

Abstract
Deleterious mutation poses a serious threat to human health and the persistence of small populations. Although adaptive recovery from deleterious mutation has been well-characterized in prokaryotes, the evolutionary mechanisms by which multicellular eukaryotes recover from deleterious mutation remain unknown. We applied high-throughput DNA sequencing to characterize genomic divergence patterns associated with the adaptive recovery from deleterious mutation using a Caenorhabditis elegans recovery-line system. The C. elegans recovery lines were initiated from a low-fitness mutation-accumulation (MA) line progenitor and allowed to independently evolve in large populations (N ∼ 1000) for 60 generations. All lines rapidly regained levels of fitness similar to the wild-type (N2) MA line progenitor. Although there was a near-zero probability of a single mutation fixing due to genetic drift during the recovery experiment, we observed 28 fixed mutations. Cross-generational analysis showed that all mutations went from undetectable population-level frequencies to a fixed state in 10–20 generations. Many recovery-line mutations fixed at identical timepoints, suggesting that the mutations, if not beneficial, hitchhiked to fixation during selective sweep events observed in the recovery lines. No MA line mutation reversions were detected. Parallel mutation fixation was observed for two sites in two independent recovery lines. Analysis using a C. elegans interactome map revealed many predicted interactions between genes with recovery line-specific mutations and genes with previously accumulated MA line mutations. Our study suggests that recovery-line mutations identified in both coding and noncoding genomic regions might have beneficial effects associated with compensatory epistatic interactions.
The accumulation of deleterious mutations under conditions of relaxed selection threatens the persistence of organisms evolving in small populations (Lynch et al. 1993; Lande 1994) and is especially relevant to small captive populations of endangered species living in benign environments (Lande 1995). The recovery from deleterious mutations also serves as an analog to adaptation to a novel environment in which previously favored alleles are now detrimental. The evolutionary mechanisms by which organisms suffering from deleterious mutation are able to recover fitness have been well-studied in bacteriophage and bacterial laboratory evolution settings that showed rapid fitness recovery and a high incidence of parallel beneficial mutation fixation in independent experimental lineages (Reynolds 2000; Maisnier-Patin et al. 2002; Poon and Chao 2005; Poon et al. 2005). For example, DNA sequencing analysis of bacteriophage ΦX174 lines that had recovered from previously accumulated deleterious mutations revealed that ∼30% of the beneficial mutations responsible for fitness recovery were back mutations (direct mutational reversals) and the remaining ∼70% were compensatory mutations at other sites in the phage genome (Poon and Chao 2005). Similarly, ∼95% of the beneficial mutations detected in Salmonella typhimurium lab populations recovering from the deleterious effects of antibiotic resistance were compensatory in nature rather than reversions (Maisnier-Patin et al. 2002). A previous fitness analysis of Caenorhabditis elegans recovery lines, initiated from mutationally degraded MA line progenitors and then allowed to evolve in large populations (N > 1000), found that many lines were able to rapidly recover fitness and suggested that compensatory mutation was most likely responsible (Estes and Lynch 2003). However, the much larger genome sizes (>100 Mb for C. elegans versus ∼5.3 kb for ΦX174) of multicellular species have thus far precluded analyses of genomic divergence patterns associated with adaptive recovery from deleterious mutation similar to those carried out in prokaryotic systems.
Here, we use replicate C. elegans lines that have suffered a nearly 50% loss in fitness due to the accumulation of deleterious mutations to examine the molecular basis of rapid fitness recovery under experimental evolution via whole genome resequencing. Using theoretical population genetic predictions we are able to rule out neutral explanations for the relatively small number of nucleotide changes that we observe within each line, and show very strong positive selection acting on a subset of these nucleotides. Although certain classes of mutation were missed by our analysis, our results show the promise of next generation sequencing approaches for the comprehensive analysis of genomic change in evolutionary studies, as well as demonstrating that compensatory mutations can be a powerful driver of evolution of genetic systems.
Results
Recovery line experimental evolution and mutational analysis
We applied Illumina high-throughput DNA sequencing (also known as Solexa sequencing) to a C. elegans recovery-line system (Fig. 1) to characterize the spectrum of mutations associated with recovery from deleterious mutation accumulation in an animal system. MA12, a C. elegans MA line derived from the N2 strain and bottlenecked as single hermaphrodite nematodes for 323 generations (Vassilieva and Lynch 1999; Estes et al. 2005), was used as the progenitor of five independent sets of recovery lines (R12A–R12E) that were allowed to evolve in large populations (N > 1000) for 60 generations. Each of the five lines was initiated from a single MA12 progenitor and rapidly regained fitness levels similar to wild type (the N2 progenitor of the MA lines) as determined by life-history assays (Fig. 2).
Figure 1.

Schematic of the MA12 recovery-line system. The seven genotypes considered are shown in circles, and arrows represent generational time. MA12 was bottlenecked as single hermaphrodite nematodes across 323 generations and each of the five MA12-derived recovery lines (R12A–R12E) evolved in the lab for 60 generations at much larger (N > 1000 nematodes) population sizes. All genotypes were analyzed by Illumina sequencing, with the exception of the two recovery lines indicated by asterisks.
Figure 2.

Fitness trajectories of individual C. elegans recovery lines. Black diamonds at generation 0 show average fitness trait values for the MA12 ancestor. Open circles represent average trait values for each of the five lines initiated from the MA12 genotype and evolved independently for 60 generations. Lines connect the same evolved populations across generations. Intrinsic rate of population increase (top panel), r (Giannelli et al. 1999), and total fecundity (middle panel) are reported as the proportion of N2 control values. The bottom panel shows lifespan; dashed line represents average lifespan of the N2 control. Bars, 1 SEM.
