

Abstract
The callipyge phenotype is a monogenic muscular hypertrophy that is only expressed in heterozygous sheep receiving the CLPG mutation from their sire. The wild-type phenotype of CLPG/CLPG animals is thought to result from translational inhibition of paternally expressed DLK1 transcripts by maternally expressed miRNAs. To identify the miRNA responsible for this trans effect, we used high-throughput sequencing to exhaustively catalog miRNAs expressed in skeletal muscle of sheep of the four CLPG genotypes. We have identified 747 miRNA species of which 110 map to the DLK1–GTL2 or callipyge domain. We demonstrate that the latter are imprinted and preferentially expressed from the maternal allele. We show that the CLPG mutation affects their level of expression in cis (∼3.2-fold increase) as well as in trans (∼1.8-fold increase). In CLPG/CLPG animals, miRNAs from the DLK1–GTL2 domain account for ∼20% of miRNAs in skeletal muscle. We show that the CLPG genotype affects the levels of A-to-I editing of at least five pri-miRNAs of the DLK1–GTL2 domain, but that levels of editing of mature miRNAs are always minor. We present suggestive evidence that the miRNAs from the domain target the ORF of DLK1, thereby causing the trans inhibition underlying polar overdominance. We highlight the limitations of high-throughput sequencing for digital gene expression profiling as a result of biased and inconsistent amplification of specific miRNAs.
The callipyge phenotype is an inherited muscular hypertrophy of sheep. It is characterized by an unusual inheritance pattern referred to as polar overdominance: Only heterozygous individuals having received the CLPG mutation from their father express the phenotype (Cockett et al. 1996). The CLPG point mutation inactivates a muscle-specific silencer controlling the expression of a subset of imprinted genes in the DLK1–GTL2 domain (i.e., the paternally expressed protein-encoding DLK1 and PEG11 [also known as RTL1] genes and the maternally expressed non-coding GTL2 [also known as MEG3], anti-PEG11 [also known as anti-RTL1], MEG8 [also known as RIAN], and MIRG genes) (Charlier et al. 2001a; Freking et al. 2002; Smit et al. 2003). Hence, padumnal heterozygotes (+Mat/CLPGPat) are characterized by ectopic expression of PEG11 (Byrne et al. 2010) and DLK1 (Davis et al. 2004) in skeletal muscle. DLK1 is thought to contribute to the callipyge phenotype as its ectopic expression increases muscle mass in transgenic mice (Davis et al. 2004). Whether ectopic expression of PEG11 is also involved in phenotypic expression remains to be established.
While showing increased levels of DLK1 mRNA in muscle—like their +Mat/CLPGPat counterparts—no DLK1 protein is observed in muscle of CLPG/CLPG animals, accounting for their wild-type phenotype (Davis et al. 2004). The absence of DLK1 protein despite increased levels of DLK1 mRNA in these animals is thought to result from the ectopic expression of the madumnal noncoding RNAs, as this feature distinguishes CLPG/CLPG from +Mat/CLPGPat individuals (Georges et al. 2003, 2004). The madumnal long, noncoding RNA genes are hosting a large array of small C/D snoRNAs and miRNAs of unknown function (Seitz et al. 2004). We have postulated that these small RNAs are the mediators of the trans effect down-regulating DLK1 in CLPG/CLPG animals thus causing polar overdominance (Supplemental Fig. 1; Georges et al. 2003, 2004). This hypothesis received strong support from the demonstration, in the same DLK1–GTL2 locus, of RNAi-mediated trans inhibition of the paternally expressed PEG11 by miRNAs processed from the maternally expressed anti-PEG11 transcript (Seitz et al. 2003; Davis et al. 2005).
To identify small RNAs that might be involved in the trans inhibition of DLK1 in skeletal muscle of CLPG/CLPG animals we have performed high-throughput sequencing (HTS) of small RNA libraries generated from skeletal muscle of sheep of the four possible CLPG genotypes. To qualify as mediators of the trans effect underlying polar overdominance (Georges et al. 2003, 2004), the corresponding small RNAs should (1) map to the DLK1–GTL2 domain; (2) be imprinted with expression from the maternal allele; (3) be subject to the cis effect of the CLPG mutation (i.e., be ectopically expressed in skeletal muscle upon maternal transmission of the mutation); and (4) have the ability to guide the RISC complex to DLK1 transcripts for inhibition.
Results
A catalog of miRNAs expressed in skeletal muscle of sheep
The callipyge phenotype is most pronounced in muscles of the hindquarters and manifests itself at ∼1 mo of age. At 8 wk of age, DLK1 protein is detected in skeletal muscle of +mat/CLPGpat but not of CLPG/CLPG animals, suggesting that the trans effect operates at that stage (Davis et al. 2004). Thus, we elected to extract RNA from longissimus dorsi (LD) of two 8-wk-old animals per CLPG genotype. Small RNA (∼18 to 30 bp) libraries were generated and sequenced on a Genome Analyzer I (Illumina). We obtained an average of 6,324,668 reads per animal (range: 5,222,920–6,685,342). Filtered, adapter-trimmed sequences (94.7%) were aligned to the bovine genome used as reference, with the exception of 390 kb of ovine sequence corresponding to the DLK1–GTL2 domain (GenBank AF354168). The resulting alignments were used to predict miRNA precursors using mirRDeep (Friedlander et al. 2008). This yielded 472 precursors, capturing 98.3% (range: 98.15%–99.32%) of trimmed reads. Sequence comparison with precursors in miRBase combined with mapping data indicate that 228 and 87 are the orthologs and paralogs, respectively, of previously reported bovine miRNAs (Glazov et al. 2009), while nine are the orthologs of miRNAs described in non-ruminant mammals. Thus, 148 precursors might correspond to previously unknown miRNAs (Supplemental Table 1). The chromosomal distribution of miRNA precursors is shown in Supplemental Figure 2. Chromosome 21, harboring the CLPG locus, stands out with 61 precursors (chromosomal average: 14.8).
As the genomic sequence of sheep is not completed, we used the bovine sequence as reference. The effect of this substitution was estimated by comparing the output of miRDeep using either the bovine or the ovine sequence of the DLK1–GTL2 domain as reference. miRDeep predicts 49 precursors in this domain when using the ovine sequence, of which three are missed when using the bovine reference. Sensitivity is thus decreased by ∼6%, but specificity does not seem affected.
We aligned all precursor pairs using BLASTN and used the bitscores (>35 bits) to identify precursors families using the MCL algorithm (Enright et al. 2002). The unique inflation parameter was set to the most aggregative value (2.0) for which the miR-376 family was recovered without contaminants (Seitz et al. 2004). Using this approach, 256 of the 472 precursors (= 54%) clustered in 62 families. The largest family (miR-2284 family) comprised 99 members. The remaining 61 families counted 2.6 members on average (range: 2–6).
While for 197 (= 41.7%) precursors we observed reads mapping either to the 5p or 3p arm of the pre-miRNA, both types of reads were observed for the remaining 275 (= 58.3%), jointly defining 747 distinct miRNA “species.” The fraction (F) of 5p over total reads distinguishes five types of precursors (Landgraf et al. 2007): 5p-mature/3p-star (1 > F > 0.87), 5p > 3p (0.87 > F > 0.50), 5p = 3p (F = 0.5), 5p < 3p (0.50 > F > 0.13), and 5p-star/3p-mature (0.13 > F > 0). The frequency distribution of F-values is shown in Supplemental Figure 3. The five types represent, respectively, 46.0%, 10.2%, 1.2%, 10.2%, and 32.4% of precursors. Precursors spawning miRNAs preferentially from the 5p arm were 1.4 times more abundant than those with 3p excess.
