooperate with PRC2 to mediate silencing through uH2A-dependent RNAPII inhibition at PRC-repressed genes.107,115,116 Although it has been suggested that uH2A prevents RNAPII phosphorylation by chromatin transcription complex, consequently blocking RNAPII elongation,117,118 how the DDR signals to RNAPII hypophosphorylation and inhibition of elongation remains to be explored.
Identification of DISC for the first time demonstrated a direct regulation of the DDR on the transcription of genes distal to the sites of DNA damage on a contiguous stretch of chromatin. Several important questions remain to be answered, including the precise mechanism of DSB silencing, how far silencing extends along chromatin from a single DSB, and what role silencing plays in maintaining genome integrity. An interesting question for future investigations is whether DSB silencing contributes to epigenetic changes emanating from persistent DSB responses following IR or telomere dysfunction, given that these lesions are known to lead to the formation of senescence-associated heterochromatin bodies. The tools are now in place to begin addressing these important questions.
Final Words
PTMs, including phosphorylation, ubiquitylation, and SUMOylation, are key elements of the DDR that provide cells multiple means to fine-tune cellular responses to DNA damage. Although the roles of ubiquitylation and SUMOylation are the emphasis of this review, compelling evidence exists that other PTMs, such as acetylation, methylation, and mono- and poly-ADP ribosylation, also actively participate in the DDR, creating a dynamic network of molecular actions that cells use to protect their genetic information.
Acknowledgments
R.A.G. gratefully acknowledges funding from 1R01CA138835-01 from the NCI, an American Cancer Society Research Scholar Grant, K08 awards 1K08CA106597-01 and 3K08CA106597-04S1 from the NCI, the Sidney Kimmel Foundation Scholar Award, and funds from the Abramson Family Cancer Research Institute and the Penn Genomes Frontiers Institute.
Footnotes
The authors declare no conflicts of interest with respect to the publication of this article.
References
- 1. Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666-72 [DOI] [PubMed] [Google Scholar]
- 2. Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160-6 [DOI] [PubMed] [Google Scholar]
- 3. Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morris JR, Boutell C, Keppler M, et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature. 2009;462:886-90 [DOI] [PubMed] [Google Scholar]
- 5. Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97-107 [DOI] [PubMed] [Google Scholar]
- 6. Matunis MJ, Coutavas E, Blobel G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J Cell Biol. 1996;135:1457-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tatham MH, Jaffray E, Vaughan OA, et al. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 2001;276:35368-74 [DOI] [PubMed] [Google Scholar]
- 8. Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem. 2000;275:6252-8 [DOI] [PubMed] [Google Scholar]
- 9. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55-72 [DOI] [PubMed] [Google Scholar]
- 10. Li W, Bengtson MH, Ulbrich A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One. 2008;3:e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399-434 [DOI] [PubMed] [Google Scholar]
- 12. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398-409 [DOI] [PubMed] [Google Scholar]
- 13. Pichler A, Knipscheer P, Saitoh H, Sixma TK, Melchior F. The RanBP2 SUMO E3 ligase is neither HECT- nor RING-type. Nat Struct Mol Biol. 2004;11:984-91 [DOI] [PubMed] [Google Scholar]
- 14. Peng J, Schwartz D, Elias JE, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921-6 [DOI] [PubMed] [Google Scholar]
- 15. Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576-83 [DOI] [PubMed] [Google Scholar]
- 17. Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275-86 [DOI] [PubMed] [Google Scholar]
- 19. Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J. 2008;411:249-60 [DOI] [PubMed] [Google Scholar]
- 20. Kim HT, Kim KP, Lledias F, et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375-86 [DOI] [PubMed] [Google Scholar]
- 21. Tatham MH, Geoffroy MC, Shen L, et al. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol. 2008;10:538-46 [DOI] [PubMed] [Google Scholar]