The genomes of the N2 (wild type) C. elegans progenitor of the MA lines, the MA12 (generation 323) progenitor of the recovery lines, and three independent recovery lines (R12A, R12B, and R12C; generation 60) were analyzed using Illumina DNA sequencing. Seven Illumina lanes were used to collect DNA sequence data for each of the five samples. Using the same general approach previously applied to base-substitution mutation identification in a set of seven C. elegans MA line genomes (Denver et al. 2009), we surveyed virtually all nonrepetitive genomic regions (>80% of the total genome) in all lines analyzed (see Methods and Supplemental Methods). We identified 68 base-substitution changes between N2 and MA12; in all 68 cases, the mutation was also detected in each of the three MA12-derived recovery lines (Supplemental Table S1). Thus, no reversions of MA12 mutations were detected in the recovery lines. These 68 mutations originated and fixed during nematode bottlenecking along the N2 to MA12 lineage. We determined the base-substitution mutation rate (μbs) for MA12 using the same method previously applied to 10 C. elegans MA lines genomes (Denver et al. 2009), and calculated a μbs value, 2.5 (±0.3) × 10−9 per site per generation, highly similar to the 10-MA line average, 2.7 (±0.4) × 10−9, from the previous study.
We extended our Illumina approach to identify base substitutions that originated and fixed during the recovery phase, as well as carrying out PCR and capillary DNA sequencing confirmation of all Illumina-identified mutant sites to rule out potential heterozygosity-related confounders (e.g., heterozygous MA12 mutations differentially fixing in recovery lines, new mutations still segregating in recovery-line populations), and confirm that the detected mutations were in a fixed (or nearly fixed) state. We detected and confirmed seven fixed base substitutions for R12A, nine for R12B, and 12 for R12C (Table 1). Due to the relatively low and fluctuating site-specific coverage levels among different strains analyzed, we were unable to effectively extend our analysis to the identification of changes in heterozygosity between MA12 and its derivative recovery lines. None of the fixed recovery line mutations detected in gene sequence occurred in genes that already harbored MA12 mutations; thus, no putative cases of intragenic compensatory mutation were identified.
Table 1.
Mutations detected in the recovery lines

aRelative to WS170 build.
bThe ancestral base present in MA12 (and N2).
cThe base present in the recovery line.
dCoding shows the coding sequence category of the mutation: EX, exon; IN, intron; UTR, untranslated region; IG, intergenic region. For exon mutations, Syn indicates synonymous mutations; for nonsynonymous mutations the specific amino acid changes are denoted.
NA, Not available.
We next analyzed the sites of recovery-line mutation fixation at nearly complete 10-generation intervals (from generation 10 to 60) using capillary DNA sequencing to investigate cross-generational patterns of mutational segregation and fixation. We visually scrutinized DNA sequence chromatogram data to search for evidence of heterozygosity; analysis of DNA sequence data from samples containing known relative molar abundances of wild-type and mutant bases suggest that we should be able to detect any segregating variants at levels >5% of the total (see Methods). Here, we assume recovery-line mutations to be in a fixed state if we were unable to detect any evidence for the wild-type base in the chromatogram data, although we cannot formally rule out the possibility that ancestral wild-type alleles are segregating at very low, undetectable frequencies. All but two mutations were observed to go from a nondetectable state in the recovery-line population to a fixed state in a single 10-generation interval (Fig. 3).
Figure 3.

Cross-generational analysis of recovery-line mutation sites. The 28 mutations identified in R12A, R12B, and R12C were analyzed at nearly complete 10-generation intervals in the recovery lines. Chromosome positions (approximate) are shown on the left for each mutation. (Green circles) Instances where the ancestral (MA12) base was detected; (red circles) the detection of recovery-line mutations in fixed states; (yellow circle) the single observed incidence of heterozygosity (Fig. 4); (gray circles) the R12B timepoint not assayed.
The expected conditional time for a new neutral mutation, unaffected by linked non-neutral mutations, to reach fixation through drift is 4Ne generations—thus, neutral mutations would be expected to take an average of 4000 or more generations to fix in recovery-line populations, unless affected by linkage to beneficial mutations. Within the 60 generations of this experiment, the probability of a neutral allele arising via mutation and becoming fixed via genetic drift alone is on the order of 10−38 (see Supplemental Methods). Even when multiplied by the ∼8.8 × 107 nucleotide sites per genome examined here, the chance that any given observed change is not the result of natural selection, either directly or via hitchhiking, is vanishingly small.
Parallel mutation fixation
Although the majority (24/28) of mutations fixed in the recovery lines were specific to a single lineage, we observed two cases in which the same base-substitution events were detected in two different recovery lines—the two substitutions were each observed in both R12A and R12C (Table 1). One of these substitutions was on C. elegans chromosome (chr) II in an intergenic region 15 bp downstream from the lips-16 annotated gene boundary; the other was on chr IV in the middle of the second intron of the Y43B11AL.1 gene (Supplemental Fig. S1). Extending PCR and capillary sequencing analysis of these two sites to the two recovery lines not analyzed by Illumina (R12D, R12E) showed that these two mutations were present and fixed in the R12A and R12C lineages alone—there was no evidence for the mutations in R12B, R12D, or R12E at any generational time interval.