Aligning the reads with the identified precursors revealed considerable 3′ length variability. As this might reflect trimming artifacts due to decreased sequencing fidelity toward the 3′-end, we will not elaborate further on it. 5′-Ends were in general more consistent, with nevertheless considerable evidence for the occurrence of isomirs (Morin et al. 2008). For 65% of the miRNAs, ≥90% of reads shared the same 5′ extremity, for 27%, ≥90% of reads shared one of two 5′ extremities, and for 6%, ≥90% of reads shared one of three 5′ extremities. In general, more than 91% of alternative 5′ extremities were within 4 bp of the most common one.
Annotating miRNAs expressed from the DLK1–GTL2 domain
Forty-nine of the precursors identified by miRDeep mapped to the DLK1–GTL2 domain. Of these, 39 corresponded to known miRNAs reported in miRBase, while 10 were unknown. Detailed examination of the SOAP (Li et al. 2008) alignments revealed 5729 reads mapping to 14 regions not recognized by miRDeep as miRNA precursors. Six of these corresponded to miRNAs reported in miRBase and were included in the catalog. In addition, 487 reads mapped to 12 predicted C/D snoRNAs within MEG8 (of note, bona fide C/D snoRNAs are ∼80 bp long and would therefore have been excluded from the small RNA libraries). We found no reads for 14 miRNAs reported in human and/or in mice (of which five conserved in sheep).
5′-Ends showed the level of variability observed in the genome-wide catalog, that is, respectively, 58%, 34%, and 8% of miRNAs with one, two, and three isomirs representing ≥90% of the reads. Remarkably, 49 precursors (89.1% of the expressed precursors) had reads mapping to both 5p and 3p arms, to be compared with the genome-wide 58.3%.
A summary of all miRNA precursors identified in the DLK1–GTL2 domain is given in Figure 1. A total of 110 distinct small RNA species (not distinguishing isomirs) were identified, mapping to 61 miRNA and 12 C/D snoRNA precursors. All detected small RNAs derive from the same strand as GTL2, anti-PEG11, MEG8, and MIRG. Using a 10-way mammalian sequence alignment of MIRG, we generated a plot of sequence conservation within 8-nt windows (Supplemental Fig. 4A). We observed a striking coincidence between the peaks of conservation and the positions of the miRNAs, supporting miRNA generation as the primary function of MIRG. A similar colocalization of conservation peaks and C/D snoRNAs is not observed for MEG8 (Supplemental Fig. 4B).
Figure 1.

Comparative map of the small RNA genes in the DLK1–GTL2 domain: snoRNAs (upper panel) and miRNAs (lower panel). Square boxes correspond to small RNAs detected in sheep (red), cow (orange), human (green), and mouse (blue). Gray lines connect orthologs in the four species and indicate their chromosomal position with respect to the four long noncoding RNA genes in the domain: GTL2, anti-PEG11, MEG8, and MIRG. The position of precursors not detected in sheep is indicated by squares nested between vertical gray lines. The numbers above and below snoRNA and miRNA columns, respectively, correspond to numbers of additional paralogs for the snoRNAs (from +1 to +8) and names of additional miRNAs. The red squares are filled when reads for the corresponding small RNA were found in the conducted HTS experiments, empty when not. (Black dots) miRNAs predicted by miRDeep (Friedlander et al. 2008) in sheep. Numbers below the black dots identify the cluster/family to which the corresponding miRNA was assigned using the BLAST/MCL algorithm (Enright et al. 2002). The family number of miR-544 is underlined as the other members map outside of the DLK1–GTL2 domain. (Black dots) snoRNAs predicted by HMMER (Durbin et al. 1998) in sheep. snoRNAs in mouse and human correspond to predictions made by Cavaille et al. (2002). snoRNAs in the cow were predicted by HMMER (Durbin et al. 1998). miRNAs in cow, human, and mouse were extracted from miRBase (Griffiths-Jones 2006).
Limits of HTS for the quantitative assessment of miRNA expression
Read numbers are assumed to faithfully reflect expression levels, allowing for accurate digital gene expression profiling. However, recent data indicate that the amplification steps during library construction may introduce substantial, protocol-specific biases (Linsen et al. 2009). To evaluate accuracy and precision of our HTS data in measuring miRNA expression, we (1) repeated the HTS experiment for seven of the eight animals (including RNA extraction, library construction, and sequencing on an Illumina GA-II instrument); (2) hybridized skeletal muscle (LD) RNA from the eight animals on Exiqon miRCURY LNA (Version 9.2—updated to miRBase 11.0) arrays (GEO GSE24146); and (3) performed QRT-PCR for eight miRNAs spanning a broad range of expression levels as determined by HTS. While the Exiqon arrays allow interrogation of 569 human miRNAs, we restricted the analysis to 265 for which the LNA probes were perfectly complementary to the orthologous ruminant miRNA.
The main conclusions of this experiment can be summarized as follows:
Spearman rank correlations (rS) between sequencing replicates were 0.80 on average, thus suggesting adequate reproducibility of HTS (Supplemental Fig. 5). Note that correlations were slightly higher when comparing pairs of animals with the same CLPG genotype within sequencing runs (average rS = 0.83) (data not shown). Examination of specific miRNAs, however, highlighted limitations of digital expression profiling by HTS. Hence, while miR-127 accounted on average for 4% of reads originating from the DLK1–GTL2 domain in the first experiment, its contribution increased to 25% on average in the second, pointing toward systematic discrepancies between the two experiments for some miRNAs. Moreover, while miR-1 represented ≥83% of reads (average 86%) in the first series of eight libraries, and ≥80% of reads (average 82%) in 5/7 libraries of the second series, it only reached 32% and 49% in the two remaining ones, thus showing substantial discrepancies even within an experiment. Finally, within sequencing experiments, the 5p/3p ratio differed significantly between individuals for nearly all miRNA precursors (chi squared test). In extreme cases, different individuals would appear to have inverted 5p/3p ratios despite sequence depths of hundreds and even thousands. In no case were these opposite 5p/3p ratios confirmed in the second experiment. A representative example (miR-382) is shown in Supplemental Figure 6. Thus, while the repeatability of HTS may seem satisfactory in general, our findings suggest that amplification efficiency of specific miRNAs may vary considerably between experiments.
rS values between expression levels (ranks) assessed using the Exiqon miRCURY LNA arrays averaged 0.86 between individuals of the same CLPG genotype. This value has to be compared with a value of 0.90 when restricting the HTS data to the 265 miRNAs interrogated with the Exiqon array. Thus, in these experiments, HTS and array hybridization were characterized by comparable reproducibility. Yet, when comparing ranks obtained with the two methods, rS values dropped to 0.63 (first sequencing experiment) and 0.68 (second sequencing experiment) (Supplemental Fig. 7A,B). Supplemental Figure 7, C and D, illustrates the impact of this correlation drop in terms of probability of reversed rank order between alternative methods as a function of observed fold difference in expression level: miRNA pairs showing a fivefold difference in expression level on the Exiqon arrays still have a probability of ∼0.15 to be ranked inversely by HTS. Of note, the RNAs hybridized on the arrays were not size-selected (thus potentially including pri-, pre-, and mature miRNA molecules), while the RNAs used to construct the libraries for HTS were size-selected to include only mature miRNAs.
Given the observed discrepancies between the HTS and array-hybridization, we performed QRT-PCR for eight miRNAs (let-7d, miR-1, miR-206, miR-127, miR-382-5p, miR-382-3p, miR-3958, miR-3959) spanning a range of expression levels using the looped RT primer approach (targeting mature miRNAs) (Chen et al. 2005). QRT-PCR experiments were conducted in duplicate on the RNA samples used for HTS. Abundance of miRNA “x” relative to let-7d was estimated as
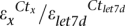 , where ε's are the experimentally determined amplification efficiencies and Ct's the threshold exceeding cycle numbers. From these analyses it appeared that the QRT-PCR results were more consistent with the array-hybridization than with HTS in terms of expression ranks and estimated fold differences in expression levels (Supplemental Fig. 8A,B). Our data strongly suggest that some miRNAs undergo preferential amplification during the HTS procedure.
