

Abstract
Racemic mefloquine is a highly effective antimalarial whose clinical utility has been compromised by its association with neuropsychiatric and gastrointestinal side effects. It is hypothesized that the cause of the side effects may reside in the (−) enantiomer. We sought to compare the safety, tolerability and pharmacokinetic profile of (+)-mefloquine with racemic mefloquine in a randomized, ascending-dose, double-blind, active and placebo-controlled, parallel cohort study in healthy male and female adult volunteers. Although differing in its manifestations, both study drugs displayed a substantially worse tolerability profile compared with placebo. The systemic clearance was slower for (−)-mefloquine than (+)-mefloquine. Thus, (+)-mefloquine has a different safety and tolerability profile compared with racemic mefloquine but its global safety profile is not superior and replacement of the currently used antimalarial drug with (+)-mefloquine is not warranted.
Introduction
Mefloquine is a quinolinemethanol antimalarial that is effective as therapy and prophylaxis for patients infected with all species of malaria parasites infecting humans, including multi-drug resistant Plasmodium falciparum found in most malaria-endemic regions with the notable exception of parts of Southeast Asia.1–3 This drug is a chiral compound with two asymmetric carbon centers that result in potentially four stereoisomers. The marketed anti-malarial drug mefloquine consist of two of these isomers, the RS and SR pair (referred hereafter as (+) and (−)). Although both enantiomers have been shown to have similar anti-malarial activities,4,5 they have been shown to have biological differences.6,7
Mefloquine remains widely used although its clinical utility is impaired by its association with neuropsychiatric side effects and gastrointestinal upset that result in a black box warning and restrictions on its use in many countries.8–12 The pharmacological basis of the central nervous system (CNS) side effects of mefloquine is not known but two of the most reported hypotheses concerning their cause relate to its action on the adenosine receptor and its effect on the cholinesterase enzyme. For both of these mechanisms, there is a significant stereoselective activity of the two enantiomers. In vitro studies show that (−)-mefloquine is 50–100-fold more potent towards adenosine receptors compared with (+)-mefloquine.6 In addition, (−)-mefloquine has considerably more anticholinesterase activity.7 It has therefore been hypothesized that (+)-mefloquine may have a considerably better CNS safety profile compared with either the racemate or (−)-mefloquine.
Whether the gastrointestinal effects, particularly nausea and vomiting, result from a CNS-mediated or a local effect is unknown. This study was considered exploratory for the investigation of the relative effect on gastrointestinal symptoms of the two study drugs. In the context of intermittent preventive therapy in pregnancy, good gastrointestinal tolerability is essential to improve adherence.
This study was designed to test the hypothesis that (+)-mefloquine is safer and better tolerated compared with racemic mefloquine with a focus on CNS and gastrointestinal (GI) side effects.
Materials and Methods
(+)-Mefloquine was produced as a capsule-shaped tablet. Each tablet contained 219 mg of the drug substance, (+)-mefloquine, as the hydrochloride salt; this is equivalent to 200 mg of free base. Each tablet was over-encapsulated with back-filling with lactose to help maintain blinding. The placebo had the same formulation as (+)-mefloquine except that it contained no drug substance. The racemic mefloquine comparator is a commercially available product that is registered by Medicines and Healthcare Products Regulatory Agency in the United Kingdom. Each tablet contained 274 mg of racemic mefloquine as the hydrochloride salt; this is equivalent to 250 mg of free base and was broken in half or in quarters before being over-encapsulated with back-filling with lactose to help maintain blinding.
This study was a randomized, ascending dose, double-blind, active and placebo-controlled, parallel cohort study in healthy male and female adult volunteers with a body mass index of 19–28. Persons were excluded if they had a history of taking mefloquine. Study approval was obtained from the Welwyn Ethics Committee (Hatfield, United Kingdom), and all persons were provided with detailed study information prior to signing an informed consent form. The primary objective was to describe the dose-concentration effect relationship of (+)-mefloquine for safety and tolerability and compare its profile with that of racemic mefloquine across a range of potentially therapeutic doses and concentrations up to the combined exposure of (+) and (−) mefloquine associated with the licensed dose of racemic mefloquine. The secondary objective was to describe the comparative pharmacokinetics of (+)-mefloquine and racemic mefloquine. Because (+)-mefloquine has a higher clearance compared with (−)-mefloquine,13 the dose of (+)-mefloquine needs to be higher compared with racemic mefloquine to achieve the same plasma exposure of mefloquine.
Persons in the first cohort were randomized on a 2:2:1 ratio based on a standard computer generated randomization code to received 500 mg of racemic mefloquine, 800 mg (+)-mefloquine, or placebo. Each person was administered a single dose that was divided into two parts; each part was taken six hours apart in keeping with widespread practice with racemic mefloquine.14 All doses were administered after a standard meal of cereal, milk, bread, butter, and jam.