Cross-generational analysis of the two sites in R12A and R12C showed distinctive fixation patterns and distinctive patterns of linkage to other recovery line-specific fixed mutations (Fig. 3). The intergenic chr II mutation was first detectable in R12A as an unfixed, segregating allele in the experimental population at generation 40 and was fixed by generation 50 (Fig. 4) along with the chr IV shared mutation, the latter being undetectable until generation 50. These two mutations were the only fixed mutation sites detected in the R12A genome at generation 50. In R12C, both of the fixed mutations shared with R12A were first detected in a fixed state at generation 60 along with 10 other R12C-specific fixed alleles. Although we cannot with 100% certainty rule out the possibility that R12A nematode(s) contaminated the R12C population between generation 50 and 60 in the lab, we believe this possibility to be highly unlikely for two reasons. First, extreme technical care was taken during the experiment to avoid the possibility of cross-contamination (see Methods). Second, the two shared mutations appeared in R12C along with 10 additional R12C-specific fixed base substitutions. Thus, the cross-contaminating nematode lineage would have had to accumulate and fix 10 additional base substitutions in 10 generations or less. Given the base-substitution mutation rate (μbs) and confidence interval for C. elegans genomic regions analyzed by Illumina (Denver et al. 2009), 2.7 (±0.4) × 10−9 per site per generation, and the numbers of sites surveyed (86.7 million, on average), a nematode is expected to acquire 0.23 (±0.4) detectable base-substitution mutations per generation. Dividing the number of observed R12C-specific mutations (N = 10) by 0.23 mutations/generation leads to an estimate of 43.5 (±11.2) expected generations for these 10 mutations to have accumulated. This suggests that 10 generations was an insufficient amount of time for these 10 mutations to have arisen in R12C. The expected number of generations for 12 mutations to accumulate, following the same logic presented above, is 52.2 (±13.5) generations. We deduce that most or all of the 12 total fixed mutations detected in R12C at generation 60 most likely accumulated during the first 50–60 generations of the recovery experiment, persisting as very low-frequency (undetectable) variants until the acquisition of a beneficial mutation on that genetic background swept them all to fixation sometime between generations 50 and 60. The two fixed mutations shared by R12A and R12C most likely arose and fixed in these two recovery lineages in an independent, parallel fashion.
Figure 4.

Detection of a segregating mutation in R12A. The three chromatograms show DNA sequence data for the single instance where we were able to detect evidence for both ancestral DNA and recovery-line mutations. The mutation at position 12,610,210 on chr II (shared with R12C), indicated by the asterisk, was first detected as a segregating variant at generation 40 in R12A then appeared in a fixed state at generation 50. There was no evidence for this mutation at generation 30. We were unable to detect a segregating variant for any of the other 27 recovery-line mutations.
Interactome analysis
We investigated the possibility of intergenic epistatic interactions between recovery-line mutations and MA12 mutations in protein-coding genes using the C. elegans interactome map (Zhong and Sternberg 2006). GeneOrienteer (http://www.geneorienteer.org) was used to calculate log-likelihood ratio scores for all possible pairwise combinations of C. elegans genes that were found to harbor a MA12 or recovery-line mutation based on numerous underlying feature data sources (yeast two-hybrid experiments, microarray data, etc.) from C. elegans, Drosophila melanogaster, and Saccharomyces cerevisiae. Consistent with the original analysis of global C. elegans genetic interactions (Zhong and Sternberg 2006), we applied a score threshold of 0.9, which exceeds the maximum contribution that any single contributing feature can achieve, to identify putative interactions between genes harboring recovery-line and MA12 mutations. Fourteen predicted interactions were identified that met or surpassed our score threshold, all involving combinations of genes mutated in MA12 and those bearing fixed recovery-line mutations (Fig. 5); no interactions were detected between mutated MA12 genes or between recovery-line genes bearing fixed mutations. One R12A fixed mutation caused a nonsynonymous change in the glp-1 gene that has predicted interactions with 8/31 genes that suffered a mutation in MA12. glp-1 encodes a transmembrane receptor protein that, along with LIN-12, comprises one of two C. elegans members of the LIN-12/Notch family of receptors and plays a key role in the control of germ cell proliferation during postembryonic development (Austin and Kimble 1987; Priess et al. 1987). One R12C fixed mutation caused a change in the 3′ untranslated region of the num-1 gene that has predicted interactions with five genes mutated in MA12. NUM-1 affects the localization and recycling of cell membrane receptor proteins (Nilsson et al. 2008). Four of the MA12-mutated genes predicted to interact with NUM-1 (fixed mutation in R12C) also have predicted interactions with GLP-1 (fixed mutation in R12A) (see Fig. 5). Three of these four genes mutated in MA12 encode transmembrane proteins: CDH-4 is a cadeherin involved in cell–cell adhesion (Schmitz et al. 2008), ITR-1 is a putative inositol (1,4,5) trisphosphate receptor that affects the defecation cycle and pharyngeal pumping (Walker et al. 2009), and Y74C10AL.2 encodes a protein bearing a conserved integral membrane protein domain (Rogers et al. 2008). Thus, we speculate that the R12A mutation in glp-1 and the R12C mutation in num-1 might have beneficial epistatic effects mediated through alteration of membrane protein activities.
Figure 5.