- 22. Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol. 2007;14:941-8 [DOI] [PubMed] [Google Scholar]
- 23. Morris JR, Solomon E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet. 2004;13:807-17 [DOI] [PubMed] [Google Scholar]
- 24. Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem. 2003;278:34743-6 [DOI] [PubMed] [Google Scholar]
- 25. Wang M, Pickart CM. Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 2005;24:4324-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains: from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grabbe C, Dikic I. Functional roles of ubiquitin-like domain (ULD) and ubiquitin-binding domain (UBD) containing proteins. Chem Rev. 2009;109:1481-94 [DOI] [PubMed] [Google Scholar]
- 28. Harper JW, Schulman BA. Structural complexity in ubiquitin recognition. Cell. 2006;124:1133-6 [DOI] [PubMed] [Google Scholar]
- 29. Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE. A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol Cell. 2006;21:873-80 [DOI] [PubMed] [Google Scholar]
- 30. Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol. 2005;12:708-14 [DOI] [PubMed] [Google Scholar]
- 31. Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009;10:466-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sobhian B, Shao G, Lilli DR, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang B, Matsuoka S, Ballif BA, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato Y, Yoshikawa A, Mimura H, Yamashita M, Yamagata A, Fukai S. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by tandem UIMs of RAP80. EMBO J. 2009;28:2461-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sims JJ, Cohen RE. Linkage-specific avidity defines the lysine 63-linked polyubiquitin-binding preference of rap80. Mol Cell. 2009;33:775-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chai Y, Berke SS, Cohen RE, Paulson HL. Poly-ubiquitin binding by the polyglutamine disease protein ataxin-3 links its normal function to protein surveillance pathways. J Biol Chem. 2004;279:3605-11 [DOI] [PubMed] [Google Scholar]
- 37. Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773-86 [DOI] [PubMed] [Google Scholar]
- 38. Kim JH, Baek SH. Emerging roles of desumoylating enzymes. Biochim Biophys Acta. 2009;1792:155-62 [DOI] [PubMed] [Google Scholar]
- 39. Johnston SC, Larsen CN, Cook WJ, Wilkinson KD, Hill CP. Crystal structure of a deubiquitinating enzyme (human UCH-L3) at 1.8 A resolution. EMBO J. 1997;16:3787-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato Y, Yoshikawa A, Yamagata A, et al. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358-62 [DOI] [PubMed] [Google Scholar]
- 41. Drag M, Salvesen GS. DeSUMOylating enzymes: SENPs. IUBMB Life. 2008;60:734-42 [DOI] [PubMed] [Google Scholar]
- 42. Popov N, Herold S, Llamazares M, Schulein C, Eilers M. Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle. 2007;6:2327-31 [DOI] [PubMed] [Google Scholar]
- 43. Wada K, Kamitani T. UnpEL/Usp4 is ubiquitinated by Ro52 and deubiquitinated by itself. Biochem Biophys Res Commun. 2006;342:253-8 [DOI] [PubMed] [Google Scholar]
- 44. Meulmeester E, Kunze M, Hsiao HH, Urlaub H, Melchior F. Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Mol Cell. 2008;30:610-9 [DOI] [PubMed] [Google Scholar]
- 45. Hu M, Li P, Song L, et al. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu M, Li P, Li M, et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041-54 [DOI] [PubMed] [Google Scholar]
- 47. Yao T, Song L, Xu W, et al. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol. 2006;8:994-1002 [DOI] [PubMed] [Google Scholar]
- 48. Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699-713 [DOI] [PubMed] [Google Scholar]
- 49. Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol. 2009;187:319-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, et al. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat Cell Biol. 2010;12:80-6 [DOI] [PubMed] [Google Scholar]
- 51. Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435-46 [DOI] [PubMed] [Google Scholar]
- 52. Stewart GS, Panier S, Townsend K, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420-34 [DOI] [PubMed] [Google Scholar]
