

Abstract
NAD+-dependent Class III histone deacetylase SIRT1 is a multiple function protein critically involved in stress responses, cellular metabolism and aging through deacetylating a variety of substrates including p53, forkhead-box transcription factors, PGC-1α, NF-κB, Ku70 and histones. The first discovered non-histone target of SIRT1, p53, is suggested to play a central role in SIRT1-mediated functions in tumorigenesis and senescence. SIRT1 was originally considered to be a potential tumor promoter since it negatively regulates the tumor suppressor p53 and other tumor suppressors. There is new evidence that SIRT1 acts as a tumor suppressor based on its role in negatively regulating β-catenin and survivin. This review provides an overview of current knowledge of SIRT1-p53 signaling and controversies regarding the functions of SIRT1 in tumorigenesis.
Keywords: SIRT1, p53, acetylation, deacetylation, cancer, senescence
Introduction
In Saccharomyces cerevisceae, the protein called Sir2, has a wide range of functions including chromatin silencing, life span extension and maintenance of genomic stability. There are seven human homologues of Sir2 in humans, called SIRT1-SIRT7, with SIRT1 largest in total sequence and most studied. SIRT1 and yeast Sir2 are members of a family of enzymes called sirtuins that are NAD+ -dependent protein deacetylases, although they are sometimes classified as class III histone deacetylases (HDACs). For example, SIRT1 not only deacetylates histones H1, H3 and H4, but also deacetylates many non-histone proteins including p53, FOXO, Ku70, p300, Rb, E2F1, NF-kB, p73 and PGC-1α[1, 2]. In virtue of these important targets, SIRT1 is linked to regulatory control of diverse normal and abnormal cellular processes ranging from stress responses, aging, and metabolism to cancer.
p53 was the first non-histone deacetylation target identified for SIRT1 [3, 4]. p53 deacetylation was originally found to be mediated by PID/HDAC1 (PID: p53 target protein in the deacetylase complexes) complex [5]. p53 acetylation was found to be extremely unstable even in the presence of the HDAC inhibitor trichostatin A (TSA) in the cell lysates [5]. The discovery of the sirtuin deacetylases suggested that one of the human isoforms could be responsible for the observed instability of p53 acetylation [6, 7]. We focused our attention on SIRT1 noting that its deacetylase activity was unable to be inhibited by the common HDAC inhibitor TSA [6]. Further work demonstrated that p53 is indeed a SIRT1 target and that SIRT1 deacetylation of p53 regulates its function [3], a finding independently confirmed by Vaziri and colleagues [4] and Langley and colleagues [8].
The tumor suppressor p53, has powerful apoptotic effects, and consequently is subject to tight regulatory control in cells. Normally, p53 protein is maintained at a low level through the MDM2-mediated ubiquitination and degradation pathway. However, when cells are exposed to stress including genotoxic stress, p53 protein is rapidly accumulated and activated for downstream biological functions. The regulatory events that affect the amount, stability and activity of p53 are in part derived from a variety of post-translational modifications, including phosphorylation, ubiquitination and acetylation. It is of interest to point out that p53 is the first functional non-histone substrate identified for the histone acetyltransferases (HATs) [9]. This discovery brought the significant new insight that HATs modulate the functions of a set of non-histone proteins through acetylation.
To date, many types of stress have been reported to enhance p53 acetylation. p53 can be acetylated by CBP/p300 at Lys 370, 372, 373, 381, 382, and 386 residues in the carboxy-terminal region. The C-terminal region acetylation of p53 activates its sequence-specific DNA binding activity and target gene expression as well as increases its stability due to inhibition of ubiquitination at acetylated lysines [10]. PCAF (p300/CBP associated factor) acetylates p53 at Lys 320. The acetylation at this site also increases its DNA binding and transcriptional activity and induces cell growth arrest [11, 12]. DNA-damage increases the acetylation of p53 at Lys 120 within the DNA-binding domain by Tip60 or hMOF acetyltransferases and this acetylation site is crucial for p53-mediated apoptosis via BAX and PUMA activation [13, 14]. More recently, a final acetylation site of p53 was identified at Lys 164 which is acetylated by CBP/p300. Acetylation at this site is required for p53-induced cell growth arrest and apoptosis although it does not affect p53 DNA binding activity [15]. Taken together, enhanced acetylation at these acetylation sites correlated well with p53 stabilization, and increased recruitment to target promoter regions for gene activation in response to various cellular stresses demonstrating that p53 acetylation is an indispensable event for p53 activation.