Predicted interactions of recovery line and MA line mutations. Each red square represents a MA line mutation; inside the square, the genotype (MA12) is on top, the mutated gene is in the middle, and the functional effect is indicated on the bottom. UTR, mutations in untranslated regions; Ex Syn, exon mutations that are synonymous. For nonsynonymous mutations in exons, the resultant amino acid change is indicated using single-letter codes. Circles represent recovery line mutations: (green) R12A; (blue) R12C. Genetic interactions predicted by GeneOrienteer are indicated by double-headed arrows, and the log-likelihood score associated with each predicted interaction is listed next to corresponding arrows.
Selective sweeps
Cross-generational DNA sequencing analysis of the recovery lines revealed that many recovery line-specific mutations fixed at common 10-generational intervals (Fig. 3). This pattern is consistent with the occurrence of series of selective sweeps over the course of the recovery experiments. In R12A, two sweep events were detected: two mutations fixed in unison between generations 40 and 50 (the two shared with R12C), then five R12A-specific mutations fixed between generations 50 and 60. In R12B, the data indicated three sweep events: one mutation fixation by generation 20, followed by seven additional mutations fixing at generation 50, followed by one mutation fixing at generation 60. In R12C, one sweep event was detected at generation 60 involving 12 mutations.
We formally explored the expected dynamics of these sweeps using simulations and a diffusion approximation of the fixation of adaptive mutations under the influences of natural selection, genetic drift, and recurrent mutation (see Methods). The first thing to note is that although most of the sweeps that we observed are confined to a 10-generation window, even under a completely deterministic model a great deal of the allele frequency change for a new adaptive mutation initially occurs below our detection threshold (Fig. 6A), indicating that the actual time for the sweeps is probably more on the order of 20–30 generations. The diffusion approach allows the probability of fixation to be calculated for every possible combination of initial allele frequency and number of generations (Fig. 6B). Here, we are most interested in integrating the probability that a new mutation (initial frequency of 1/2000) will be fixed over the total length of the experiment and/or observation window. Because of the very small probabilities involved, the diffusion approximation substantially underestimates the probability of fixation during early generations, but performs increasingly better over time, especially for strong selection (Fig. 6C). Since we observed at least one selective sweep in each of the replicates, the expected number of fixed mutations for a given time interval under a given set of parameter values must be at least 1.0. Assuming no interference between fixation events, moving from the single locus results to whole genome expectations requires multiplying the probability of fixation by the number of possible sites under selection. There are two domains of parameters that are consistent with the timescale of the response displayed by our populations. First, if the majority of the genome has the potential to contribute to the recovery observed here, then the probability of fixation (Fig. 6C) can be multiplied by a very large number (∼8.8 × 107), allowing mutations under moderately strong selection (s ≈ 0.3) to contribute to the response even though it is very unlikely that any particular mutation would become fixed. However, the consistency of the sweep dynamics and our observations of repeated fixations in several of the lines and an apparently limited set of interacting components suggest that this is unlikely. Fixation of a smaller subset of the genome (say 1 in 10,000 nucleotides) is feasible in the timescale observed here, but only if selection is strong. Thus, in order to satisfy the conditions (1) that we always observe fitness recovery in our experiments and (2) that the sweeps that we observed occur within a most a few dozen generations, we find that selection on at least one of the mutations is likely to be very strong: on the order of a 70%–90% increase in fitness relative to the ancestral genotype (Fig. 6C).
Figure 6.

Population genetic analysis of the fixation of new mutations under positive selection and complete self-fertilization. (A) Deterministic sweep of a new adaptive mutation in an infinitely large self-fertilizing population under different strengths of selection. There can be a significant lag before the mutation reaches a high enough frequency to be detectable at the sensitivity threshold present in this experiment (dashed line). (B) Solution to the diffusion equation for finding the probability of fixation of a segregating allele (s = 0.5, Ne = 1000). The probability of fixation for a new mutation over the course of the experiment is calculated by summing over the cumulative probability of fixation for an initial allele frequency of 1/2000. (C) Cumulative probability of fixation over a given number of generations for varying levels of positive selection. Solid lines show simulation results that simultaneously include mutation, drift, and selection (Ne = 1000; μ = 2.6 × 10−9). Points below each line show the results of the diffusion approximation in which mutation is treated separately from drift and selection. The diffusion approximation tends to underestimate the probability of fixation, especially during early generations and under weaker selection.
Discussion
Evolutionary implications
Our study provides a broad-based view of genomic divergence patterns associated with the adaptive recovery from deleterious mutation in C. elegans. None of the recovery response is attributable to detectable reversion mutations, and no cases of putative intragenic compensatory mutations were identified. Thus, intergenic compensatory mutations likely drive all of the change that we observe, suggesting that this might be a common avenue for genetic change within complex multicellular organisms. Second, virtually all of the base-substitution polymorphisms detected in the recovery lines went from zero or near-zero (undetectable) population-level frequencies to a state of fixation (or near fixation) in a few dozen generations, suggesting the occurrence of selective sweeps in the adaptively evolving lab populations. The fact that the changes that we are able to observe all occur toward the end of our experiment suggests that these lab populations are likely subject to a constant genetic churn in which early sweeps are replaced by subsequent adaptive changes. If this is a common occurrence, then future studies will need to completely sequence individuals from multiple time points in order to fully characterize the underlying evolutionary dynamics.