The decision to escalate was made after a review of the safety and pharmacokinetic data that included adverse events (AEs), vital signs, 12-lead electrocardiographs (ECGs), and laboratory safety data up to and including the seven day post-dose assessments. Pharmacokinetic data were provided up to the 72-hour blood draw. Persons were given a different identification code for the purposes of the interim data review to protect the blind in the follow-up period. There was a minimum interval of 14 days between dosing of sequential cohorts when the dose was to be escalated. Stopping rules were in place in case of severe or serious adverse events. The highest proposed dose of racemic mefloquine and (+)-mefloquine was based on the exposures associated with the licensed dose for the treatment of malaria.
The primary endpoint for the safety analysis was the relative incidence of a composite of CNS AEs between the treatment cohorts. Secondary safety endpoints were other AEs, nausea and vomiting scores, ECG parameters, vital signs, profile of mood states (POMS), power of attention composite endpoint (comprising a composite of simple reaction time, choice reaction time, and digit vigilance), cognitive function tests (including immediate word recall, picture presentation, simple reaction time, digit vigilance, choice reaction time, spatial working memory, numeric working memory, delayed word recall, word recognition, picture recognition, Bond-Lader Visual Analog Scale of Mood and Alertness, tracking, postural stability), and sleep evaluation (via actigraphy and the Leeds Sleep Evaluation Questionnaire).15,16 The Short Form POMS consists of 30 adjectives that are rated by persons on a five-point scale. Six factors have been derived from these states: tension-anxiety, depression-dejection, anger-hostility, fatigue-inertia, vigor-activity, and confusion-bewilderment. The Leeds Sleep Evaluation Questionnaire comprises 10 self-rating 100-mm-line analog questions concerned with aspects of sleep and early morning behavior.
Blood samples were collected for assay of plasma pre-dose; 6 hours (pre-second part of divided dose where applicable); 8, 10, 12, 14, 18, and 24 hours (day 1, 0 hours); 36 and 48 hours (day 2, 0 hours); and 72 hours (day 3, 0 hours). In addition, samples were collected for assay of plasma levels at days 7, 14, and 42. The endpoints for the pharmacokinetic parameters were maximum concentration (Cmax), time after administration of drug when maximum plasma concentration is reached (Tmax), half-life (t½), area under curve (AUC0–t), AUC0–∞, and bioavailability (Cl/F).
Power calculation.
Assuming a background rate of reported CNS AEs of 12.5% and a CNS AE rate with racemic mefloquine of 40%,8–12,17,18 at the 5% level, with a two-sided test, to obtain 80% power between two parallel cohorts, 40 volunteers per treatment group were required. It was planned to have 5 cohorts of 20 persons; in each, cohort, 8 persons were to receive racemic mefloquine, 8 persons were to receive (+)-mefloquine, and 4 persons were to receive placebo.
Results
Study protocol.
After an uneventful dosing of cohort 1, the doses were increased as per protocol for cohort 2 (1,600 mg of (+)-mefloquine and 1,000 mg of racemic mefloquine). However, after a higher than expected incidence of AEs in cohort 2, an ad hoc Independent Data Monitoring Committee consisting of a clinician with expertise in neuropsychiatry, an experienced pharmaceutical physician, and one statistician advised that dosing could continue but because of the reports of moderate-to-severe dizziness and vomiting in cohort 2, an intermediate dose between cohorts 1 and 2 was administered. Doses of 1,200 mg of (+)-mefloquine and 750 mg of racemic mefloquine were chosen for cohort 3, which was divided into two sub-cohorts for logistical reasons. After dosing of the first sub-group of cohort 3, a similar AE profile was observed as had been seen in cohort 2. After further review by the Independent Data Monitoring Committee, the decision was made to cancel further dosing based on the recommendation that the objective of being able to demonstrate that (+)-mefloquine was substantially safer than racemic mefloquine was unlikely to be achieved.
Disposition of study participants.
One hundred fifty-three persons were screened, and 46 persons were randomized. Of these 46 persons, all received study treatment but only 44 persons completed the study. Two persons given 1,600 mg of (+)-mefloquine were withdrawn or withdrew before completion of the study: Person 31 was withdrawn because of an AE of sinus tachycardia, which was considered to be possibly related to the study treatment. The AE was diagnosed by ECG prior to the administration of the second part of the dose (which was not administered) and resolved spontaneously three hours later. Person 28 withdrew because she was unable to swallow the full complement of capsules. Both of these persons were included in the safety and pharmacokinetic populations. Demographics of the study population are shown in Table 1.
Table 1.