Reversal of the acetylation modification can modulate the function of p53, weakening the biologic effects of acetylation described in the prior section. Two different protein systems can mediate p53 deacetylation, as mentioned previously, SIRT1 and the PID/HDAC1 complex. Biochemical analysis identified PID as a direct interacting protein for p53 in the HDAC1 complex. PID thereby recruits p53 to the HDAC1 complex for deacetylation [5]. p53 deacetylation by PID/HDAC1 complex strongly represses p53 transactivation activity and modulates cell growth arrest and apoptosis. As previously discussed, SIRT1 was found to deacetylate p53 and regulate p53 functions in cells. Overexpression of SIRT1 strongly attenuated p53 transcription-dependent apoptosis upon DNA-damage and oxidative stress, and expression of SIRT1-H363Y in cells, a dominant-negative mutant of SIRT1 (DNSIRT1), significantly increased the p53 acetylation level. Consistent with the positive effects of acetylation on p53 transcriptional activities, DNSIRT1 increased expression levels of p53 targets p21 and Bax. Furthermore, DNSIRT1 was able to sensitize cells to apoptosis caused by oxidative stress[3], DNA damage[3,4], and ionizing radiation [4].
SIRT1 regulates both of the two types of known p53-mediated apoptotic pathways. p53 transcription-dependent apoptosis requires the expression of apoptosis-related target genes including BAX, PUMA, and NOXA. p53 transcription-independent apoptosis is initiated by the release of cytochrome C from the mitochondrial intramembrane by a direct interaction between mitochondrial p53 and antiapoptotic BCL (B Cell Lymphoma, members of which bearing at least one of four conserved Bcl-2 homology domains but opposite effect on apoptosis process) proteins [16]. Han and colleagues showed that SIRT1 can facilitate the mitochondrial-dependent apoptotic response by controlling p53 subcellular localization in mouse embryonic stem (mES) cells through blocking cytoplasmic p53 nuclear translocation [17]. They found that when intracellular ROS is elevated in SIRT1-deficient mES cells in which the SIRT1 alleles were sequentially targeted with knockout vectors designed to delete exons 5 and 6[18,19], p53 was acetylated at Lys 379 (equivalent to human Lys 382), a known target for SIRT1 [20] and this modification led to nuclear localization of p53, indicating that SIRT1 blocks nuclear translocation of p53 induced by oxidative stress via its deacetylation [17]. Meanwhile, intracellular ROS elevation resulted in translocation of p53 into mitochondria and induced apoptosis in wild-type, but not in SIRT1-deficient mES cells. The above findings suggest that SIRT1 can redirect p53 from the cytosol to the mitochondria in response to increased ROS. The biological consequence of which is transcription-independent p53-induced apoptosis (Figure 1). From these studies, it appears that SIRT1 activity levels during oxidative stress can regulate cell fate by p53 deacetylation. SIRT1 deacetylation blocks nuclear translocation, however, deacetylation increases the accumulation of cytosolic p53 and enhances its passage to mitochondria. Thus, SIRT1 blocks transcription dependent apoptosis by p53, but increases p53-mediated transcription independent apoptosis.
Figure 1.
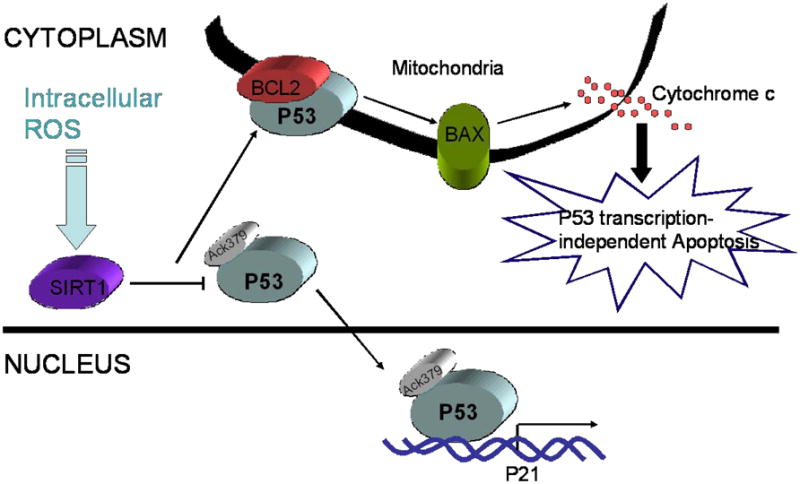
SIRT1 promotes p53 transcription-independent apoptosis. In mouse embryonic stem cells, intracellular ROS activates cytoplasm SIRT1 protein. SIRT1 binds to and deacetylates p53 at K379 (human K382) and blocks p53 nuclear translocation. Deacetylated p53 translocates onto the mitochondrial outer-membrane and releases proapototic BCL protein BAX by interacting with anti-apoptotic BCL proteins including BCL2, BCLxl. Activated BAX leads to the release of Cytochrome c from mitochondrial to cytoplasm. p53 transcription-independent apoptosis initiated.