The timescale of simultaneous fixation requires very strong selection and suggests that the majority of the changes that we observed are generated via hitchhiking of very low frequency background mutations and/or that epistasis between multiple loci generates the fitness effects that we observe. The strength and genomic impacts of the sweeps observed here may help to explain the extreme haplotype structure observed within natural populations of C. elegans (Cutter 2006; Rockman and Kruglyak 2009). On the other hand, the fact that a number of mutations appear to readily hitchhike along with these sweeps indicates that local adaptation should drive strong genetic divergence among C. elegans isolates. Instead, we see very little evidence for genetic variation or population structure on a worldwide scale (Barriere and Felix 2005; Haber et al. 2005; Cutter 2006; Rockman and Kruglyak 2009). This observation provides support for the view that most extant C. elegans populations may have diverged from one another relatively recently (Phillips 2006; Cutter et al. 2008).
Parallel mutation and compensatory epistatic interactions
Two identical fixed mutations were detected in R12A and R12C that most likely arose and fixed in these two recovery lineages in an independent, parallel fashion. The likelihood of two identical mutations occurring in two different recovery lines is ∼0.0008 (see Methods). Although this probability is very low, it is possible that these sites might experience higher mutation rates than genome-wide averages (Denver et al. [2009] was unable to effectively account for potential hotspots). Further, parallel mutation has previously been observed in similar experimental evolution studies in prokaryotes (Maisnier-Patin et al. 2002; Poon and Chao 2005). The observation of two sites fixing the exact same mutation type in independent recovery lines suggests that these mutations might have beneficial effects that were directly acted upon by natural selection. The observation of parallel mutation in the recovery lines might reflect a limited number of beneficial mutations available as potential substrates for adaptive recovery from MA12 mutations (Orr 2005). This interpretation is consistent with our population-genetic analysis of selective sweep dynamics that suggested very strong selection on a very small fraction of the C. elegans recovery-line genomes.
Both parallel mutations, however, occurred in genomic regions that are not predicted to encode functional protein products, suggesting that any positive effects would be mediated through regulatory or DNA structural effects. The chr IV shared mutation occurred in an intron of the Y43B11AL.1 gene—the only functional information available for this gene from WormBase (Rogers et al. 2008) is that its product encodes F-box domains (involved in protein–protein interactions). The chr II shared mutation is just downstream from the lips-16 gene whose product is predicted to encode a lipase function and affect fat content. Three of the detected mutations that accumulated in the MA12 genome prior to recovery (presumably deleterious) were in genes (nonsynonymous change in ZK682.2, intron change in H08M01.2, intron change in mgl-3) that play roles in maintaining fat content, as determined by RNAi experiments (Greer et al. 2008). Further, one R12A-specific fixed mutation resulted in a nonsynonymous change in the inx-4 gene whose product affects fat content in RNAi experiments (Greer et al. 2008). We speculate that the chr II mutation shared by R12A and R12C that is downstream from lips-16, as well as the inx-4 mutation specific to R12A, might have beneficial effects associated with lipid metabolism manifested through epistatic compensatory interactions with MA12 mutations. The presence of both the chr II and chr IV shared mutations in R12A and R12C indicates that any putative beneficial effects of these mutations might require epistatic interactions between these two loci, though there is no functional information in support of this possibility. More evidence for epistatic compensatory interactions underlying adaptive evolution in the recovery lines resulted from our interactome analysis (Fig. 5) that revealed strong evidence for interactions between genes that suffered (presumably deleterious) MA line mutations during the bottleneck phase and genes that acquired fixed recovery-line mutations.
Although we were able to detect rare mutations that fixed in recovery-line lineages and characterize selective sweep events in our cross-generational analysis, we were unable to determine whether mutations identified were beneficial versus selectively neutral (or slightly deleterious) mutations that hitchhiked to fixation due to the complete linkage of all sites in the primarily self-reproducing nematodes. Our survey was also only able to identify base-substitution mutations—it is possible that other mutation types (e.g., insertion-deletion mutations, large rearrangement events) left undetected were the primary drivers of fitness recovery. Backcrossing individual mutations identified in this study onto MA12 genetic backgrounds, followed by comparative fitness studies, would provide an avenue for understanding the effects of various mutations fixed in the recovery lines. We have repeatedly attempted such an analysis and, unfortunately, have been unable to successfully perform crosses with the low-fitness MA12 nematodes.
Another limitation to this study is the fact that the mutations responsible for the very rapid recovery of fitness in the first 10–20 generations were most likely missed since all but one of the fixed mutations detected here occurred at later generations. Given that all five MA12-derived recovery lines independently regained wild-type fitness levels by generation 20 (Fig. 2), we speculate that the mutations responsible for most of the fitness recovery were of high-mutation rate types and/or involved highly plastic epistatic changes, rather than the base-substitution changes analyzed here. For example, gene duplication and deletion dynamics involving large gene families have the potential to have profound and rapid consequences on fitness via changes in dosage. Likewise, changes in ribosomal DNA cluster copy number are shown to occur at very high rates and have broad-based epigenetic effects on global chromatin and gene regulation in Drosophila (Paredes and Maggert 2009). Although unlikely, it is also possible that highly deleterious mutations present in a heterozygous state in the MA12 progenitor were largely responsible for the greatly reduced fitness of this MA line—individuals homozygous for the wild-type alleles at these sites would have an immediate large fitness advantage in the recovery lines. Expanding our mutation survey to encompass heterozygous MA line mutation sites and repetitive DNA units will be required to pinpoint the nature of the beneficial changes responsible for the rapid regain of fitness in the recovery lines. We also note that although we detected very fast selective sweeps with associated selection coefficients of ≥0.3, two of our fitness measures (intrinsic rate of population increase, total fecundity) did not reveal fitness increases expected at these generational intervals (Fig. 2). Our third fitness measure, lifespan, did show increases across analyzed generational intervals. It is possible that our fitness measures lacked the sensitivity required to detect these fitness increases; competition assays involving recovery lines from different generations might provide a more powerful option for future analyses.