Demographics of the study population*
| Cohort | No. | Statistic | Age (years) | Height (cm) | Body weight (kg) | BMI (kg/m2)† |
|---|---|---|---|---|---|---|
| (+)-mefloquine, 800 mg | 8 (4 F, 4 M) | Mean | 33.6 | 169 | 69.36 | 24.26 |
| Median | 36.5 | 168 | 68.8 | 23.95 | ||
| (+)-mefloquine, 1,600 mg | 7 (5 F, 2 M) | Mean | 27.4 | 166 | 62.56 | 22.7 |
| Median | 26 | 166 | 63.4 | 21.7 | ||
| (+)-mefloquine, 1,200 mg | 4 (2 F, 2 M) | Mean | 39.3 | 174.3 | 69.8 | 22.78 |
| Median | 39 | 175.5 | 67 | 22.7 | ||
| Placebo | 9 (4 F, 5 M) | Mean | 32.6 | 172.7 | 73.98 | 24.58 |
| Median | 32 | 176 | 72.3 | 25 | ||
| Racemic mefloquine, 500 mg | 8 (6 F, 2 M) | Mean | 30.4 | 168 | 64.65 | 22.96 |
| Median | 29 | 167 | 66 | 22.25 | ||
| Racemic mefloquine, 1,000 mg | 6 (4 F, 2 M) | Mean | 28.8 | 165.7 | 65.07 | 23.52 |
| Median | 22 | 163 | 64.45 | 23.15 | ||
| Racemic mefloquine, 750 mg | 4 (3 F, 1 M) | Mean | 34.3 | 167.8 | 62.78 | 22.33 |
| Median | 31.5 | 166 | 62.7 | 22.65 |
BMI = body mass index.
Mean (SD) BMI for all male and female participants was 23.7 (2.0) kg/m2 and 23.3 (2.1) kg/m2, respectively.
Pharmacokinetics.
Across all three cohorts, peak concentrations of (+)-mefloquine after administration of racemic mefloquine and (+)-mefloquine were observed at 14–18 hours post-dose in the different cohorts. A secondary peak of (+)-mefloquine was observed for some persons, a finding that is consistent with entero-hepatic recirculation. After reaching Cmax, the concentrations decreased in a mono-exponential manner; mean terminal phase half-lives ranged from 214 to 290 hours in the different cohorts. This trend was also apparent for (−)-mefloquine after administration of racemic mefloquine at all three dose levels. For (−)-mefloquine, Tmax was slightly later (18, 16, and 21 hours in the three cohorts) and T½ was longer (519, 676 and 467 hours in the three cohorts) than its antipode. When we compared mefloquine enantiomers after administration of racemic mefloquine, Cmax, AUC0–t, and AUC all appeared approximately 1.6-, 3.0-, and 3.8-fold higher for the (−) enantiomer respectively, and CL/F was on average 3.3 times faster for (+)-mefloquine as previously reported.
Tmax, T½, and CL/F, and dose-normalized Cmax, AUC0–t, and AUC0–∞ across cohorts for (+)-mefloquine after administration of (+)-mefloquine or racemic mefloquine appeared to be comparable between males and females study participants. For (−)-mefloquine, Tmax was comparable between male and female participants, but T½ was on average 30% longer and CL/F was 42% lower in female participants after administration of racemic mefloquine in all cohorts. Additionally, dose-normalized Cmax, AUC0–t, and AUC0–∞ of (−)-mefloquine were on average up to two-fold higher in female participants.
Pharmacokinetic parameter summary for (+) and (−)-mefloquine after administration of racemic mefloquine and (+)-mefloquine to male and female participants combined are shown in Table 2.
Table 2.
Pharmacokinetic parameters summary for (+)-mefloquine and (−)-mefloquine after administration of racemic mefloquine and (+)-mefloquine to male and female study participants*
| Summary statistic | Cmax (ng/mL) | Tmax (hr)† | AUC0–t (mg.hr/mL) | AUC (mg.hr/mL) | t½ (hr) | CL/F (L/hr) |
|---|---|---|---|---|---|---|
| Cohort 1: (−)-mefloquine after administration of 500 mg of racemic mefloquine (n = 6–8) | ||||||
| Mean | 496.9 | 18.0 | 241.4 | 295.4 | 519.3 | 0.930 |
| SD | 96.4 | 10.0–72.0 | 68.4 | 96.6 | 129.8 | 0.330 |
| Cohort 1: (+)-mefloquine after administration of 500 mg of racemic mefloquine (n = 8) | ||||||
| Mean | 277.6 | 14.0 | 80.0 | 90.8 | 279.4 | 3.199 |
| SD | 62.9 | 10.0–24.0 | 29.1 | 35.6 | 115.3 | 1.405 |
| Cohort 1: (+)-mefloquine after administration of 800 mg of (+)-mefloquine (n = 8) | ||||||
| Mean | 613.8 | 14.0 | 181.3 | 189.3 | 213.8 | 4.505 |
| SD | 141.9 | 10.0–36.0 | 50.0 | 57.9 | 49.6 | 1.079 |
| Cohort 2: (−)-mefloquine after administration of 1,000 mg of racemic mefloquine (n = 6) | ||||||
| Mean | 1085.5 | 16.0 | 535.8 | 875.7 | 675.9 | 0.733 |
| SD | 318.1 | 14.0–24.0 | 182.3 | 479.6 | 266.9 | 0.388 |
| Cohort 2: (+)-mefloquine after administration of 1,000 mg of racemic mefloquine (n = 6) | ||||||
| Mean | 663.2 | 16.0 | 179.5 | 194.0 | 259.1 | 2.938 |