SIRT1-p53 and cancer
The tumor suppressor p53 can exert anti-proliferative effects, including growth arrest, apoptosis, and cell senescence in response to various types of cell stress. Mutations within the p53 gene have been documented in more than half of all human tumor types [21]. Accumulating evidence further indicates that tumors that retain wild-type p53 have other defects in the p53 pathway which are important in cancer formation [22]. Given that SIRT1 can negatively regulate p53 localization to the nucleus and its function as a transcription factor, it is worth considering if SIRT1 can have a role in the formation of cancers. Several studies support the notion that SIRT1 could be involved in carcinogenesis: first, SIRT1 knockout mice exhibit p53 hyperacetylation and increased radiation-induced apoptosis consistent with SIRT1 inhibition of p53 function, raising the possibility that SIRT1 can facilitate tumor growth by antagonism of p53 [20]; second, inhibition of SIRT1 activity by Sirtinol, an inhibitor of SIRT1, induces senescence-like cell growth arrest with attenuated Ras-MAPK signaling in human cancer cells [23]; third, DBC1 (deleted in breast cancer 1) acts as a native inhibitor of SIRT1 in human cells. Repression of SIRT1 by DBC1 leads to increased level of p53 acetylation and upregulation of p53 function [24, 25]. The finding of SIRT1 up-regulation in various cancers such as leukemia, prostate cancer, skin cancer and colon cancers [26–29], also supports the hypothesis of SIRT1 playing a role in the formation of cancers.
On the other hand, studies also indicate that SIRT1 can act as a tumor suppressor. SIRT1 suppressed intestinal tumorigenesis and colon cancer growth in a β-catenin-driven mouse model of colon cancer [30]. Deacetylation of β-catenin promoted cytoplasmic localization of the otherwise nuclear-localized oncogenic form of β-catenin, reminiscent of its effects on p53 and significantly reduced colon cancer tumor formation and proliferation [30].
SIRT1 suppresses survivin in BRCA1 (breast cancer 1, a tumor suppressor mutated in some breast cancers)-associated breast cancer. Survivin is a small molecular weight protein that is an inhibitor of apoptosis, and is overexpressed in various cancers. Moreover, it is a validated target for anti-cancer drug discovery.[31]. BRCA1-associated breast cancers have low levels of SIRT1 and high levels of survivin. BRCA1 is found at the SIRT1 promoter in mammary tumor cell lines with wild-type BRCA1 and increases SIRT1 expression [32]. SIRT1 in turn inhibits survivin expression by deacetylation of histone H3 within nucleosomes at the survivin promoter [32]. The discovery that SIRT1 down regulates survivin provides a potential strategy to inhibit survivin in cancer cells. Thus SIRT1 activators could be interesting drugs for survivin overexpressing cancers. The report that significantly lower levels of SIRT1 were detected in breast and hepatic carcinoma compared to the adjacent normal tissue provides evidence that SIRT1 may function as a tumor suppressor [33]. The seemingly contradictory findings for numerous studies examining the effect of SIRT1 in biological processes linked to cancers underscore the importance of further studies to explore the link between SIRT1 in human cancer and in tumorigenesis.
SIRT1 knockout mice have allowed further understanding of the roles of SIRT1 in regulating p53 and its biological functions. SIRT1-deficient mice were employed to study SIRT1 functions by several independent groups [18–20, 33]. SIRT1−/− mice were generated which lack part of the catalytic domain of SIRT1. These SIRT1−/− mice and embryos were smaller than their littermate controls. The majority of the mice died before birth or during the early postnatal period [18–20, 33]. These findings, combined with the observation that SIRT1 is strongly expressed in embryonic mouse heart and brain, suggest that SIRT1 is important for embryogenesis. Supporting a role for SIRT1 in regulating p53 acetylation levels, SIRT1-deficient thymocytes displayed p53 hyperacetylation following adriamycin treatment and enhanced ionizing radiation-induced apoptosis [20]. A question thus emerged as to whether p53 hyperacetylation results in the embryonic lethality of SIRT1−/− mice. Wang et al. found that SIRT1−/−; p53−/−double knockout mice still died in embryonic stages, indicating that SIRT1 deficiency likely causes other essential pathway defects [33]. Intriguingly, double heterozygotic SIRT1+/−; p53+/− mice survived but exhibited a remarkably high incidence (76%) of tumors, while only two of 21 SIRT1+/− mice and 3 of 23 p53+/− mice developed tumors [33]. Further analysis of tumor tissues in double heterozygotic mice showed severe genomic impairment including extensive aneuploidy and diverse chromosomal aberrations. 73% of the tumors had lost p53 expression and 44% tumors had little or no SIRT1. These findings suggest that genomic integrity and stability require cooperation of p53 and SIRT1. As cancer has been considered as a set of diseases driven by epigenetic changes and genetic modifications in oncogenes and/or tumor suppressors, it seems likely that elaborate and complex signal networks protect genome integrity in cells. With insufficiency of SIRT1 or p53 alone, compensatory signal pathways may be activated or enhanced to inhibit tumorigenesis. With loss of both SIRT1 and p53, cells appear to lose control of genomic integrity, and these cells could be at greater risk for development of tumors. In this context, SIRT1 seems to act as a tumor suppressor.