, where ε's are the experimentally determined amplification efficiencies and Ct's the threshold exceeding cycle numbers. From these analyses it appeared that the QRT-PCR results were more consistent with the array-hybridization than with HTS in terms of expression ranks and estimated fold differences in expression levels (Supplemental Fig. 8A,B). Our data strongly suggest that some miRNAs undergo preferential amplification during the HTS procedure.
Effect of CLPG genotype on relative expression levels of miRNAs in the DLK1–GTL2 domain
The previous findings call for caution when interpreting variations in expression levels of individual miRNAs. To overcome this limitation, we examined the effect of CLPG genotype on the expression level of the miRNAs from the DLK1–GTL2 domain considered as a group.
We first confirmed the previously described cis effect of the CLPG mutation on neighboring genes in the sequenced RNA samples. QRT-PCR experiments were conducted using primer sets specific for mature DLK1 and GTL2 transcripts, and for two internal controls (RPLP0, RPS18) selected with geNorm out of five housekeeping genes (Vandesompele et al. 2002). The expected CLPG effects were clearly observed (Supplemental Fig. 9). Expression levels of DLK1 were increased ∼12-fold and approximately fourfold in, respectively, +Mat/CLPGPat and CLPG/CLPG animals when compared to +/+, while being slightly decreased (∼0.6) in CLPGMat/+Pat. Expression levels of GTL2 were increased ∼30-fold, ∼14-fold, and approximately fivefold in CLPG/CLPG, CLPGMat/+Pat, and +Mat/CLPGPat, when compared to +/+ animals. These results were undistinguishable from the ones that were previously reported using samples originating from other animals (Charlier et al. 2001a; Davis et al. 2004, 2005). The previously observed approximately fivefold increase of GTL2 expression in +Mat/CLPGPat when compared to +/+ animals, and approximately twofold increase of GTL2 expression in CLPG/CLPG when compared to CLPGMat/+Pat animals remains particularly intriguing and points toward a trans effect of the padumnal CLPG mutation on the expression level of the madumnal noncoding RNA genes (Charlier et al. 2001a).
We then analyzed the HTS data. Read numbers corresponding to a given miRNA species (i.e., mapping either to the 5p or 3p arm of a precursor) were first adjusted to account for the different numbers of total “mappable” reads per individual. The relative expression level for a given animal was expressed as log2(i/m), where i corresponds to the adjusted number of reads for that individual and m is the experiment-specific average number of adjusted reads for that miRNA across the seven individuals that were sequenced twice. Average log2(i/m) across miRNAs differed considerably between individuals, including for miRNAs outside of the CLPG locus. This was thought to reflect experimental issues rather than genuine biological differences (Supplemental Fig. 10). Therefore, log2(i/m) values were corrected for the average log2(i/m) value across miRNAs mapping outside of the DLK1–GTL2 domain (for that individual). We then tested the effect of CLPG genotype on the corrected relative expression levels by ANOVA, using both sequencing experiments jointly. Figure 2A shows the corresponding log(1/p) values. The effect of CLPG genotype on the relative expression level of miRNAs from the DLK1–GTL2 domain is clearly visible from the localized cluster of significant log(1/p) values. Six (miR-379, miR-411a, miR-495, miR-154b, miR-655, and miR-299) of the 99 “regular” miRNAs (i.e., excluding small RNAs derived from C/D snoRNAs) exhibited P-values <6 × 10−5, corresponding to the Bonferroni-corrected 5% threshold, while 47 were characterized by nominal P-values <0.05. Figure 2B shows, for each of the eight studied animals, the average relative expression levels over all “regular” miRNAs of the DLK1–GTL2 domain. The order is identical to that observed for the long noncoding RNA genes including GTL2: CLPG/CLPG > CLPGMat/+Pat > +Mat/CLPGPat > +/+. The magnitude of the effect, however, was smaller: Expression levels were increased ∼6.4-fold, ∼4.4-fold, and ∼2.0-fold in CLPG/CLPG, CLPGMat/+Pat, and +Mat/CLPGPat when compared to +/+ animals.
Figure 2.

(A) Log10(1/p) values of the effect of CLPG genotype on the expression level of 851 small RNAs in skeletal muscle of eight 8-wk-old sheep. Expression levels were estimated from the number of Illumina GA reads from two independent HTS experiments. The statistical significance of the CLPG effect was estimated by ANOVA. Gray vertical bars correspond to miRNAs outside of the DLK1–GTL2 domain, red vertical bars to miRNAs from the DLK1–GTL2 domain, and orange vertical bars to small RNAs derived from C/D snoRNA precursors. Horizontal black lines correspond to the nominal (plain line) and Bonferroni-adjusted (dotted line) 5% significance thresholds. Horizontal blue bars mark the different chromosomes (right Y-axis). (UN) Unassigned sequence contigs. (B) Average expression level, relative to the mean expression level of seven individuals sequenced twice (HTS1 and HTS2), of 99 “regular” miRNAs (i.e., excluding small RNAs derived from C/D snoRNAs) from the DLK1–GTL2 domain in skeletal muscle of eight sheep sorted by CLPG genotype (gray: +/+; blue: +Mat/CLPGPat; red: CLPGMat/+Pat; purple: CLPG/CLPG). Error bars correspond to 1.96 × the standard error of the estimate.
The previous figures pertain to “regular” miRNAs from the DLK1–GTL2 domain. As can be seen from Figure 2A, the effect of CLPG genotype on the expression level of small RNAs derived from C/D snoRNAs were not significant, suggesting that MEG8 might escape the cis effect of the CLPG mutation, contradicting previous findings (Charlier et al. 2001a). Examination of the effect of CLPG genotype on C/D snoRNA-derived small RNAs, however, revealed the expected trend in all but one +Mat/CLPGPat individual (Supplemental Fig. 11). Expression levels of C/D snoRNA-derived species (average number of reads: 70; median: 6) were low when compared to miRNAs (average number of reads: 21,300; median: 249). Low levels, combined with the aberrant behavior of one individual, explain the nonsignificance of the CLPG effect on the expression level of small RNAs derived of C/D snoRNAs, which we nevertheless believe exists.
The effect of CLPG genotype on relative miRNA expression levels was also evaluated from the array data (Supplemental Fig. 12A,B). The effect of CLPG genotype was equally clear, manifesting itself as a clustered rise in log(1/p) values. Expression levels were increased ∼4.8-fold, ∼3.1-fold, and ∼2.2-fold in CLPG/CLPG, CLPGMat/+Pat, and +Mat/CLPGPat, when compared to +/+ animals. Hence, the ranking was as expected (CLPG/CLPG > CLPGMat/+Pat > +Mat/CLPGPat > +/+), yet the magnitude of the effect was slightly lower than the HTS estimates.
Imprinting status of miRNAs in the DLK1–GTL2 domain
The obvious interpretation of miRNA expression levels in +Mat/CLPGPat and CLPGMat/+Pat intermediate between +/+ and CLPG/CLPG (Fig. 2B) is that most miRNAs from the DLK1–GTL2 domain are not imprinted, yet affected by the CLPG cis effect. All previous evidence, however, indicates that the maternally expressed noncoding RNA genes, including the embedded C/D snoRNA and miRNA genes, are exclusively expressed from the maternal allele. In the mouse, tested C/D snoRNAs and miRNAs from the DLK1–GTL2 domain were expressed in mice with maternal uniparental disomies of chromosome 12 (mUPD12) but not with paternal UPD12 (Cavaille et al. 2002; Seitz et al. 2003, 2004). The same small RNA genes were expressed in mice inheriting a deletion of the IG-DMR imprinting control element when on the paternal allele, but not when on the maternal allele (Lin et al. 2003; Seitz et al. 2004). In human, MEG8 (also known as RIAN), from which the C/D snoRNA genes are processed (Cavaille et al. 2002), was not expressed in patients with pUPD14 (Kagami et al. 2008). We have previously shown that in sheep muscle, anti-PEG11 and MEG8 (hosting miRNAs and C/D snoRNAs, respectively) are exclusively expressed from the maternal allele, irrespective of CLPG genotype (Charlier et al. 2001a,b).