Adaptive mutation rate
We can reasonably assume that each of the six selective sweeps detected in the cross-generational analysis was caused by positive selection acting on at least one beneficial mutation. Thus, a minimum of six beneficial mutations arose in the three recovery-line populations analyzed (two in R12A, three in R12B, and one in R12C), each underlying one of the six sweeps detected. This leads to a lower-bound adaptive genomic mutation rate estimate (Ua) of 3.8 × 10−5 per nematode per generation (see Supplemental Methods). Given the current total genomic mutation rate (Ut) estimate for C. elegans, 2.1 per genome per generation (Denver et al. 2004), this suggests that as few as one mutation in 55,263 (Ua/Ut) is adaptive. Our C. elegans Ua estimate is remarkably similar to a recent Ua estimate for Escherichia coli, 2.0 × 10−5, based on laboratory evolution studies (Perfeito et al. 2007). It is also consistent with our theoretical estimate of how much of the genome must be under strong positive selection in order for fixation to occur within the timeframe observed here. Our Ua estimate for C. elegans, however, is likely to be an underestimate for two reasons. First, as discussed above, some selective sweep events likely went undetected, especially in earlier recovery generations where there might have been insufficient time for detectable base substitutions to accumulate. Second, the effects of clonal interference, the loss of competing beneficial mutations in the population as a consequence of selective sweeps at other loci, likely resulted in underestimation of the numbers of beneficial mutations arising in each recovery line. Thus, although whole-genome resequencing provides an unprecedented opportunity to identify the specific genetic changes responsible for fitness recovery, understanding the role of beneficial mutations in shaping natural patterns of genomic variation remains a formidable problem in evolutionary analysis.
Methods
Experimental evolution and life-history analyses
We selected five C. elegans MA lines for the current study that were shown to completely recover ancestral levels of fitness in a previous experiment (Estes and Lynch 2003). These lines were thawed, expanded for a single generation, and subdivided into five replicate populations. Each replicate was initiated from a single MA12 nematode and then independently maintained in large population sizes under standard laboratory conditions for 60 overlapping generations following Estes and Lynch (2003). The ancestral N2 control (progenitor of the MA lines) underwent the same treatment concurrently. Approximately 1000 individuals were transferred each generation, with populations expanding to roughly 10,000 individuals in between transfers (see Supplemental Methods for a discussion of effective population size). Extreme care was taken to avoid cross-contamination among experimental lines by keeping plates well separated on trays and through ethanol/flame sterilization of the metal core boring tool used for transfers. Finally, samples from each replicate were frozen at 10-generation intervals during the experimental evolution phase. The evolutionary trajectories of the other four recovery lines will be reported elsewhere. Because they exhibited the greatest total fitness increase during experimental evolution, we chose the MA12 recovery lines for Illumina analysis.
Life-history assays were conducted as described in Estes and Lynch (2003). Briefly, for each line and each generational time point, total progeny production, population growth rate (r), and longevity were measured for 10–15 single individuals obtained from frozen stock. Single worms were transferred to fresh plates daily and progeny production measured by directly counting the progeny produced over the entire reproductive period. Intrinsic population growth rate, r, was calculated for each line by solving ∑e-rx l(x) m(x) = 1 for r, where l(x) is the proportion of worms surviving to day x and m(x) is the fecundity at day x. Longevity was taken to be the total number of days lived from the L1 stage. Assays were carried out on standard OP50 E. coli-seeded NGM agar plates at 20°C.
We tested for recovery of ancestral levels of fitness and for evolution of the ancestral control using analyses of variance for each fitness trait with population treatment (MA, recovery, ancestral control, evolved control) as a fixed effect. To test for differences between pairs of treatment group means, least-squares contrasts (Tukey's HSD for all pairwise comparisons; Zar 1999) were performed on the data for each life-history trait.
Male frequency analysis
To approximate the amount of sexual recombination that may have been occurring during experimental evolution of the C. elegans recovery lineages (A–E), we estimated the proportion of males produced by the MA12 ancestor and by each recovery line following 60 generations of laboratory evolution. Male frequency was scored by counting 200 individuals from five replicate plates per line. Male frequency in the MA12 line was estimated to be 0.7%; however, these males were apparently incapable of mating given our lack of success in backcrossing experiments. Male frequencies were even lower in the generation-60 recovery lines (Supplemental Table S2). We thus conclude that outcrossing-based sexual recombination most likely played a very minimal (if any) role in shaping the evolutionary trajectories of the recovery lines.
DNA sequence analysis
We screened for base-substitution mutations in MA12, R12A, R12B, and R12C by applying the same Illumina high-throughput sequencing method previously used to accurately identify base substitutions in seven C. elegans MA lines genomes (Denver et al. 2009). The details of our analytical approach are provided in the Supplemental Methods. For the current study it was especially important to account for the potential effects of heterozygous MA12 mutant sites that might be differentially fixed/segregated in different recovery lines. For Illumina sample preparations, nematode lab populations were initiated from frozen stocks (∼50–100 nematodes) and allowed to expand for two generations in order to amass a sufficient number of animals for Illumina DNA extraction protocols. Our level of Illumina coverage (∼7× for unique regions, on average) was insufficient to distinguish mutations fixed in the recovery lines from those still segregating in the population and coexisting with ancestral alleles. To address these concerns, we carried out PCR and conventional direct capillary DNA sequencing analysis for all 28 detected MA12 recovery-line changes (Supplemental Fig. S2) and in all cases there was no evidence of heterozygosity in MA12 or the recovery line. Many thousands of nematodes were used for DNA extractions used in PCR/capillary sequencing assays. The primers used to PCR-amplify and capillary-sequence these sites are provided in Supplemental Table S3.