| SD | 156.8 | 12.0–24.0 | 62.9 | 73.8 | 71.9 | 1.161 |
| Cohort 2: (+)-mefloquine after administration of 1,600 mg of (+)-mefloquine (n = 5) | ||||||
| Mean | 1065.5 | 14.0 | 347.2 | 384.5 | 289.9 | 4.584 |
| SD | 341.4 | 12.0–48.0 | 106.5 | 130.1 | 80.7 | 1.582 |
| Cohort 3: (−)-mefloquine after administration of 750 mg of racemic mefloquine (n = 4) | ||||||
| Mean | 519.2 | 21.0 | 271.5 | 352.6 | 466.6 | 1.101 |
| SD | 110.2 | 18.0–24.0 | 50.1 | 70.9 | 102.87 | 0.251 |
| Cohort 3: (+)-mefloquine after administration of 750 of mg racemic mefloquine (n = 4) | ||||||
| Mean | 338.3 | 14.0 | 93.2 | 100.7 | 222.4 | 4.087 |
| SD | 58.0 | 12.0–36.0 | 30.4 | 33.4 | 96.0 | 1.476 |
| Cohort 3: (+)-mefloquine after administration of 1,200 mg of (+)-mefloquine (n = 4) | ||||||
| Mean | 833.3 | 18.0 | 234.3 | 243.8 | 221.0 | 5.159 |
| SD | 280.3 | 12.0–36.0 | 55.4 | 58.1 | 22.8 | 1.345 |
Cmax = maximum concentration; Tmax = time after administration of drug when maximum plasma concentration is reached; AUC = area under curve; t½ = half-life; Cl/F = bioavailability.
Values are medians and ranges.
Adverse events.
During the trial, there were 139 AEs. Of these AEs, 127 (91.4%) were treatment emergent adverse events (TEAEs). Female participants in the study reported 102 (80.3%) of the TEAEs, and the male participants reported only 25 (19.7%) of the TEAEs. Overall, the incidence of AEs in persons receiving (+)-mefloquine doses was more than three times greater than in persons receiving placebo (odds ratio [OR] = 3.1, 95% confidence interval [CI] = 0.19–51.24) and approximately 14% more prevalent in persons receiving racemic mefloquine than in persons receiving placebo (OR = 1.14, 95% CI = 0.09–14.91). Comparison of the incidence rates between female and male participants for all groups showed that female participants had an OR of nearly 15 for a CNS-related AE (OR = 14.97, 95% CI = 2.51–89.1) compared with male participants, irrespective of dose. The most frequently reported AEs are shown in Table 3, and the incidence of most frequently reported AEs by sex is shown in Table 4.
Table 3.
Most frequently reported adverse events in the study population
| Treatment/dose (no.) | Dizziness, no. (%) | Headache, no. (%) | Nausea, no. (%) | Vomiting, no. (%) |
|---|---|---|---|---|
| 800 mg of (+)-mefloquine (8) | 1 (12.5) | 4 (50.0) | 0 (0.0) | 0 (0.0) |
| 1,200 mg of (+)-mefloquine (4) | 3 (75.0) | 1 (25.0) | 3 (75.0) | 3 (75.0) |
| 1,600 mg of (+)-mefloquine (7) | 3 (42.9) | 5 (71.4) | 4 (57.1) | 3 (42.9) |
| 500 mg of racemic mefloquine (8) | 2 (25.0) | 2 (25.0) | 1 (12.5) | 0 (0.0) |
| 750 mg of racemic mefloquine (4) | 2 (50.0) | 0 (0.0) | 1 (25.0) | 0 (0.0) |
| 1,000 mg of racemic mefloquine (6) | 4 (66.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) |
| Placebo (9) | 1 (11.1) | 2 (22.2) | 3 (33.3) | 1 (11.1) |
Table 4.
Incidence of most frequently reported adverse events by sex in the study population
| Treatment/dose (no.) | Dizziness* | Nausea/vomiting | ||
|---|---|---|---|---|
| M | F | M | F | |
| (+)-mefloquine | 1/8 (12.5%) | 7/11 (63.6%) | 2/8 (25.0%) | 5/11 (45.4%) |
| Racemic mefloquine | 2/5 (40.0%) | 7/13 (53.8%) | 0/5 (0%) | 5/13 (38.5%) |
| Placebo | 0/5 (0%) | 1/4 (25.0%) | 1/5 (20.0%) | 2/4 (50.0%) |
Includes reports of dizziness, lightheadedness, loss of balance, and lack of coordination.
Most AEs were reported as mild or moderate in intensity. Although there were similar incidence of mild TEAEs reported by persons receiving racemic mefloquine (31 events in 18 persons), (+)-mefloquine (32 events in 19 persons), and placebo (22 events in 12 persons), there appeared to be a greater number of moderate and severe events reported by persons receiving (+)-mefloquine. In addition, more moderate and severe events were reported by female participants. There were 24 moderate events and 5 severe events reported by persons receiving (+)-mefloquine, 12 moderate events and 0 severe events reported by persons receiving racemic mefloquine, and 9 moderate events and 1 severe events in persons receiving placebo. The incidence of AEs appeared to increase in association with doses of (+)-mefloquine, although the low numbers of persons exposed at each dose makes this finding difficult to evaluate.