To more directly assess the imprinting of the miRNAs from the DLK1–GTL2 domain in sheep and the effect of the CLPG mutation on it, we searched for SNPs in the vicinity of pre-miRNAs for which at least one of the four studied CLPGMat/+Pat or +Mat/CLPGPat animals would be heterozygous. We found nine such SNPs tagging six pre-miRNAs. Seven of these SNPs were within 160 bp from the corresponding pre-miRNA (miR-379, miR-134, miR-485, miR-453, miR-154b), one was in the loop (miR-453), and one was at position 20 of the miRNA* (miR-377). One +Mat/CLPGPat animal was homozygous for all SNPs and hence noninformative, but the other three were heterozygous for most (Table 1). For each SNP the allele associated with the CLPG mutation was determined by sequencing a CLPG/CLPG animal.
Table 1.
Imprinting status of miRNAs in the ovine DLK1–GTL2 domain and effect of the CLPG genotype

aName of the tested miRNA. miR-379 to miR-154b, percent of the madumnal/padumnal alleles found in cDNA; miR-377, number of HTS reads corresponding to the madumnal/padumnal allele.
bPosition of the interrogated SNP with respect to the corresponding pre-miRNA.
cSNP alleles associated with the CLPG and + allele.
dCLPG genotype of corresponding animal.
For miR-377 and knowing that the SNP mapped to the miRNA*, we determined imprinting status from HTS data. We exclusively detected reads corresponding to the madumnal allele, both in the CLPGMat/+Pat and +Mat/CLPGPat animals, thus supporting tight imprinting, exclusive madumnal expression, and no effect of CLPG genotype on imprinting. For miRNAs with SNPs lying outside of the mature miRNAs, we amplified the pri-miRNA with primers within 179 bp from the pre-miRNA, directly sequenced the resulting amplicons, and measured the allelic ratio using PeakPicker (Ge et al. 2005). For four of the five miRNAs, the results were identical to miR-377 (Table 1): tight imprinting, near exclusive madumnal expression, and no effect of CLPG genotype. For miR-485, however, we observed relaxation of imprinting in one CLPGMat/+Pat animal and one +Mat/CLPGPat animal, for which ∼20% of transcripts were derived from the padumnal allele. There was no evidence for relaxation of imprinting of miR-485 in the other informative CLPGMat/+Pat animal (Table 1). It is noteworthy that miR-134 located 588 bp upstream, and miR-154b located 4293 bp downstream of miR-485 did not show evidence for relaxation of imprinting in the same individuals.
Effect of CLPG genotype on absolute expression levels of miRNAs in the DLK1–GTL2 domain
The previous analyses are not informative about absolute miRNA expression levels: Are they just present in minute, biologically irrelevant amounts? Or do they make a significant contribution to the pool of miRNAs in muscle? Analysis of the two sequencing experiments indicate that, after exclusion of miR-1 whose read numbers were inflated (see above), the percentage of reads originating from miRNA precursors in the DLK1–GTL2 domain was 4% in +/+ animals, but increased to 10%, 11%, and 21% of the total in +Mat/CLPGPat, CLPGMat/+Pat, and CLPG/CLPG animals. The Exiqon arrays only measure the abundance of some miRNAs present in a tissue. However, as the proportion of miRNAs mapping to the DLK1–GTL2 domain was virtually identical in the HTS (99/826 = 12%) and hybridization data (34/265 = 12.8%), the ratio of the sum of fluorescence intensities for miRNAs in the domain over the sum of fluorescence intensities over all miRNAs would also provide an estimate of the cellular abundance of miRNAs originating from the DLK1–GTL2 domain. miRNAs from the domain accounted for 3.5%, 9.0%, 15.6%, and 22.3% of the Hy3 fluorescence on the Exiqon arrays in +/+, +Mat/CLPGPat, CLPGMat/+Pat, and CLPG/CLPG animals. Both approaches thus provided comparable estimates, indicating that the DLK1–GTL2 domain contributes a sizeable fraction of the miRNA population, especially in CLPG/CLPG animals in which they are predicted to mediate the trans inhibition of DLK1.
While claims about expression levels of individual miRNAs are hazardous for the reasons mentioned before, the QRT-PCR and array experiments strongly suggest that miRNAs from the DLK1–GTL2 domain are characterized by an at least 30-fold range of expression levels (Supplemental Figs. 8, 13).
Effect of CLPG genotype on relative expression levels of miRNAs outside the DLK1–GTL2 domain
Skeletal muscles that express the callipyge hypertrophy have a profoundly altered physiology. Ectopic expression of DLK1 (Davis et al. 2005) triggers a cascade of secondary events leading to muscular hypertrophy (e.g., Vuocolo et al. 2007). These may involve altered miRNA expression. To detect such secondary miRNA perturbations, we tested the effect of CLPG genotype on relative expression levels of miRNAs outside of the domain. We first considered the HTS and array hybridization data separately. When accounting for multiple testing, no miRNA outside of the DLK1–GTL2 domain appeared to be significantly affected by the CLPG genotype (Fig. 2; Supplemental Fig. 12). In an attempt to increase power, we combined HTS and array data for the 265 miRNAs with information on both platforms. Even then, no miRNA outside of the domain was significant (Supplemental Fig. 14). We conclude that the molecular events connecting ectopic expression of DLK1 and muscular hypertrophy do not involve altered miRNA expression.
Editing of miRNAs from the DLK1–GTL2 domain in skeletal muscle
It was recently observed that a cluster of miRNAs mapping to the DLK1–GTL2 domain (human miR-368, miR-376a1 [5′ and 3′], miR-376b and miR-376a2 [5′ and 3′]; murine miR-376a [5′ and 3′], miR-376b [5′ and 3′], and miR-376c) undergo extensive A-to-I editing in human and mice, particularly in the central nervous system (Kawahara et al. 2007). The most extensively edited sites correspond to position “+3” or “+4” of the 5p miRNAs and position “+6” of the 3p miRNAs. Analyses performed in knockout (KO) mice suggest that 5p editing is ADAR2 (also known as ADARB1) dependent, while 3p editing is ADAR1 (also known as ADAR) dependent. Editing seemed not to affect processing, as equally high levels were observed in pri-miRNAs and derived mature miRNAs. By changing the seed, editing was predicted to alter the target spectrum.
As editing of miRNA seeds from the DLK1–GTL2 domain in sheep may likewise alter affinity for DLK1, we systematically searched for it. We first examined whether the precursors of mir-376a,b,c undergo editing in skeletal muscle of mice. We RT-PCR-amplified the corresponding pri-miRNAs from cDNA of brain, kidney, and skeletal muscle (quadriceps femoris) from an FVB mouse and sequenced the corresponding PCR products. Strong (∼80%) and moderate (∼60%) editing of the “+44” 3p position (residue “+6” of the mature miRNA) was observed for the three studied miRNAs in, respectively, brain and kidney, hence recapitulating part of the results of Kawahara et al. (2007). Contrary to these investigators, we found no evidence for editing of the 5p arms, whether in or upstream of the mature miRNA sequence. No editing was observed in skeletal muscle of mice (data not shown).