Several potential tumor suppressors, including HIC1, microRNA34a, and DBC1, have been reported to modulate p53-mediated apoptosis by directly repressing SIRT1 expression and activity [24,25,34,35]. Tumor suppressor HIC1 (hypermethylated in cancer 1) causes promoter hypermethylation and epigenetic silencing in many types of human cancers [36–40]. HIC1+/− mice develop various malignant tumors, including epithelial cancers, lymphomas and sarcomas. In many of these tumors, the wild type HIC1 allele is silenced by hypermethylation epigenetic modification [41]. HIC1 is a target of p53 transactivation [42, 43]. A study of mice heterozygous for both HIC1 and p53 suggested that HIC1 functionally cooperates with p53 to suppress age-dependent tumorigenesis [41]. Chen and colleagues put forward a model of the HIC1-SIRT1-p53 regulatory loop [34] in which under normal conditions, stress-induced rapid accumulation of p53 activates the HIC1 gene. The POZ (poxvirus and zinc finger, a conserved protein-protein interaction motif) domain of HIC1 binds to SIRT1 to form a transcriptional repression complex. The SIRT1/HIC1 complex is recruited to the SIRT1 promoter to suppress SIRT1 transcription. Reduced SIRT1 levels are apparently responsible for increased acetylation level of p53, thereby facilitating its functions of cell cycle arrest, DNA repair and apoptosis. SIRT1 expression was increased due to age associated promoter hypermethylation and epigenetic silencing of HIC1 expression. As a consequence of age dependent derepression of SIRT1 transcription, it is hypothesized that p53 function is less active in aged cells since deacetylation of p53 provides negative regulation on its nuclear functions. Downregulated p53 activity is expected to produce a defective apoptotic response to diverse types of genotoxic stress. Thereby, this could be one of the mechanisms by which aged cells can increase cancer risk in mammals [34].
SIRT1 might also act as a tumor promoter by repressing p53-induced expression of microRNA 34a. Several groups have reported the abnormal expression of microRNA family members in human cancers and the involvement of microRNAs in tumorigenesis, progression and metastasis [44, 45]. MicroRNA 34a (miR34a), a p53 target gene, has been identified as a potential tumor suppressor gene [46]. A recent study reported that miR34a binds to the 3′-UTR of SIRT1 mRNA to repress its translation [35]. Consistently, miR34a enhanced p53 acetylation and activation of p53-dependent apoptosis by increased expression of the p53 target PUMA in HCT116 cells. Overexpression of SIRT1 rescued cells from miR34a-induced apoptosis [35]. Collectively these data indicate that miR34a functions as a potential tumor suppressor by its antagonism of SIRT1 mediated p53 negative regulation, which, in turn increases miR34a expression via upregulation of p53 transcriptional activity. These relationships establish a positive feedback loop for miR34a action via SIRT1 and p53..
As previously mentioned, DBC1 (deleted in breast cancer 1) serves as a negative regulator of SIRT1 [24, 25]. The DBC1 gene was initially identified to be homozygously deleted in chromosome 8p21 in a breast cancer specimen. Frequent silencing or downregulation of DBC1 has also been observed in non-small cell lung cancers, leukemia and bladder cancers [47, 48]. Co-immunoprecipitation assay demonstrated that DBC1 binding to SIRT1 disrupts the interaction between SIRT1 and p53 [24, 25]. DBC1-mediated repression of SIRT1 leads to increased p53 acetylation and p53-induced apoptotic responses to genotoxic stress [25]. Conversely, down-regulation of DBC1 enhances SIRT1-dependent stress-resistance and cell survival [25, 26]. A recent study discovered that DBC1 and its homologs contain a catalytically inactive version of the Nudix hydrolase domain which is known to bind NAD+. Hence, they predicted that DBC1 and its homologs integrate of diverse regulatory signal pathways involving chromatin protein modification and NAD+ metabolism [49]. A study by Li and colleagues suggested that DBC1 disrupted the SUV39H1-SIRT1 complex (SUV39H1 is a histone H3K9-specific methyltransferase essential for heterochromatin formation) and inactivated both enzymes [50]. Therefore, DBC1 may modulate SIRT1 activity by both potentially interacting with NAD+ metabolism and by direct interactions with SIRT1 for regulating its function and its downstream targets. The current data invites the possibility that DBC1 could play a significant role in regulating SIRT1-related activities in metabolism, cancer and aging (Figure 2).