There were no obvious or consistent dose-related changes for any laboratory test results except for a transient isolated elevation in the bilirubin level in one person receiving racemic mefloquine. In terms of cardiovascular risk, persons receiving up to 1,200 mg of (+)-mefloquine or 1,000 mg of racemic mefloquine did not show any changes in ECG parameters, including QTc prolongation.
Gastrointestinal side effects.
Persons were asked to complete a visual analog scale to indicate the extent of nausea experienced after administration of study treatment. Scores for persons receiving placebo were consistently low throughout the study. Both (+)-mefloquine and racemic mefloquine were associated with development of nausea and marked measures of nausea in the days after drug administration. Changes from baseline treatment differences indicate that at 24 hours (P = 0.0314), 36 hours (P = 0.0265), 48 hours (P = 0.0314), and 72 hours (P = 0.0223), persons receiving (+)-mefloquine were more nauseated than persons receiving racemic mefloquine. Compared with placebo, persons receiving (+)-mefloquine reported statistically significant higher nausea scores; the differences between placebo and racemic mefloquine were not statistically significant. These results are consistent with reported nausea and vomiting AEs reported above. It is noteworthy that in most persons experiencing nausea and vomiting, the onset of symptoms was relatively late, typically starting 12–18 hours post-first administration, and peaking at 24 hours post-drug administration, which is consistent with a central mechanism of action.
Cognitive function.
There were decreases in power of attention at the highest dose levels of (+)-mefloquine and racemic mefloquine but not with placebo. These decreases were large in magnitude and statistically significant on day 1, but had completely passed by 48 hours post-dose. In contrast, there were no consistent changes between the study drug groups to the scores for the composite continuity of attention, quality of working memory, quality of episodic memory or speed of memory. A more consistent pattern was seen over the various self-ratings of mood (POMS) and sleep parameters. The highest dose of racemic mefloquine tested (1,000 mg) consistently disrupted mood and resulted in decrements in alertness, calmness and vigor, and increases in confusion, fatigue, and tension (Figure 1). For the composite total mood disturbance, the changes for racemic mefloquine compared with baseline were statistically significant at day 1 (P = 0.005) and day 7 (P = 0.026). The sleep questionnaire further identified evidence of reduced sleep quality, difficulties in getting to sleep (Figure 2), difficulties wakening, and poorer behavior after waking. The middle dose (750 mg) also showed some support for this pattern. Some of these findings waned after a few days, but some were seen over the initial two-week assessment period, although none of these disturbances persisted to day 42. In contrast, (+)-mefloquine and placebo were relatively free from such effects.
Figure 1.
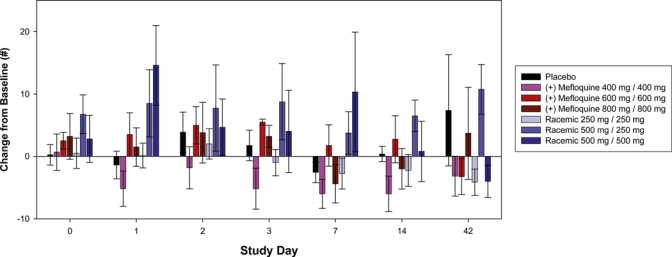
Total mood disturbance change from baseline in the study population. Error bars show mean ± SEM. This figure appears in color at www.ajtmh.org.
Figure 2.
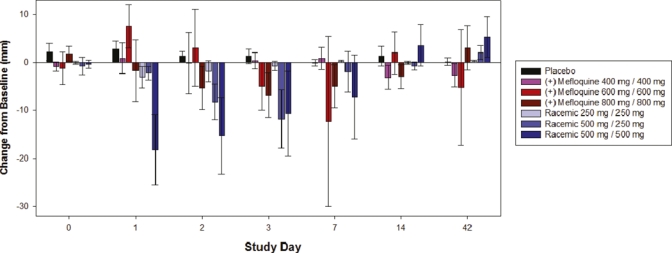
Leeds sleep evaluation questionnaire ease of getting to sleep change from baseline in the study population. Error bars show mean ± SEM. This figure appears in color at www.ajtmh.org.
With respect to the ability to stand upright without swaying (postural stability), the pattern of results indicated possible impairments in performance for racemic mefloquine and potential improvements for some doses of (+)-mefloquine; no differences were observed with placebo. However, these findings were driven by individual persons and were not large overall. In addition, the results are confounded by the inability of some persons to perform this test because of the AE of dizziness, in which the most severely affected persons could not be included in the test and analysis.