We then scanned the pri-miRNAs corresponding to 56 precursors from the DLK1–GTL2 domain using RNA extracted from skeletal muscle (LD) of one CLPG/CLPG and one +/+ sheep. Within the miR-376 cluster, we did observe substantial levels of editing of the “+44” 3p position (corresponding to position “+6” of the mature miRNA) of miR-376e (0%–25%), miR-376c (also known as miR-368; 5%–45%), miR-376a2 (0%–50%), miR-376b (0%–95%), but not of miR-654 and miR-376a1. No editing was observed in the 5p arm for any of these pri-miRNAs. Outside of the miR-376 cluster, we observed strong editing of three other pri-miRNAs: at the equivalent “+44” 3p position (corresponding in this case to position “+5” of the mature miRNA) for miR-381 (10%–82%), at 5p position “+5” (corresponding to position “+5” of the mature miRNA) for miR-411a (0%–20%), and at 5p position −4 outside of the mature miRNA sequence for miR-369 (0%–40%). Note that neither miR-381 nor miR-411a shows obvious similarity with members of the miR-376 cluster. Based on these results, we evaluated the level of editing of miR-376e, miR-376c, miR-376a2, miR-376b, miR-381, and miR-411a in 16 additional animals representing the four possible CLPG genotypes at 2 and 8 wk of age. Editing of miR-376c, miR-376b, and miR-381 was observed in some of the new animals, but not of miR-376e, miR-376a2, and miR-411a. Unexpectedly, we observed a highly significant (p ≤ 1.3 × 10−5) effect of CLPG genotype on the level of editing of miR-376c, miR-376b, and miR-381: +/+ animals had markedly higher levels of pri-miRNA editing than the three other genotypes (Fig. 3A).
Figure 3.

(A) Percentage of A-to-I editing of pri-miRNAs at the “+5” position (pre-miR-411a) or “+44” position (pre-miR-376e, pre-miR-376c, pre-miR-376a2, pre-miR-381) in longissimus dorsi of 18 animals representing the four possible CLPG genotypes [gray: +/+ (5); blue: +Mat/CLPGPat (4); red: CLPGMat/+Pat (4); purple: CLPG/CLPG (5)]. The first two animals of each CLPG genotype were analyzed at 2 wk, the others at 8 wk. (B) Percentage of A-to-I editing of mature miRNAs at position “+5” (miR-411a = 5p), “+6” (miR-376e, miR-376c, miR-376a2, miR-376b = 3p), and “+5” (miR-381 = 3p). Animals are ordered as in A. (*) Animals without HTS data. The numbers above each column correspond to the total number of reads (edited + non-edited) available for analysis. The black horizontal lines correspond to the average level of A-to-G substitution observed for miRNAs derived from the 5p arm at position “+5” (miR-411a), from the 3p arm at position “+6” (miR-376e, miR-376c, miR-376a2, miR-376b), and from the 3p arm at position “+5” (miR-381).
To verify whether editing of the pri-miRNAs resulted in equivalent proportions of edited mature miRNAs (as observed by Kawahara et al. [2007]), we evaluated the level of editing of mature miR-376e, miR-376c, miR-376a2, miR-376b, miR-381, and miR-411a in the HTS libraries (Fig. 3B). In general, editing levels of mature miRNAs were below 10%. For the three miRNAs with high levels of pri-miRNA editing (i.e., miR-376c, miR-376b, miR-381), levels dropped considerably in the fully processed miRNAs. This was most striking for miR-376b with virtually total absence of edited reads. For miR-376c and miR-381, editing levels dropped by a factor of ∼10 when compared to the precursors. Thus, for these miRNAs, editing either inhibits pri/pre-miRNA processing and/or reduces the stability of the miRNA. The effect of CLPG genotype on editing levels was still apparent for miR-376c and miR-381. For miR-376a2, editing levels appeared higher after than before miRNA processing (although still well below 10%). In this case, editing may thus promote processing and/or stability. Finally, for miR-411a, mature editing levels were consistently of the order of 1%, which was well above background (average of 0.02% across 14 5p miRNAs with an A residue at position “+5”). Such levels would not have been reliably detected at the pri-miRNA level.
Evaluating the affinity of miRNAs in the DLK1–GTL2 domain for DLK1
Having generated an exhaustive catalog of miRNAs expressed in skeletal muscle of CLPG/CLPG animals allowed us to test the miRNA-mediated DLK1 trans inhibition hypothesis with unprecedented power.
For each of the 114 miRNA species from the DLK1–GTL2 domain, we singled out the most abundant isomir (or pair of isomirs in ex aequo cases, leading to 127 distinct sequences) and quantified its affinity for DLK1 using two established metrics. The first one (“G-species score”) follows Grimson et al. (2007) and counts the occurrences of 6-mer (Watson-Crick [WC] reverse complement of miRNA residues 2 to 7), 7-mer-m8 (WC reverse complement of miRNA residues 2 to 8), 7-mer-A1 (WC reverse complement of miRNA residues 2 to 7 plus 3′ A anchor), and 8-mer matches (WC reverse complement of miRNA residues 2 to 8 plus 3′ A anchor) in DLK1. Thus, an 8-mer match would increase the “G-species score” by 4, that of a 7-mer (without 8-nt match) by 2, and that of 6-mer match by 1. The second one (“M-species score”) sums scores (≥140) obtained with the more liberal miRNA-target miRanda identification engine (John et al. 2004). G-species scores were summed to generate a “G-quadrille score,” and “M-species scores” were summed to generate an “M-quadrille score.” “Quadrille scores” evaluate the affinity for DLK1 of the miRNAs considered as a team. Moreover, the same scores were generated for human and mouse (using species-specific miRNA sequences reported in miRBase), and corresponding scores were summed across species to generate “multiorganism (MO) scores.” The latter should be more effective at identifying an unusual affinity for DLK1 if conserved across species. Whether the mechanisms underlying the trans inhibition of DLK1 observed in sheep are shared with other species remains unknown.
To evaluate the statistical significance of the obtained metrics, we compared them with their distribution obtained on 10,000 random shuffles of the DLK1 sequence. Shuffling was conducted such as to maintain the original trinucleotide composition of the target gene (cf. Supplemental Methods). While miRanda is expected to inflate the number of target predictions, their statistical significance should be well controlled by this approach (i.e., there is no reason why the true DLK1 sequence should yield better miRanda scores than the shuffled sequences).
We first tested the approach using the PEG11 ORF as positive control (1000 shufflings). PEG11 is indeed targeted by at least six miRNA species derived from five pre-miRNAs in anti-PEG11 in ovine, human, and mouse (miR-431, miR-433, miR-127, miR-432 [human and sheep], miR-434 [mouse], miR-136) (Davis et al. 2005). The targeting of PEG11 by these miRNAs is “plant-like,” relying on WC complementarity over the entire length, resulting in target slicing (Davis et al. 2005). For proper comparison with the presumably “animal-like” situation of DLK1, we only considered the miRNA seed sequences (residues 1 to 8) when computing the G-species scores (replacing the 3′ A-anchor constraint [applying to 8-mer and 7-mer-A1 matches] by 3′ W&C complementarity). miRanda scores were computed as before. When considering the ovine sequence (ORF) alone, both quadrille scores were significant (G: p = 0.003; M: p = 0.025), hence detecting the presence of one or more miRNAs targeting PEG11. Considered individually, however, none of the miRNA exceeded the Bonferroni-corrected 5% threshold. Two (of 12) miRNAs processed from anti-PEG11 (miR-431-5p and miR-136-3p) achieved nominal significance (p = 0.003) for both G- and M-species scores. Intriguingly, the ovine-specific miR-3959-3p, although processed from MIRG, achieved equivalent significance (p = 0.003) (Supplemental Fig. 15A). When considering the three species simultaneously (hence exploiting evolutionary conservation known to exist), the significance of the two quadrille scores increased (G and M: p < 0.001). Moreover, six (of 12) miRNA species processed from anti-PEG11 yielded the highest possible signal (nominal p < 0.001; Bonferroni-corrected p ∼ 0.10). Interestingly, miR-411a-5p processed from MIRG achieved the same top score, while the signal for miR-3959 remained essentially unchanged (nominal p = 0.003) as this Laurasiatheria-specific miRNA is not shared with human and mouse (Supplemental Fig. 15B). The high miR-411a scores reflect one 7-mer and 10 6-mer matches in mouse, one 7-mer and six 6-mers in human, and two 7-mers and two 6-mers in sheep. Three matches were conserved in the three species, and one in two species. The high ovine miR-3959 score is due to one 8-mer and one 7-mer match. Thus, application of our method to PEG11 indicated that (1) significant quadrille scores, but not species scores, could be obtained without exploiting evolutionary conservation; (2) significant quadrille and species scores could be obtained when exploiting conservation. Most interestingly, this analysis strongly suggests that the paternally expressed PEG11 is not only targeted by fully complementary miRNAs processed from the maternally expressed anti-PEG11, but also by miRNAs processed from the maternally expressed MIRG pri-miRNAs, which recognize their target via seed-dominated complementarity.