Figure 2.
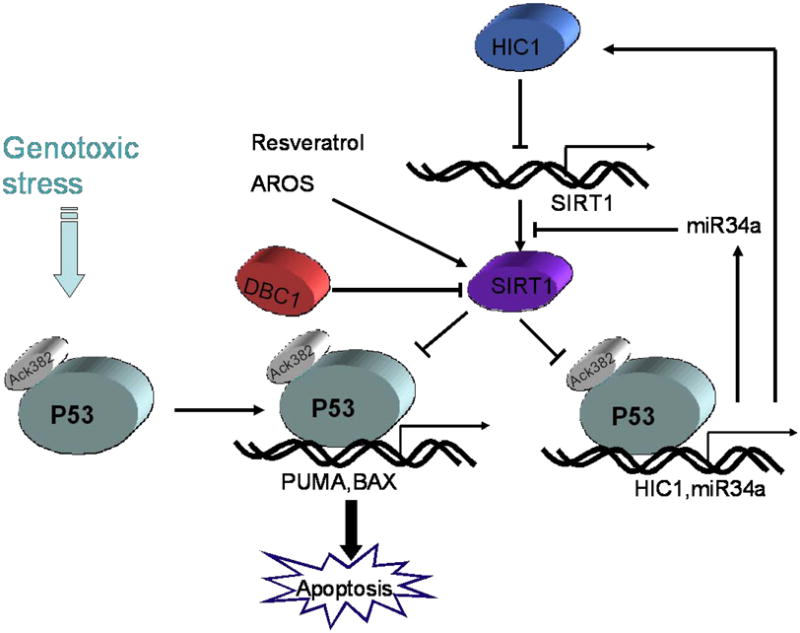
SIRT1 promotes tumorigenesis. Upon genotoxic stress, p53 is acetylated at K382, which enhanced DNA binding and transactivation activity. HIC1, miR34a, and DBC1 suppress SIRT1 activity. Multilayered inhibition of SIRT1 allows the activation status of p53 for p21, PUMA and BAX genes expression which induced cell cycle arrest, apoptosis, and senescence. Resveratrol and AROS, Along with other SIRT1 activators, enhance SIRT1 deacetylation on p53 and suppress p53 transcription-dependent cell cycle arrest and apoptosis by genotoxic stress.
The multifunctional role of SIRT1, especially the evidence for both tumor promoter and tumor suppressor functions, has attracted great interest in SIRT1 directed drugs for tumor therapy. Both SIRT1 inhibitors and activators have been discovered [51, 52]. The study of these small molecules will help to understand the molecular mechanism underlying biological functions of SIRT1. The most studied natural compound for SIRT1 activator is resveratrol although the activation effects of resveratrol on SIRT activity are controversial [53]. Activation of SIRT1 by resveratrol has been claimed to partially mimic caloric restriction and resveratrol delays aging parameters in mice [54]. In spite of the appeal of SIRT1 activation for improving metabolic parameters in mammals, an increased cancer risk is a concern if activation of SIRT1 leads to unhealthy repression of p53 activity. It would be highly desirable to find an optimal condition or drug that would activate SIRT1 and not increase human cancer risk.
The concept of inhibiting SIRT1 as an anti-tumor strategy has been explored by several groups. Recent findings by Lain and colleagues on a novel small-molecule sirtuin inhibitors, tenovin-1 and the more water-soluble derivative, tenovin-6, which act on SIRT1 and SIRT2, suggest new approaches to cancer therapy [55]. Sequential target identification studies demonstrated that tenovins acted by inhibiting deacetylase activity of SIRT1 and SIRT2, thereby significantly increasing acetylation level of p53K382, histoneH4K16 and tubulinK40. Lain et al. [55] further demonstrated that tenovin-6 can slow tumor growth in a ARN8 melanoma xenograft model in SCID mice. The anti-tumor activity of these two compounds suggests that inhibition of SIRT1 activity to activate p53 may be a useful anticancer approach. The concept that sirtuin inhibition can be a potentially interesting anti-cancer approach has been supported by research with other described sirtuin inhibitors, including cambinol which induced hyperacetylation of BCL6 and p53, with increased apoptosis in Burkitt lymphoma cells [56]. In addition selermide was shown to reactivate transcription of proapoptotic genes epigenetically repressed in cancer cells [57].