Sleep quality in terms of parameters involving ease of getting to sleep, and behavior after waking showed signs of disruption with racemic mefloquine but not (+)-mefloquine, and there was some statistical support for these effects. For the variable getting to sleep, analysis of variance showed statistically significant treatment × visit interaction (P = 0.0011). On days 1 and 2, the highest dose of racemic mefloquine (1,000 mg) significantly disrupted getting to sleep compared with placebo (P = 0.0006 and P = 0.0034, respectively). On day 3, the middle dose of (+) mefloquine (1,200 mg) and racemic mefloquine (750 mg) also resulted in significant difficulties in getting to sleep compared with placebo (P = 0.0461 and P = 0.0419 respectively), as did the highest dose of racemic mefloquine at this time point (P = 0.0345). On day 7, the middle dose of (+)-mefloquine (1,200 mg) significantly disrupted getting to sleep compared with placebo (P = 0.0483), although this effect was largely observed in only one volunteer.
Correlation between exposure and adverse events.
Mean plasma exposure as measured by Cmax and AUC0–∞ was greater in those persons reporting CNS and GI AEs compared with persons who did not report these AEs (Table 5) This trend appeared more marked for overall exposure than for Cmax. For example, the mean AUC0–∞ for persons reporting either a GI or a CNS AE was just more than two times as high compared with persons who did not experience these symptoms (598.4 versus 291.9 μg.hour/mL).
Table 5.
Mean mefloquine exposure in study participants reporting gastrointestinal and central nervous system adverse events*
| Characteristic | Central nervous system adverse event | Gastrointestinal adverse event | Central nervous system or gastrointestinal adverse event |
|---|---|---|---|
| Mean Cmax (ng/mL) with (and without) adverse event | 1,070.1 (871.9) | 1,195.2 (855.9) | 1,172.6 (740.1) |
| Mean AUC (mg.hr/mL) with (and without) adverse event | 520.9 (389.4) | 563.7 (397.2) | 598.4 (291.9) |
Cmax = maximum concentration; AUC = area under curve. An increase in total mood disturbance score is consistent with a worsening of total mood. No specific correlation was made between the magnitude of effect and its impact on daily living.
Discussion
The clinical utility of racemic mefloquine has been compromised by its association with CNS and gastrointestinal side effects. These effects were also seen in the present study and the most common AEs were detrimental effects on mood and sleep, nausea, and the ability to focus attention (power of attention). New treatment guidelines for the administration of mefloquine suggest dividing the dose over a three-day period and as part of an artemisinin-based combination, which has resulted in the drug being better tolerated than if it was given in one dose or as a split dose.19 However, a modest incidence of dizziness is still observed when mefloquine and artemisinin are administered in a three-day regimen (up to 6.6%).20 Additionally, the use of mefloquine for intermittent preventive therapy in infants and in pregnancy is restricted by high rates of nausea and vomiting after single or split administration of full therapeutic doses. Pregnant women are more susceptible to stimulants of nausea and vomiting, and a drug that is associated with a higher incidence of these side effects than mefloquine would be unacceptable for this population. In Africa, mefloquine is still not recommended for use in children or pregnant women because of the high incidence of GI side effects, especially vomiting, which in children seems to affect drug levels in blood and therefore limit its antiparasitic efficacy.21,22
In terms of its CNS profile, it has been hypothesized that at least some of the neuropsychiatric effects could be associated to the binding of mefloquine to adenosine receptors in the CNS and/or its effect on cholinesterase, an enzyme responsible for the breakdown of acetylcholine. The adenosine A2a receptor is co-expressed with the dopamine D2 receptor within the striatum and suppresses its activity.23–25 The A2a antagonists thus increase the activity of the D2 receptor and have been explored as anti-Parkinsonian targets because of their stimulation of locomotor activity.26 The A2a antagonists under clinical development, such istradefylline, have been linked to nausea in humans.27,28 Similarly, cholinesterase inhibitors such as donezepil and galantamine, which are used to treat the cognitive symptoms of Alzheimer's disease by maintaining acetylcholine levels, have also been linked to nausea and vomiting.29 The adenosine hypothesis appeared to be supported by the absence of CNS side effects observed in studies conducted with (+)-mefloquine in persons with rheumatoid arthritis. However, these studies reported a lower dose resulting in a lower exposure (up to 400–600 ng/mL) and a more gradual profile to Cmax based on a loading-dose regimen.
In summary, this study has demonstrated that (+)-mefloquine appears to have a more favorable profile compared with racemic mefloquine with respect to mood and sleep, which is consistent with the observation that adenosine receptor binding is associated with sleep and mood disturbances. However, at the doses required for therapeutic efficacy, (+)-mefloquine was associated with a higher than expected incidence of undesirable side effects, in particular, nausea, dizziness, and headache. The mechanism for these side effects is still unclear, but at least in the case of dizziness, mood change, sleep disturbance, and late-onset nausea and vomiting, the mechanism appears to be centrally mediated. Furthermore, the differential neuropsychiatric side effect profile of both substances suggests heterogeneity in neuroreceptor binding of the two enantiomers.