We then applied the same approach to DLK1 including the 5′-UTR, ORF, and 3′-UTR as it is established that miRNAs may target these different gene compartments (Baek et al. 2008; Selbach et al. 2008; Tay et al. 2008; Chi et al. 2009). When relying solely on ovine information, the most noteworthy result was the nearly significant G-quadrille score (p = 0.052) on the DLK1 ORF, hence suggesting an unusual affinity of the ovine miRNA team for this segment of DLK1 (Fig. 4A). This signal was primarily driven by miR-377-3p (nominal p = 0.0012; one 8-mer and two 7-mer-m8 matches), miR-1193-3p (nominal p = 0.0098; one 8-mer and two 7-mer-m8 matches), and miR-370-5p (nominal p = 0.0098; one 8-mer and one 7-mer-m8 match; Fig. 4B). Note that none of these miRNAs achieves Bonferroni-adjusted significance. There was no convincing evidence for miRNA targeting of the DLK1 5′- or 3′-UTR.
Figure 4.



(A) Statistical significance [log(1/p)] of the affinity of ovine miRNAs in the DLK1–GTL2 domain for the 5′-UTR, coding sequence (ORF), and 3′-UTR of the ovine DLK1. The affinity was measured using either G- (blue) or M-scores (orange) as defined in the text. Bars are dark colored for highly expressed and light colored for lowly expressed miRNAs. The last pair of bars (“quad”) at the right of the graph corresponds to the quadrille scores, the remaining bars to the species scores and are labeled accordingly. P-values were determined using the sequence-shuffling test described in the text. Species scores require a Bonferroni correction for 127 independent tests. (B) Position in the DLK1 mRNA of target sites (8-mers, 7-mers, and 6-mers as defined by Grimson et al. [2007]) for the same set of miRNA species. (C) Same as in A except that the scores are “multiorganism (MO) scores” combining information from sheep, human, and mouse.
When adding the human and murine information, the significance of the MO G-quadrille score for the ORF increased slightly (p = 0.036), although none of the individual miRNAs clearly stood out (Fig. 4C). When applied to the 3′-UTR, the MO M-species score for miR-376c-3p achieved Bonferroni-corrected significance (nominal p = 0.0004; Bonferroni-corrected p = 0.05). This signal was due to a miRanda target site shared between mouse and sheep (Fig. 4C).
Weighting miRNA scores by expression level estimated from HTS reads and including all isomirs in such analyses did not yield stronger signals (data not shown).
Discussion
We herein establish a catalog of miRNAs expressed in skeletal muscle of sheep. Using miRDeep (Friedlander et al. 2008), we detected 747 small RNA species mapping to 472 miRNA precursors. Of these, 324 were classified as orthologs or close paralogs of known miRNAs, leaving 148 candidate novel miRNAs. It is noteworthy that expression levels of new miRNAs were considerably lower than those of known miRNAs (Supplemental Fig. 16).
As the ovine genome sequence is not completed, we used the bovine as reference for most of the genome. Comparison of the miRDeep performances on 390 kb of contiguous sequence available in both bovine and sheep indicates a possible loss of ∼6% sensitivity but not of specificity. Reanalyzing the sequence data with the ovine reference may thus increase the number of detected miRNAs. Within the DLK1–GTL2 domain, six known miRNA precursors were missed by miRDeep despite the occurrence of HTS reads. Assuming that the DLK1–GTL2 miRNA catalog of sheep is near complete, this corresponds to a sensitivity of 49/55 = 0.89. This figure is identical to that obtained by the miRDeep developers in Caenorhabditis elegans (Friedlander et al. 2008).
When focusing on the annotation of miRNAs in the DLK1–GTL2 domain, we noted about 500 reads mapping to 12 predicted C/D snoRNA genes in MEG8. This is reminiscent of Ender et al. (2008), who reported human AGO1-4-associated small RNAs mapping to C/D and H/ACA snoRNA precursors. More specifically, Ender et al. (2008) demonstrated Drosha-independent (also known as RNASEN), Dicer-dependent (also known as DICER1) processing of miRNAs derived from the bona fide ACA45 snoRNA (also known as SCARNA15), thereby revealing an alternative pathway for the generation of functional miRNAs. It is not yet known which of the C/D snoRNAs in MEG8 (Cavaille et al. 2002) are genuine, associating with core components of the C/D RNP (e.g., FBL, NHP2L1, NOP56, NOP58). Contrary to Ender et al. (2008), in four of six cases in which reads were derived from both arms, the two miRNA species were characterized by 2-nt 3′ overhangs compatible with Drosha-dependent processing of the pri-miRNA (Supplemental Fig. 17). Further work is needed to exclude the trivial possibility that the corresponding miRNAs derive from erroneously annotated C/D snoRNAs.
We confirm that miRNAs from the DLK1–GTL2 domain are imprinted in skeletal muscle of sheep and preferentially expressed from the maternal allele. As for the other genes in the domain, the imprinting status of the miRNAs is not affected by the CLPG genotype. Of note, the imprinting status of the genes in the DLK1–GTL2 domain cannot be directly tested in CLPG/CLPG animals as the padumnal and madumnal alleles are identical. However, we have demonstrated that the IG-DMR is differentially methylated in CLPG/CLPG animals as in other genotypes, supporting regular imprinting (data not shown). Previous studies in skeletal muscle of sheep revealed tight imprinting control for both paternally and maternally expressed genes. For one of the miRNAs (miR-485), we observed relaxation of imprinting in two (one CLPGMat/+Pat and one +Mat/CLPGPat) out of three studied individuals: Molecules derived from the paternal allele represented ∼15% of the total. Flanking miRNAs (located 600 bp upstream [miR-134] and 4000 bp downstream [miR-453]) did not show evidence of relaxation in the same samples. This strongly suggests that miR-485 is at least in part processed from a transcription unit independent of the one generating the two other miRNAs. Rather than being one unique large pri-miRNA, MIRG may thus encompass multiple transcription units controlled by distinct promoters. Along the same lines, it has recently been suggested that miR-433 and the adjacent miR-127 are processed from distinct pri-miRNAs (Song and Wang 2008) rather than from a unique anti-PEG11 precursor shared with miR-431, miR-434, and miR-136 (Davis et al. 2005).
We find that the miRNAs from the DLK1–GTL2 domain are affected by the CLPG mutation in the same manner as the maternally expressed long noncoding RNA genes (Charlier et al. 2001a). The main effect is in cis causing an ∼3.2-fold increase in miRNA expression from a CLPG versus wild-type maternal chromosome ([CLPGMat/+Pat]/[+/+] ≈ 3, 7; [CLPG/CLPG]/[+Mat/CLPGPat] ≈ 2, 7). As for the other genes affected by the CLPG mutation, this is thought to result from the inactivation of a muscle-specific cis-acting silencer element. In addition, we confirm a trans effect consisting in the ∼1.8-fold higher expression of miRNAs from the maternal allele when the paternal allele is CLPG versus wild type ([+Mat/CLPGPat]/[+/+] ≈ 2.1; [CLPG/CLPG]/[CLPGMat/+Pat] ≈ 1.5). While the MAT → PAT trans effect is known (PEG11) (Davis et al. 2005) or hypothesized (DLK1) (Georges et al. 2003, 2004) to reflect miRNA-mediated trans inhibition, the molecular mechanisms underlying this PAT → MAT trans effect remain elusive. One possible explanation is that the silencer element that is inactivated by the CLPG mutation has the capacity to exert its effect in trans on the homologous chromosome. Such mechanisms have been attributed to enhancers in Drosophila and other organisms and may underlie transvection (Kennison and Southworth 2002).