We performed a controlled capillary DNA sequencing experiment to evaluate allele frequencies required for detection in our cross-generational analyses. Using initial “wild-type” and “mutant” (containing base substitution) PCR product samples of known concentrations estimated through standard spectroscopy methods (NanoDrop), we made a series of samples where the molar ratios of input wild-type and mutant PCR products varied from 15% to 0.5%. We evaluated chromatogram data from samples containing 15%, 10%, 5%, 1%, and 0.5% mutant PCR products and found that the mutant peak was discernible in the 15%, 10%, and 5% samples but not readily distinguishable from the baseline (“noise”) in the 1% and 0.5% samples. This result is generally consistent with similar studies aimed at identifying and characterizing low-frequency heteroplasmic mitochondrial DNA mutations (Theves et al. 2006). We conclude that any chromatograms that did not reveal any evidence for minority peaks (wild type or mutant, depending on the situation) correspond to DNA samples where the site is in a fixed state, or the frequency of the minority allele is <5%.
Mutation rate calculations
Individual MA line-specific mutation rates were calculated with the equation μbs = m/(nT), where μbs is the base-substitution mutation rate (per nucleotide site per generation), m is the number of observed mutations, n is the number of nucleotide sites, and T is the time in generations, as previously described (Denver et al. 2004). The standard errors for individual mutation rates were calculated as [μbs/(nT)]1/2, as described (Denver et al. 2004). Values for n were defined as the total number of sites surveyed that met our criteria for consideration of a possible mutation site (Supplemental Methods).
To address the matter of parallel mutation, we first estimated the probability of a particular mutation not occurring during the recovery experiment (Pnm) as (1 − μbs)TNe where Ne is the estimated effective population size (1000); Pnm = 0.9998. Given a fixed mutation in one recovery-line population, the probability of the same exact mutation occurring in any of the other four recovery lines is (1 – Pnm) × 4, resulting in a value of 0.0008.
Population genetic analysis
We used a combination of exact calculations, simulations, and a diffusion model to assess the probability of fixation of recurrent mutations in both the presence and absence of natural selection. In keeping with the mating system of C. elegans (particularly the N2 laboratory strain), we assume complete selfing throughout. Because of the very small probabilities involved, it is actually quite difficult to solve the case for complete neutrality for the timescales and population sizes used in this study. We outline upper bound calculations in the Supplemental Methods and Supplemental Fig. S3.
For the case of positive selection operating on one or more loci, we use the standard Kolmogorov backward equation for genetic drift:
(Crow and Kimura 1970, equation 8.8.3.1). We follow Caballero et al. (1991) and Caballero and Hill (1992) in defining the instantaneous mean change in allele frequency under partial selfing as
where s is the strength of selection for the new mutation with homozygous fitness 1 + s versus the initial genotype with fitness 1.0, h is the dominance coefficient of the new mutation, and F is the inbreeding coefficient (set to 1.0 for the case of complete selfing). The variance of this process generated by genetic drift is given by
where Ne is the effective size of the population (here taken to be 1000). The diffusion equation was solved numerically using Mathematica (Wolfram Research) under the boundary conditions u(0, t) = 0, u(1, t) = 1, and u(x, 0) = [x2 + x(1 – x)F]Ne (code provided in Supplemental Methods).
The probability of fixation at any time point was calculated by obtaining the value of u(x0, t), with x0 set to the initial frequency of a new mutation (1/2000). Treating each mutation as independent of one another, the number of mutations expected to reach fixation over a period of T generations was calculated as 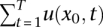 . For the case of complete selfing, the dominance parameter h has no influence on the probability of fixation.
. For the case of complete selfing, the dominance parameter h has no influence on the probability of fixation.
These theoretical approximations were tested using a simulation of the entire mutation and fixation process. Mutation, genetic drift, and selection were sequentially imposed on a population initially fixed for the less fit allele. The number of generations needed for the alternative high fitness allele to become fixed within the population was recorded, with the probability of fixation at a given generation calculated as the fraction of 108 replicate populations in which the high fitness allele became fixed. The results presented here assume a population size of 1000 individuals. Results for other population sizes are presented in the Supplemental Figure S4.
Acknowledgments
We thank L. Albergotti, H. Bui, A. Coleman-Hulbert, K. Hicks, B. Leon, S. Martha, S. Smith, T. Tague, and A. Woodbury for laboratory assistance. We thank M. Dasenko at the OSU Center for Genome Research and Biocomputing for DNA sequencing support. This work was supported by NSF grants DEB-062521 to S.E. and DEB-0743871 to S.E. and D.R.D., PSU Faculty Enhancement Grant to S.E., OSU Computational and Genome Biology Initiative support to D.R.D., and NSF grants DEB-0236180 and DEB-0641066 to P.C.P.
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA023539.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.108191.110.