SIRT1-p53 and senescence
Besides inducing cell growth arrest and apoptosis, p53 activation also induces cellular senescence in response to oxidative stress. The promyelocytic leukemia protein (PML) nuclear bodies are discrete nuclear substructures and represent the natural accumulation sites of PML proteins [8]. The size and number of PML nuclear bodies are altered during the cell cycle and in response to various cellular and environmental stresses [58]. There are seven splice variants of PML named PML I–VII in mammalian cells. PML IV interacts with HDACs in the regulation of p53 function and induction of premature senescence [8, 59–61]. Upon PML IV upregulation or oncogenic Ras expression, SIRT1 is recruited to nuclear bodies and interacts directly with PML IV and p53 to repress p53-dependent transactivation by deacetylation of p53 [8]. The recruitment of SIRT1 also rescues PML-induced cellular senescence by inhibiting p53 acetylation [8]. This phenomenon of senescence inhibition by SIRT1 also exists in human endothelial cells [62]. Endothelial cells with down-regulated SIRT1 exhibit increased acetylation of p53 and premature senescence-like phenotype. Conversely, overexpression of SIRT1 prevents cells from physiological alteration toward senescence [62]. Therefore, SIRT1 seems to exert a protective role against p53-dependent senescence under multiple cellular stresses although it was also reported that SIRT1 could limit replicative life span in response to chronic genotoxic stress [63].
The activity of NAD+ biosynthetic enzymes, which can indirectly regulate sirtuins by their effects on NAD and metabolites, can also affect cell senescence. For example, Nicotinamide phosphoribosyltransferase (Nampt) is a rate-limiting enzyme for NAD+ biosynthesis from nicotinamide in mammalian cells, and limits on its activity have been found to be associated with the development of senescence in some cell types. The intracellular levels of NAD+ and nicotinamide have been identified as important factors for regulating SIRT1 deacetylase activity. A recent study by van der Veer and colleagues [64] demonstrated an extension of human vascular smooth muscle cell (SMC) lifespan by overexpression of Nampt. Nampt is known to increase cellular NAD+ and caused increased SIRT1 activity and reduced fraction of p53 bearing acetylation on Lys382 [64]. Low Nampt activity was observed in both replicative senescence and FK866-induced premature senescence in SMCs. In contrast, overexpression of Nampt resulted in deferred senescence and significantly prolonged lifespan, concomitant with enhanced SIRT1 activity and degradation of increased deacetylated p53. Moreover, exogenous p53 introduced via adenovirus to co-transfected SMCs (Nampt and p53) abolished Nampt protection from senescence [64]. The data from this study support a mechanism wherein increased Nampt activity produces more NAD+ from nicotinamide in cells, which in turn activates SIRT1 deacetylase activity. Increased SIRT1 activity represses p53 activity to prevent p53-dependent cellular senescence. The above findings provide evidence that the SIRT1-p53 axis also determines cell fate, growth or senescence, in response to various types of stress in mammalian cells and this function can be regulated by modulating the NAD+ level in cells for SIRT1 activation and p53 repression.
SIRT1-p53 and neurodegeneration
Aging is associated with progressive loss of neurons and a variety of neurodegenerative disorders, such as Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS), diseases which currently lack treatment or prevention strategies. Neuroprotection can be understood to be constituted from the natural survival and maintenance mechanisms acting within neural tissues, as well as the medical strategies that can be used to enhance these mechanisms or to inhibit cell death mechanisms. From the latter, medical scientists hope to slow or prevent neurodegeneration in the Central Nervous System. Several studies have suggested that sirtuins could play roles in natural forms of neuroprotection and regulation of cell death in neurons, and could be targeted for therapy. For example, SIRT1 activation has been suggested to be one mechanism by which increased nuclear NAD+ biosynthesis prevents axonal degeneration in Wallerian degeneration slow mice [65], although another group has controverted this view [66]. Recent studies found that SIRT1 has an important function in neuronal survival via p53 deacetylation [67, 68]. Mouse models for AD or ALS and primary neurons display increased SIRT1 expression upon neurotoxic insults and SIRT1 promotes neuronal survival [68]. Either SIRT1 overexpression or resveratrol treatment promoted neuronal survival and prevented neurodegeneration in mouse models. Deacetylation of p53K382 is required for this neuroprotective effect [68].
Hasegawa and colleagues recently reported that Necdin reduced p53-induced DNA damage response via SIRT1 mediated p53 deacetylation in cortical neurons [65]. Necdin is a member of melanoma antigen (MAGE) family protein that promotes neuronal differentiation and survival by interacting with p53 and repressing p53-mediated apoptosis. Necdin is expressed predominantly in post-mitotic neurons and it has been shown to interact with p53 and SIRT1. The three proteins form a stable complex in cells as shown in both co-immunoprecipitation and immunostaining assays [67]. An increased interaction was correlated with a reduced amount of acetylated p53 in cortical neurons [67]. Necdin represses p53-dependent neuronal apoptosis in response to camptothecin (topoisomerase-I inhibitor, a strong DNA damaging compound) via SIRT1 deacetylase activity. Moreover, either Necdin deficiency or SIRT1 knockdown enhanced p53 acetylation and apoptosis in neuron cells[67]. These observations implythat SIRT1 plays an important rolein Necdin -mediated neuron survival through modulating p53 acetylation. The above findings suggestthat SIRT1 has an important function in neuronal survival via p53 deacetylation. Further work is needed to explore the possibility that activation of SIRT1 may be beneficial for treatment of neurodegenerative disorders.