- 53. Feng L, Huang J, Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009;23:719-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shao G, Patterson-Fortin J, Messick TE, et al. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23:740-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23:729-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202-5 [DOI] [PubMed] [Google Scholar]
- 57. Polanowska J, Martin JS, Garcia-Muse T, Petalcorin MI, Boulton SJ. A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J. 2006;25:2178-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20:1721-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brzovic PS, Keeffe JR, Nishikawa H, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A. 2003;100:5646-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reid LJ, Shakya R, Modi AP, et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci U S A. 2008;105:20876-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morris JR. SUMO in the mammalian response to DNA damage. Biochem Soc Trans. 2010;38:92-7 [DOI] [PubMed] [Google Scholar]
- 63. Thomson TM, Guerra-Rebollo M. Ubiquitin and SUMO signalling in DNA repair. Biochem Soc Trans. 2010;38:116-31 [DOI] [PubMed] [Google Scholar]
- 64. Hardeland U, Bentele M, Lettieri T, Steinacher R, Jiricny J, Schar P. Thymine DNA glycosylase. Prog Nucleic Acid Res Mol Biol. 2001;68:235-53 [DOI] [PubMed] [Google Scholar]
- 65. Scharer OD, Jiricny J. Recent progress in the biology, chemistry and structural biology of DNA glycosylases. Bioessays. 2001;23:270-81 [DOI] [PubMed] [Google Scholar]
- 66. Ulrich HD. SUMO modification: wrestling with protein conformation. Curr Biol. 2005;15:R257-9 [DOI] [PubMed] [Google Scholar]
- 67. Sacher M, Pfander B, Hoege C, Jentsch S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat Cell Biol. 2006;8:1284-90 [DOI] [PubMed] [Google Scholar]
- 68. Golebiowski F, Matic I, Tatham MH, et al. System-wide changes to SUMO modifications in response to heat shock. Sci Signal. 2009;2:ra24. [DOI] [PubMed] [Google Scholar]
- 69. Yurchenko V, Xue Z, Gama V, Matsuyama S, Sadofsky MJ. Ku70 is stabilized by increased cellular SUMO. Biochem Biophys Res Commun. 2008;366:263-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yurchenko V, Xue Z, Sadofsky MJ. SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Mol Cell Biol. 2006;26:1786-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet. 2008;40:17-22 [DOI] [PubMed] [Google Scholar]
- 72. Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med. 2003;348:2339-47 [DOI] [PubMed] [Google Scholar]
- 73. Brzovic PS, Meza JE, King MC, Klevit RE. BRCA1 RING domain cancer-predisposing mutations: structural consequences and effects on protein-protein interactions. J Biol Chem. 2001;276:41399-406 [DOI] [PubMed] [Google Scholar]
- 74. Morris JR, Pangon L, Boutell C, Katagiri T, Keep NH, Solomon E. Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum Mol Genet. 2006;15:599-606 [DOI] [PubMed] [Google Scholar]
- 75. Maeda D, Seki M, Onoda F, Branzei D, Kawabe Y, Enomoto T. Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S. cerevisiae. DNA Repair (Amst). 2004;3:335-41 [DOI] [PubMed] [Google Scholar]
- 76. Mo YY, Yu Y, Ee PL, Beck WT. Overexpression of a dominant-negative mutant Ubc9 is associated with increased sensitivity to anticancer drugs. Cancer Res. 2004;64:2793-8 [DOI] [PubMed] [Google Scholar]
- 77. Potts PR, Yu H. Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol Cell Biol. 2005;25:7021-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhao X, Blobel G. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci U S A. 2005;102:4777-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nikkila J, Coleman KA, Morrissey D, et al. Familial breast cancer screening reveals an alteration in the RAP80 UIM domain that impairs DNA damage response function. Oncogene. 2009;28:1843-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Solyom S, Patterson-Fortin J, Pylkas K, Greenberg RA, Winqvist R. Mutation screening of the MERIT40 gene encoding a novel BRCA1 and RAP80 interacting protein in breast cancer families. Breast Cancer Res Treat. 2010;120:165-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Novak DJ, Sabbaghian N, Maillet P, Chappuis PO, Foulkes WD, Tischkowitz M. Analysis of the genes coding for the BRCA1-interacting proteins, RAP80 and Abraxas (CCDC98), in high-risk, non-BRCA1/2, multiethnic breast cancer cases. Breast Cancer Res Treat. 2009;117:453-9 [DOI] [PubMed] [Google Scholar]