The cis effect on miRNA expression is consistent with that observed for the long noncoding RNA genes but is considerably weaker (∼3.2-fold vs. ∼9.5-fold). The reason for this difference is unknown. An explanation might be the saturation of the miRNA processing machinery.
Although HTS-based digital expression profiling may not be as quantitative as initially assumed, the combined HTS, array, and QRT-PCR data strongly suggest that miRNA expression levels differ at least ∼30-fold. This could be due to differential processing efficiency of the precursors and/or stability of the processed miRNAs, but may also reflect the dependence on distinct promoters of unequal strength. The latter hypothesis is supported by the miR-485 imprinting data (see above), as well as by the recent identification of private host-gene-independent promoters for intronic miRNAs (Ozsolak et al. 2008).
Contrary to skeletal muscle of mouse, we observed substantial levels of A-to-I editing for 4/6 pri-miRNAs from the miR-376 family (miR-376e, miR-376c, miR-376a2, miR-376b) and for three unrelated pri-miRNAs (miR-381, miR-411a, miR-369) from the DLK1–GTL2 domain in skeletal muscle of sheep. We noted a significant effect of CLPG genotype on pri-miRNA editing. Editing in +/+ animals, characterized by the lowest miRNA expression levels, was ∼4.2-fold higher than in the other genotypes. The reasons underlying this observation remain unclear, but could involve saturation of the editing machinery or down-regulation of components of the editing machinery by miRNAs from the domain. Contrary to Kawahara et al. (2007), editing levels were lower in the mature miRNA population when compared to precursors: Edited molecules never made up >10% of reads.
The primary aim of this study was to lay the grounds for the identification of miRNAs that might account for the translational down-regulation of DLK1 observed in CLPG/CLPG animals, that is, the MAT → PAT trans effect. We approached this by posing that if an exceptional affinity of miRNAs from the DLK1–GTL2 domain for DLK1 could be clearly demonstrated in silico, this would very strongly support the hypothesis. The absence of such statistically significant affinity does not preclude actual interaction in vivo, but is neutral with respect to the hypothesis. We would not pretend that our data reveal an unambiguous affinity of the DLK1–GTL2 miRNAs for DLK1, yet it is intriguing that both the G-quadrille score for the ORF and the miR-376c-3p MO M-species score for the 3′-UTR achieved 5% significance. This suggests that the DLK1–GTL2 miRNAs might, indeed, effectively down-regulate DLK1 in CLPG/CLPG animals. It is worthwhile re-stating in this regard that miRNAs from the domain account for an estimated ∼20% of cellular miRNAs in these animals. We are in the process of testing this prediction biochemically using both reporter assays and AGO-based (also known as EIF2C) target coimmunoprecipitation (e.g., Takeda et al. 2010), prioritizing miRNAs on the basis of the results presented in Figure 4.
While miRNAs from the domain are strong candidate direct mediators of the MAT → PAT trans effect on DLK1, alternative possibilities should not be excluded. Among those figure (1) an indirect effect of the miRNAs from the domain, as well as (2) miRNA-independent mechanisms. It is interesting with regard to the latter that no clear function has yet been assigned to GTL2.
Individual miRNAs are predicted to target 200 to 300 genes on average (Grimson et al. 2007). What are the targets of the highly conserved miRNAs in the DLK1–GTL2 domain? To start addressing this question, we assembled the list of 2832 bovine genes having at least one conserved 8-mer or 7-mer target site in their 3′-UTRs for any of the 24 out of 153 “conserved miRNA families” with representatives in the DLK1–GTL2 domain (Friedman et al. 2009). We then looked for the enrichment of specific gene ontology (GO) terms among these genes (Ashburner et al. 2000). To that end, we randomly sampled 10,000 (GO Slim analysis) or 200,000 (whole GO analysis) sets of 2832 genes from the complete TargetScan list of 8458 bovine miRNA-targeted genes and compared the hit number of each GO term for the list of target genes of the DLK1–GTL2 miRNAs with the distribution of hit numbers across the 10,000 (respectively, 200,000) random sets of genes. The resulting P-values were Bonferroni-corrected for the 6930 terms sampled out of the whole GO graph, or for the 55 terms of the GO Slim graph. Supplemental Table 2A shows the eight most enriched terms in both analyses, corresponding to a Bonferroni-corrected P-value ≤0.027 for the GO slim analysis, or a nominal P-value ≤10−4 for the GO whole analysis. The outcome of this analysis strongly suggests that the miRNAs from the DLK1–GTL2 domain are devoted to the targeting of regulators of the gene circuitry operating at the transcriptional, translational, and post-translational level, primarily in the nervous system. The list of genes corresponding to these top hits is provided in Supplemental Table 2, B and C.
Methods
Construction of small RNA libraries and high-throughput sequencing
Small RNA libraries were constructed using the “Small RNA sample preparation kit” following the instructions of the manufacturer (Illumina). Briefly, 10 μg of total RNA extracted with TRIzol (Invitrogen) was size-fractionated by denaturing polyacrylamide gel electrophoresis (PAGE; 15%), and molecules ranging from 18 to 30 nt were eluted. RNA adapters were successively ligated to the 5′- then 3′-end of the isolated small RNAs, and ligation products of the desired length (40–60 bp then 70–90 bp) were recovered by sequential PAGE (15% then 10%). Small RNAs appended with 5′ and 3′ adapters were reverse-transcribed with Superscript II (Invitrogen) and amplified with Phusion DNA polymerase (Finnzymes Oy). Resulting amplicons were PAGE (6%) gel-purified, hybridized on a flow cell lane, clustered, and sequenced (36 cycles) using standard procedures (Illumina). Libraries corresponding to eight animals (two of each CLPG genotype) were first sequenced on a GA-I (Illumina) by Fasteris SA. The experiment was subsequently repeated for seven animals using a GA-II instrument (Illumina) at the GIGA-R core facilities.
Bioinformatics analysis of small RNA reads
The bioinformatics procedures applied for preprocessing of HTS reads; prediction, curation, and annotation of miRNA precursors; prediction of C/D snoRNAs in the DLK1–GTL2 domain; gene annotation and conservation analyses in the domain; quantitative analyses of HTS reads; comparison of HTS, Exiqon, and TaqMan data; analysis of non-miRNA HTS reads; evaluation of miRNA affinity for DLK1; and GO analyses of targets of miRNAs encoded in the domain are described in detail in the Supplemental Methods.
Exiqon array hybridization
Skeletal muscle RNA samples from the same eight animals (two of each CLPG genotype) extracted with TRIzol (Invitrogen) were hybridized on Exiqon miRCURY LNA Arrays (v.9.2) at Exiqon (Vedbaek). Briefly, RNA quality was evaluated on an Agilent Bioanalyzer 2100. Individual samples were labeled with Hy3 using the miRCURY Hy3/Hy5 power labeling kit, and cohybridized on the arrays with a Hy5-labeled equimolar mix of the eight samples. Arrays were scanned in an ozone-free environment. Fluorescence intensities were normalized with the global Lowess (LOcally WEighted Scatterplot Smoothing) regression algorithm using all probes except those corresponding to miRNAs from the DLK1–GTL2 domain.
Quantitative RT-PCR
QRT-PCR analyses of miRNAs were conducted using predesigned (miR-1, miR-127, miR-206, miR-382, miR-382*, let-7d) or custom (miR-3958 and miR-3959) TaqManMicroRNA assays (ABI) on an 9700HT (ABI) instrument. Assay-specific amplification efficiencies were determined using serial RNA dilutions.