References
- Austin J, Kimble J 1987. glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell 51: 589–599 [DOI] [PubMed] [Google Scholar]
- Barriere A, Felix M-A 2005. Natural variation and population genetics of Caenorhabditis elegans. WormBook 26: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Hill WG 1992. Effects of partial inbreeding on fixation rates and variation of mutant genes. Genetics 131: 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Keightley PD, Hill WG 1991. Strategies for increasing fixation probabilities of recessive mutations. Genet Res 58: 129–138 [Google Scholar]
- Crow JF, Kimura M 1970. An introduction to population genetics theory. Burgess, Minneapolis, MN [Google Scholar]
- Cutter AD 2006. Nucleotide polymorphism and linkage disequilibrium in wild populations of the partial selfer Caenorhabditis elegans. Genetics 172: 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Wasmuth JD, Washington NL 2008. Patterns of molecular evolution in Caenorhabditis preclude ancient origins of selfing. Genetics 178: 2093–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Thomas WK 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430: 679–682 [DOI] [PubMed] [Google Scholar]
- Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledo JI, Howe DK, Lewis SC, Okamoto K, Thomas WK, Lynch M, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci 106: 16310–16314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes S, Lynch M 2003. Rapid fitness recovery in mutationally degraded lines of Caenorhabditis elegans. Evolution 57: 1022–1030 [DOI] [PubMed] [Google Scholar]
- Estes S, Ajie BC, Lynch M, Phillips PC 2005. Spontaneous mutational correlations for life-history, morphological and behavioral characters in Caenorhabditis elegans. Genetics 170: 645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli F, Anagnostopoulos T, Green PM 1999. Mutation rates in humans. II. Sporadic mutation-specific rates and rate of detrimental human mutations inferred from hemophilia B. Am J Hum Genet 65: 1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer ER, Perez CL, Van Gilst MR, Lee BH, Ashrafi K 2008. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab 8: 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber M, Schungel M, Putz A, Muller S, Hasert B, Schulenburg H 2005. Evolutionary history of Caenorhabditis elegans inferred from microsatellites: Evidence for spatial and temporal genetic differentiation and the occurrence of outbreeding. Mol Biol Evol 22: 160–173 [DOI] [PubMed] [Google Scholar]
- Lande R 1994. Risk of population extinction from fixation of new deleterious mutations. Evolution 48: 1460–1469 [DOI] [PubMed] [Google Scholar]
- Lande R 1995. Mutation and conservation. Conserv Biol 9: 782–791 [Google Scholar]
- Lynch M, Burger R, Butcher D, Gabriel W 1993. The mutational meltdown in asexual populations. J Hered 84: 339–344 [DOI] [PubMed] [Google Scholar]
- Maisnier-Patin S, Berg OG, Liljas L, Andersson DI 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol Microbiol 46: 355–366 [DOI] [PubMed] [Google Scholar]
- Nilsson L, Conradt B, Ruaud AF, Chen CC, Hatzold J, Bessereau JL, Grant BD, Tuck S 2008. Caenorhabditis elegans num-1 negatively regulates endocytic recycling. Genetics 179: 375–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA 2005. The probability of parallel evolution. Evolution 59: 216–220 [PubMed] [Google Scholar]
- Paredes S, Maggert KA 2009. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci 106: 17829–17834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito L, Fernandes L, Mota C, Gordo I 2007. Adaptive mutations in bacteria: High rate and small effects. Science 317: 813–815 [DOI] [PubMed] [Google Scholar]
- Phillips PC 2006. One perfect worm. Trends Genet 22: 405–407 [DOI] [PubMed] [Google Scholar]
- Poon A, Chao L 2005. The rate of compensatory mutation in the DNA bacteriophage ϕX174. Genetics 170: 989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon A, Davis BH, Chao L 2005. The coupon collector and the suppressor mutation: Estimating the number of compensatory mutations by maximum likelihood. Genetics 170: 1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priess JR, Schnabel H, Schnabel R 1987. The glp-1 locus and cellular interactions in early C. elegans embryos. Cell 51: 601–611 [DOI] [PubMed] [Google Scholar]
- Reynolds MG 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156: 1471–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L 2009. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet 5: e1000419 doi: 10.1371/journal.pgen.1000419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers A, Antoshechkin I, Bieri T, Blasiar D, Bastiani C, Canaran P, Chan J, Chen WJ, Davis P, Fernandes J, et al. 2008. WormBase 2007. Nucleic Acids Res 36: D612–D617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Wacker I, Hutter H 2008. The Fat-like cadherin CDH-4 controls axon fasciculation, cell migration and hypodermis and pharynx development in Caenorhabditis elegans. Dev Biol 316: 249–259 [DOI] [PubMed] [Google Scholar]
- Theves C, Keyser-Tracqui C, Crubezy E, Salles JP, Ludes B, Telmon N 2006. Detection and quantification of the age-related point mutation A189G in the human mitochondrial DNA. J Forensic Sci 51: 865–873 [DOI] [PubMed] [Google Scholar]
- Vassilieva LL, Lynch M 1999. The rate of spontaneous mutation for life-history traits in Caenorhabditis elegans. Genetics 151: 119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DS, Vazquez-Manrique RP, Gower NJ, Gregory E, Schafer WR, Baylis HA 2009. Inositol 1,4,5-trisphosphate signalling regulates the avoidance response to nose touch in Caenorhabditis elegans. PLoS Genet 5: e1000636 doi: 10.1371/journal.pgen.1000636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar J 1999. Biostatistical analysis. Prentice Hall, Upper Saddle River, NJ [Google Scholar]
- Zhong W, Sternberg PW 2006. Genome-wide prediction of C. elegans genetic interactions. Science 311: 1481–1484 [DOI] [PubMed] [Google Scholar]