- 82. Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735-48 [DOI] [PubMed] [Google Scholar]
- 84. Chenevix-Trench G, Spurdle AB, Gatei M, et al. Dominant negative ATM mutations in breast cancer families. J Natl Cancer Inst. 2002;94:205-15 [DOI] [PubMed] [Google Scholar]
- 85. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Keogh MC, Kim JA, Downey M, et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497-501 [DOI] [PubMed] [Google Scholar]
- 87. Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nicassio F, Corrado N, Vissers JH, et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr Biol. 2007;17:1972-7 [DOI] [PubMed] [Google Scholar]
- 89. Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529-42 [DOI] [PubMed] [Google Scholar]
- 90. Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Smogorzewska A, Matsuoka S, Vinciguerra P, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Oestergaard VH, Langevin F, Kuiken HJ, et al. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28:798-809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim JM, Parmar K, Huang M, et al. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009;16:314-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li SJ, Hochstrasser M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol. 2000;20:2367-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Taylor DL, Ho JC, Oliver A, Watts FZ. Cell-cycle-dependent localisation of Ulp1, a Schizosaccharomyces pombe Pmt3 (SUMO)-specific protease. J Cell Sci. 2002;115:1113-22 [DOI] [PubMed] [Google Scholar]
- 96. Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135-41 [DOI] [PubMed] [Google Scholar]
- 97. Bienko M, Green CM, Crosetto N, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821-4 [DOI] [PubMed] [Google Scholar]
- 98. Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491-500 [DOI] [PubMed] [Google Scholar]
- 99. Papouli E, Chen S, Davies AA, et al. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell. 2005;19:123-33 [DOI] [PubMed] [Google Scholar]
- 100. Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005;436:428-33 [DOI] [PubMed] [Google Scholar]
- 101. Bredemeyer AL, Helmink BA, Innes CL, et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature. 2008;456:819-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ziv Y, Bielopolski D, Galanty Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870-6 [DOI] [PubMed] [Google Scholar]
- 103. Zhang Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev. 2003;17:2733-40 [DOI] [PubMed] [Google Scholar]
- 104. Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-depdent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2020;141:970-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Janicki SM, Tsukamoto T, Salghetti SE, et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang H, Wang L, Erdjument-Bromage H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873-8 [DOI] [PubMed] [Google Scholar]
- 107. Stock JK, Giadrossi S, Casanova M, et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9:1428-35 [DOI] [PubMed] [Google Scholar]
- 108. Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845-54 [DOI] [PubMed] [Google Scholar]
- 109. Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mathas S, Kreher S, Meaburn KJ, et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:5831-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Khanna KK. Cancer risk and the ATM gene: a continuing debate. J Natl Cancer Inst. 2000;92:795-802 [DOI] [PubMed] [Google Scholar]
- 112. Lee K, Zhang Y, Lee SE. Saccharomyces cerevisiae ATM orthologue suppresses break-induced chromosome translocations. Nature. 2008;454:543-6 [DOI] [PubMed] [Google Scholar]
- 113. Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697-708 [DOI] [PubMed] [Google Scholar]
- 114. O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chopra VS, Hong JW, Levine M. Regulation of Hox gene activity by transcriptional elongation in Drosophila. Curr Biol. 2009;19:688-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhou W, Zhu P, Wang J, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29:69-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400:284-8 [DOI] [PubMed] [Google Scholar]
- 118. Saunders A, Werner J, Andrulis ED, et al. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science. 2003;301:1094-6 [DOI] [PubMed] [Google Scholar]