Editing
The level of pri-miRNA editing was determined by sequence analysis of genomic- and cDNA-derived PCR products. Genomic DNA and total RNA were extracted using TRIzol (Invitrogen). Reverse transcription was carried out using Supercript III (Invitrogen) on 1 μg of total RNA pre-treated with Turbo DNase (Ambion) and cDNA PCR-amplified using GOLD Taq (ABI; 35 cycles). The primer sequences used for mouse and sheep are provided in Supplemental Table 3. Amplicons were sequenced on a 3730 instrument (ABI), and the degree of editing was estimated from the electropherograms using PeakPicker (Ge et al. 2005).
Acknowledgments
This work was funded by grants from the Fonds National de la Recherche Scientifique, the University of Liège, the European FW6 program (CALLIMIR), the Communauté Française de Belgique (ARC Mirage and ARC Biomod), and the Belgian Science Policy Organisation (SSTC Genefunc PAI). Carole Charlier is Chercheur Qualifié au Fonds National de la Recherche Scientifique. We are grateful for the support of the GIGA-R sequencing core facility.
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence and miRNA expression data from this study have been submitted to NCBI's GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession nos. AF354168 and GSE24146, respectively. All new ovine miRNAs corresponding to the DLK1–GTL2 locus also have been submitted to miRBase (http://www.mirbase.org).]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.108787.110.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne K, Colgrave ML, Vuocolo T, Pearson R, Bidwell CA, Cockett NE, Lynn DJ, Fleming-Waddell JN, Tellam RL 2010. The imprinted retrotransposon-like gene PEG11 (RTL1) is expressed as a full-length protein in skeletal muscle from Callipyge sheep. PLoS ONE 5: e8638 doi: 10.1371/journal.pone.0008638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaille J, Seitz H, Paulsen M, Ferguson-Smith AC, Bachellerie JP 2002. Identification of tandemly-repeated C/D snoRNA genes at the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet 11: 1527–1538 [DOI] [PubMed] [Google Scholar]
- Charlier C, Segers K, Karim L, Shay T, Gyapay G, Cockett N, Georges M 2001a. The callipyge mutation enhances the expression of coregulated imprinted genes in cis without affecting their imprinting status. Nat Genet 27: 367–369 [DOI] [PubMed] [Google Scholar]
- Charlier C, Segers K, Wagenaar D, Karim L, Berghmans S, Jaillon O, Shay T, Weissenbach J, Cockett N, Gyapay G, et al. 2001b. Human-ovine comparative sequencing of a 250-kb imprinted domain encompassing the callipyge (clpg) locus and identification of six imprinted transcripts: DLK1, DAT, GTL2, PEG11, antiPEG11, and MEG8. Genome Res 11: 850–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. 2005. Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res 33: e179 doi: 10.1093/nar/gni178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB 2009. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 460: 479–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockett NE, Jackson SP, Shay TL, Farnir F, Berghmans S, Snowder GD, Nielsen DM, Georges M 1996. Polar overdominance at the ovine callipyge locus. Science 273: 236–238 [DOI] [PubMed] [Google Scholar]
- Davis E, Jensen CH, Schroder HD, Farnir F, Shay-Hadfield T, Kliem A, Cockett N, Georges M, Charlier C 2004. Ectopic expression of DLK1 protein in skeletal muscle of padumnal heterozygotes causes the callipyge phenotype. Curr Biol 14: 1858–1862 [DOI] [PubMed] [Google Scholar]
- Davis E, Caiment F, Tordoir X, Cavaille J, Ferguson-Smith A, Cockett N, Georges M, Charlier C 2005. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol 15: 743–749 [DOI] [PubMed] [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison G 1998. Biological sequence analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK [Google Scholar]
- Ender C, Krek A, Friedlander MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G 2008. A human snoRNA with microRNA-like functions. Mol Cell 32: 519–528 [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freking BA, Murphy SK, Wylie AA, Rhodes SJ, Keele JW, Leymaster KA, Jirtle RL, Smith TP 2002. Identification of the single base change causing the callipyge muscle hypertrophy phenotype, the only known example of polar overdominance in mammals. Genome Res 12: 1496–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N 2008. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26: 407–415 [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge B, Gurd S, Gaudin T, Dore C, Lepage P, Harmsen E, Hudson TJ, Pastinen T 2005. Survey of allelic expression using EST mining. Genome Res 15: 1584–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges M, Charlier C, Cockett N 2003. The callipyge locus: Evidence for the trans interaction of reciprocally imprinted genes. Trends Genet 19: 248–252 [DOI] [PubMed] [Google Scholar]
- Georges M, Charlier C, Smit M, Davis E, Shay T, Tordoir X, Takeda H, Caiment F, Cockett N 2004. Toward molecular understanding of polar overdominance at the ovine callipyge locus. Cold Spring Harb Symp Quant Biol 69: 477–483 [DOI] [PubMed] [Google Scholar]
- Glazov EA, Kongsuwan K, Assavalapsakul W, Horwood PF, Mitter N, Mahony TJ 2009. Repertoire of bovine miRNA and miRNA-like small regulatory RNAs expressed upon viral infection. PLoS ONE 4: e6349 doi: 10.1371/journal.pone.0006349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S 2006. miRBase: The microRNA sequence database. Methods Mol Biol 342: 129–138 [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP 2007. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell 27: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS 2004. Human microRNA targets. PLoS Biol 2: e363 doi: 10.1371/journal.pbio.0020363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami M, Yamazawa K, Matsubara K, Matsuo N, Ogata T 2008. Placentomegaly in paternal uniparental disomy for human chromosome 14. Placenta 29: 760–761 [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K 2007. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennison JA, Southworth JW 2002. Transvection in Drosophila. Adv Genet 46: 399–420 [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J 2008. SOAP: Short oligonucleotide alignment program. Bioinformatics 24: 713–714 [DOI] [PubMed] [Google Scholar]
- Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC 2003. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 35: 97–102 [DOI] [PubMed] [Google Scholar]
- Linsen SE, de Wit E, Janssens G, Heater S, Chapman L, Parkin RK, Fritz B, Wyman SK, de Bruijn E, Voest EE, et al. 2009. Limitations and possibilities of small RNA digital gene expression profiling. Nat Methods 6: 474–476 [DOI] [PubMed] [Google Scholar]
- Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. 2008. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18: 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE 2008. Chromatin structure analyses identify miRNA promoters. Genes Dev 22: 3172–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, Ferguson-Smith AC, Cavaille J 2003. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet 34: 261–262 [DOI] [PubMed] [Google Scholar]
- Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaille J 2004. A large imprinted microRNA gene cluster at the mouse Dlk1–Gtl2 domain. Genome Res 14: 1741–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63 [DOI] [PubMed] [Google Scholar]
- Smit M, Segers K, Carrascosa LG, Shay T, Baraldi F, Gyapay G, Snowder G, Georges M, Cockett N, Charlier C 2003. Mosaicism of Solid Gold supports the causality of a noncoding A-to-G transition in the determinism of the callipyge phenotype. Genetics 163: 453–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Wang L 2008. MiR-433 and miR-127 arise from independent overlapping primary transcripts encoded by the miR-433-127 locus. PLoS ONE 3: e3574 doi: 10.1371/journal.pone.0003574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H, Charlier C, Farnir F, Georges M 2010. Demonstrating polymoprhic miRNA-mediated gene regulation in vivo: Application to the g+6223G→A mutation of Texel sheep. RNA 16: 1854–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I 2008. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455: 1124–1128 [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: research0034–research0034.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuocolo T, Byrne K, White J, McWilliam S, Reverter A, Cockett NE, Tellam RL 2007. Identification of a gene network contributing to hypertrophy in callipyge skeletal muscle. Physiol Genomics 28: 253–272 [DOI] [PubMed] [Google Scholar]