In conclusion, (+)-mefloquine has a different safety and tolerability profile compared with racemic mefloquine but its global safety profile is not superior to that of racemic mefloquine and its efficacy is likely to be no more than equivalent. Thus, these results do not warrant the development of (+)-mefloquine as an alternative to the currently used antimalarial drugs. Lastly, the AE profile confirms the restricted utility of mefloquine as a preventive therapy in pregnancy.
Acknowledgments
We thank the volunteers for participating in this study; Professor Keith Wesnes and Barbra Samuels (United Bioscience) for advice and input on and analysis of the cognitive function tests; Dr. David McGibney and Professor Dr. Norma Selve for input into the protocol design; Professor Brian Greenwood (London School of Health and Tropical Medicine) for his critical comments on this manuscript; and Dr. Brian Gennery, Dr. Paola Dazzan, and Dr. Richard Kay for work on the Independent Data Monitoring Committee and to Dr. Hugh Wiltshire for his critical appraisal of the pharmacokinetic data.
Footnotes
Financial support: This study was supported by the Medicines for Malaria Venture.
Disclosure: Julie Lotharius, Stephan Duparc, and Jörg Möhrle are employees of the Medicines for Malaria Venture, which funded the study. Robert Tansley has stock in Treague Ltd., makers of (+)-mefloquine and is a former employee of this company. These statements are made in the interest of full disclosure and not because the authors consider this to be a conflict of interest.
Authors' addresses: Robert Tansley, Cambridge, United Kingdom. Julie Lotharius, Stephan Duparc, and Jörg Möhrle, Medicines for Malaria Venture, International Centre Cointrin, Geneva, Switzerland. Anthony Priestley, LCG Bioscience, Bourn, Cambridge, United Kingdom. Fiona Bull, Treague Ltd., Cambridge, United Kingdom.
References
- 1.Croft AM, Garner P. Mefloquine for preventing malaria in non-immune adult travellers. Review. Cochrane Database Sys Rev. 2000;4:CD000138. doi: 10.1002/14651858.CD000138. [DOI] [PubMed] [Google Scholar]
- 2.Giao PT, de Vries PJ. Pharmacokinetic interactions of antimalarial agents. Clin Pharmacokinet. 2001;40:343–373. doi: 10.2165/00003088-200140050-00003. [DOI] [PubMed] [Google Scholar]
- 3.Baird JK. Effectivenss of antimalarial drugs. N Engl J Med. 2005;352:1565–1577. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 4.Karle JM, Olmeda R, Gerena L, Milhous WK. Plasmodium falciparum: role of absolute stereochemistry in the antimalarial activity of synthetic amino alcohol antimalarial agents. Exp Parasitol. 1993;76:345–351. doi: 10.1006/expr.1993.1042. [DOI] [PubMed] [Google Scholar]
- 5.Basco LK, Gillotin C, Gimenez F, Farinotti R, Le Bras J. In vitro activity of the enantiomers of mefloquine, halofantrine and enpiroline against Plasmodium falciparum. Br J Clin Pharmacol. 1992;33:517–520. doi: 10.1111/j.1365-2125.1992.tb04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerebrus Ltd . Patent WO 98/39003. 1998. [Google Scholar]
- 7.Ngiam TL, Go ML. Stereospecific inhibition of cholinesterases be mefloquine enantiomers. Chem Pharm Bull (Tokyo) 1987;35:409–412. doi: 10.1248/cpb.35.409. [DOI] [PubMed] [Google Scholar]
- 8.Barrett P, Emmins P, Clarke PD, Bradley DJ. Comparison of the adverse events associated with the use of mefloquine and combination of chloroquine and proguanil as antimalarial prophylaxis: postal and telephone surveys of travellers. BMJ. 1996;313:525–528. doi: 10.1136/bmj.313.7056.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbett EL, Doherty JF, Behrens RH. Adverse events associated with mefloquine. BMJ. 1996;313:1552. doi: 10.1136/bmj.313.7071.1552b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steffen R, Fuchs E, Schildkneckht J, Naef U, Funk M, Schlgenhauf P, Phillips-Howard P, Nevill C, Stürchler D. Mefloquine compared with other malaria chemoprophylactic regimens in tourists visiting east Africa. Lancet. 1993;341:1299–1302. doi: 10.1016/0140-6736(93)90814-w. [DOI] [PubMed] [Google Scholar]
- 11.Lobel HO, Miani M, Eng T, Bernard KW, Hightower AW, Campbell CC. Long-term malaria prophylaxis with weekly mefloquine. Lancet. 1993;341:848–851. doi: 10.1016/0140-6736(93)93058-9. [DOI] [PubMed] [Google Scholar]