Perspective
The seemingly controversial roles of SIRT1 in tumorigenesis underscore the importance for further studies including: 1) more investigation on the expression level of SIRT1 in human cancer cells and tissues; 2) development of transgenic animals with over expression or down-regulation of SIRT1 for tumorigenesis analysis; 3) investigation of the influence of other targets of SIRT1 such as Ku70, NF-kB and Foxo on interactions between SIRT1 and p53 in human cancers. 4) Studies of the effects of small molecules which regulate SIRT1 activity (positively or negatively) to develop novel effective gene-targeting drugs against tumors, neuronal disorders, aging and other diseases. As SIRT1 and p53 are important regulators of aging and cellular senescence, further research on SIRT1-p53 signaling will be likely to provide specific clues for understanding the complicated relationship between cellular senescence, aging and tumorigenesis.
Acknowledgments
We thank the members of Luo lab for help in preparing the manuscript. J. Yi is sponsored by the China Scholarship Council. J. Luo is supported by grant from NIH/NIA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer. Nat Rev Cancer. 2009;9:123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng C. SIRT1, is it a tumor promoter or tumor suppressor. Int J Biol Sci. 2009;5:147–152. doi: 10.7150/ijbs.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 4.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 5.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 6.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 7.Imai S, Johnson FB, Marciniak RA, McVey M, Park PU, Guarente L. Sir2: an NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging, Cold Spring Harb. Symp Quant Biol. 2000;65:297–302. doi: 10.1101/sqb.2000.65.297. [DOI] [PubMed] [Google Scholar]
- 8.Langley E, Pearson M, Faretta M, Bauer U, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 10.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 11.Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- 12.Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 15.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol. 2004;24:6728–6741. doi: 10.1128/MCB.24.15.6728-6741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han M, Song E, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McBurney MW, Yang X, Jardine K, Bieman M, Th’ng J, Lemieux M. The Absence of SIR2α Protein Has No Effect on Global Gene Silencing in Mouse Embryonic Stem Cells1 1 National Cancer Institute of Canada and the Canadian Institutes of Health Research. Molecular Cancer Research. 2003;1:402–409. [PubMed] [Google Scholar]
- 19.McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng H, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 22.Lain S, Lane D. Improving cancer therapy by non-genotoxic activation of p53. Eur J Cancer. 2003;39:1053–1060. doi: 10.1016/s0959-8049(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 23.Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y, Kaneki M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25:176–185. doi: 10.1038/sj.onc.1209049. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 25.Zhao W, Kruse J, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 27.Huffman DM, Grizzle WE, Bamman MW, Kim J, Eltoum IA, Elgavish A, Nagy TR. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 28.Stünkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto-Tellez M, Ni B, Entzeroth M, Wood J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2:1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 29.Hida Y, Kubo Y, Murao K, Arase S. Strong expression of a longevity-related protein, SIRT1, in Bowen’s disease. Arch Dermatol Res. 2007;299:103–106. doi: 10.1007/s00403-006-0725-6. [DOI] [PubMed] [Google Scholar]
- 30.Firestein R, Blander G, Michan S, Oberdoerffer R, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 32.Wang R, Zheng Y, Kim H, Xu X, Cao L, Luhasen T, Lee M, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man Y, Hung M, Finkel T, Deng C. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang R, Sengupta K, Li C, Kim H, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng Z, Appella E, Wang XW, Ried T, Deng C. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morton RA, Watkins JJ, Bova GS, Wales MM, Baylin SB, Isaacs WB. Hypermethylation of chromosome 17P locus D17S5 in human prostate tissue. J Urol. 1996;156:512–516. doi: 10.1097/00005392-199608000-00073. [DOI] [PubMed] [Google Scholar]
- 37.Fujii H, Biel MA, Zhou W, Weitzman SM, Baylin SB, Gabrielson E. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene. 1998;16:2159–2164. doi: 10.1038/sj.onc.1201976. [DOI] [PubMed] [Google Scholar]
- 38.Eads CA, Lord RV, Wickramasinghe K, Long TI, Kurumboor SK, Bernstein L, Peters JH, DeMeester SR, DeMeester TR, Skinner KA, Laird PW. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001;61:3410–3418. [PubMed] [Google Scholar]
- 39.Hayashi M, Tokuchi Y, Hashimoto T, Hayashi S, Nishida K, Ishikawa Y, Nakagawa K, Tsuchiya S, Okumura S, Tsuchiya E. Reduced HIC-1 gene expression in non-small cell lung cancer and its clinical significance. Anticancer Res. 2001;21:535–540. [PubMed] [Google Scholar]
- 40.Koul S, Houldsworth J, Mansukhani MM, Donadio A, McKiernan JM, Reuter VE, Bosl GJ, Chaganti RS, Murty VV. Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol Cancer. 2002;1:8. doi: 10.1186/1476-4598-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fleuriel C, Touka M, Boulay G, Guérardel C, Rood BR, Leprince D. HIC1 (Hypermethylated in Cancer 1) epigenetic silencing in tumors. Int J Biochem Cell Biol. 2009;41:26–33. doi: 10.1016/j.biocel.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, Cavenee WK, Kuerbitz SJ, Baylin SB. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–577. doi: 10.1038/nm0695-570. [DOI] [PubMed] [Google Scholar]
- 43.Deltour S, Pinte S, Guérardel C, Leprince D. Characterization of HRG22, a human homologue of the putative tumor suppressor gene HIC1. Biochem Biophys Res Commun. 2001;287:427–434. doi: 10.1006/bbrc.2001.5624. [DOI] [PubMed] [Google Scholar]
- 44.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 46.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Izumi H, Inoue J, Yokoi S, Hosoda H, Shibata T, Sunamori M, Hirohashi S, Inazawa J, Imoto I. Frequent silencing of DBC1 is by genetic or epigenetic mechanisms in non-small cell lung cancers. Hum Mol Genet. 2005;14:997–1007. doi: 10.1093/hmg/ddi092. [DOI] [PubMed] [Google Scholar]
- 48.San José-Enériz E, Agirre X, Román-Gómez J, Cordeu L, Garate L, Jiménez-Velasco A, Vázquez I, Calasanz MJ, Heiniger A, Torres A, Prósper F. Downregulation of DBC1 expression in acute lymphoblastic leukaemia is mediated by aberrant methylation of its promoter. Br J Haematol. 2006;134:137–144. doi: 10.1111/j.1365-2141.2006.06131.x. [DOI] [PubMed] [Google Scholar]
- 49.Anantharaman V, Aravind L. Analysis of DBC1 and its homologs suggests a potential mechanism for regulation of sirtuin domain deacetylases by NAD metabolites. Cell Cycle. 2008;7:1467–1472. doi: 10.4161/cc.7.10.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Chen L, Kabra N, Wang C, Fang, Chen J. Inhibition of SUV39H1 methyltransferase activity by DBC1. J Biol Chem. 2009;284:10361–10366. doi: 10.1074/jbc.M900956200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alcaín FJ, Villalba JM. Sirtuin inhibitors. Expert Opin Ther Pat. 2009;19:283–294. doi: 10.1517/13543770902755111. [DOI] [PubMed] [Google Scholar]
- 52.Alcaín FJ, Villalba JM. Sirtuin activators. Expert Opin Ther Pat. 2009;19:403–414. doi: 10.1517/13543770902762893. [DOI] [PubMed] [Google Scholar]
- 53.Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barger JL, Kayo T, Vann JM, Arias EB, Wang J, Hacker TA, Wang Y, Raederstorff D, Morrow JD, Leeuwenburgh C, Allison DB, Saupe KW, Cartee GD, Weindruch R, Prolla TA. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE. 2008;3:e2264. doi: 10.1371/journal.pone.0002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJR, Pass G, Woods J, Lane DP, Westwood NJ. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA, Bedalov A. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–4377. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- 57.Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, Varela-Rey M, Rotili D, Nebbioso A, Ropero S, Montoya G, Oyarzabal J, Velasco S, Serrano M, Witt M, Villar-Garea A, Imhof A, Inhof A, Mato JM, Esteller M, Fraga MF. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28:781–791. doi: 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 58.Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nat Cell Biol. 2000;2:E85–90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- 59.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 60.Wu WS, Vallian S, Seto E, Yang WM, Edmondson D, Roth S, Chang KS. The growth suppressor PML represses transcription by functionally and physically interacting with histone deacetylases. Mol Cell Biol. 2001;21:2259–2268. doi: 10.1128/MCB.21.7.2259-2268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 63.Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, Kaushal D, Cheng HL, Fischer MR, Stokes N, Murphy MM, Appella E, Alt FW. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 64.van der Veer E, Ho C, O’Neil C, Barbosa N, Scott R, Cregan SP, Pickering JG. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. 2007;282:10841–10845. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 65.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 66.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai L. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]