- 12.Schlagenhauf P, Tschopp A, Johnson R, Nothdurft HD, Beck B, Schwartz E, Herold M, Krebs B, Veit O, Allwinn R, Steffen R. Tolerability of malaria chemoprophylaxis in non-immune travellers to sub-Saharan Africa: multicentre, randomised, double-blind 4 arm study. BMJ. 2003;3327:1078–1084. doi: 10.1136/bmj.327.7423.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gimenez F, Pennie RA, Koren G, Crevoisier C, Wainer IW, Farinotti R. Stereoselective pharmacokinetics of mefloquine in healthy caucasians after multiple doses. J Pharm Sci. 1994;83:824–827. doi: 10.1002/jps.2600830613. [DOI] [PubMed] [Google Scholar]
- 14.Na Bangchang K, Molunto P, Banmairuroi V, Thanavibul A, Karbwang J. Pharmacokinetics of mefloquine when given as a single and two divided-dose regimens. Int J Clin Pharmacol Res. 1995;XV:215–220. [PubMed] [Google Scholar]
- 15.Simpson PM. The construct validity of the cognitive drug research computerised assessment system. J Psychopharmacol. 1994;8((Suppl)):A20. [Google Scholar]
- 16.Parrott AC, Hindmarsh I. The LSEQ in psychopharmacological investigations. Psychopharmacology (Berl) 1978;71:173–179. doi: 10.1007/BF00434408. [DOI] [PubMed] [Google Scholar]
- 17.Boudreau EF, Flecenstein L, Pang LW, Childs GE, Schroeder AC, Ratnaratorn B, Phintuyothin P. Mefloquine kinetics in cured and recrudescent patients with acute falciparum malaria and in healthy volunteers. Clin Pharmacol Ther. 1990;48:399–409. doi: 10.1038/clpt.1990.168. [DOI] [PubMed] [Google Scholar]
- 18.Schlagenhauf P, Lobel H, Steffen R, Johnson R, Popp K, Tschopp A, Letz R, Crevoisier C. Tolerance of mefloquine by Swissair trainee pilots. Am J Trop Med Hyg. 1997;56:235–240. doi: 10.4269/ajtmh.1997.56.235. [DOI] [PubMed] [Google Scholar]
- 19.World Health Organization Guidelines for the Treatment of Malaria. Second Edition. 2010. http://www.who.int/malaria/publications/atoz/9789241547925/en/index.html Available at. Accessed April 2, 2010.
- 20.Rueangweerayut R, Pyae Phyo A, Uthaisin C, Socheat D, Quang Binh T, Tinto H, Penali L, Valecha N, Abdulla S, Thi Tien N, Borghini-Fuhrer I, Shin CS. Efficacy and safety of pyronaridine/artesunate fixed-dose combination compared with mefloquine plus artesunate in patients with acute uncomplicated Plasmodium falciparum malaria: results of a pivotal Phase III trial. Trop Med Int Health. 2009;14((Suppl 2)):45. [Google Scholar]
- 21.Slutsker LM, Khoromana CO, Payne D, Allen CR, Wirima JJ, Heymann DL, Patchen L, Steketee RW. Mefloquine therapy for Plasmodium falciparum malaria in children under 5 years of age in Malawi: in vivo/in vitro efficacy and correlation of drug concentration with parasitological outcome. Bull World Health Organ. 1990;68:53–59. [PMC free article] [PubMed] [Google Scholar]
- 22.Briand V, Bottero J, Noël H, Masse V, Cordel H, Guerra J, Kossou H, Fayomi B, Ayemonna P, Fievet N, Massougbodji A, Cot M. Intermittent treatment for the prevention of malaria during pregnancy in Benin: a randomized, open-label equivalence trial comparing sulfadoxine-pyrimethamine with mefloquine. J Infect Dis. 2009;200:991–1001. doi: 10.1086/605474. [DOI] [PubMed] [Google Scholar]
- 23.Ferre S, Fuxe K, von Euler G, Johansson B, Fredholm BB. Adenosine-dopamine interactions in the brain. Neuroscience. 1992;51:501–512. doi: 10.1016/0306-4522(92)90291-9. [DOI] [PubMed] [Google Scholar]
- 24.Ongini E, Fredholm BB. Pharmacology of adenosine A2A receptors. Trends Pharmacol Sci. 1996;17:364–372. [PubMed] [Google Scholar]
- 25.Ferre S, Fedholm BB, Morelli M, Popoli P, Fuxe K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997;20:482–487. doi: 10.1016/s0166-2236(97)01096-5. [DOI] [PubMed] [Google Scholar]
- 26.Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacoli. 2009;193:589–615. doi: 10.1007/978-3-540-89615-9_18. [DOI] [PubMed] [Google Scholar]
- 27.Hauser RA, Hubble JP, Truong DD. Randomized trial the adenosine A(2A) receptor antagonist istradefyline in advanced PD. Neurology. 2003;12:297–303. doi: 10.1212/01.wnl.0000081227.84197.0b. Istradefylline US-001 Study Group. [DOI] [PubMed] [Google Scholar]
- 28.Shiozaki S, Ichikawa S, Nakamura J, Kitamura S, Yamada K, Kuwana Y. Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology (Berl) 1999;147:90–95. doi: 10.1007/s002130051146. [DOI] [PubMed] [Google Scholar]
- 29.Alva G, Cummings JL. Relative tolerability of Alzheimer's disease treatments. Psychiatry (Edgmont) 2008;5:27–36. [PMC free article] [PubMed] [Google Scholar]