

Abstract
An increasing number of examples in the literature suggest that the in vivo duration of drug action not only depends on macroscopic pharmacokinetic properties like plasma half-life and the time needed to equilibrate between the plasma and the effect compartments, but is also influenced by long-lasting target binding and rebinding. The present review combines information from different research areas and simulations to explore the nature of these mechanisms and the conditions in which they are most prevalent. Simulations reveal that these latter phenomena become especially influential when there is no longer sufficient free drug around to maintain high levels of receptor occupancy. There is not always a direct link between slow dissociation and long-lasting in vivo target protection, as the rate of free drug elimination from the effect compartment is also a key influencing factor. Local phenomena that hinder the diffusion of free drug molecules away from their target may allow them to consecutively bind to the same target and/or targets nearby (denoted as ‘rebinding’) even when their concentration in the bulk phase has already dropped to insignificant levels. The micro-anatomic properties of many effect compartments are likely to intensify this phenomenon. By mimicking the complexity of tissues, intact cells offer the opportunity to investigate both mechanisms under the same, physiologically relevant conditions.
Keywords: pharmacokinetics, pharmacodynamics, drug, radioligand, receptor target, dissociation, rebinding, diffusion, cell membranes, intact cells
Introduction
There are increasing examples in the literature illustrating that the long-lasting clinical action of drugs not only depends on their macroscopic pharmacokinetic properties like their plasma half-life and the time needed to equilibrate between the plasma and the effect compartments, but also on their ability to bring about long-lasting target binding. Less known but equally important are local phenomena that cause the drug molecules to accumulate near the target and/or hinder their free three-dimensional (3D) diffusion away from that target. Such hindrance may allow the same drug molecule to consecutively bind to the same target and/or targets nearby even when the free drug concentration further away has already dropped to insignificant levels. Information about the pharmacodynamic and local pharmacokinetic mechanisms that may contribute to long-lasting drug action is widely dispersed in the literature. Indeed, these topics have not only caught attention of pharmacologists but also of, among others, biophysicians, physiologists, neurochemists and immunologists. The present review combines relevant information from these different research areas in an effort to have a better understanding of the nature of these mechansisms and the conditions in which they are most conspicuous.
Contribution of pharmacokinetics to long-lasting clinical efficacy
The efficacy and duration of drug action depends on both pharmacokinetic and pharmacodynamic factors. Pharmacokinetics (PK) is the discipline in pharmacology that investigates what the body does to the drug and, to this end, it focuses on the temporal evolution of a drug and its metabolites in serum as well as other body compartments like target and non-target tissues/organs. PK models mainly deal with the extent and rate of absorption, distribution (the dispersion of the drug and its potential metabolites between blood plasma and other body compartments), metabolism (the transformation of drug into active or inactive metabolites) and excretion. Pharmacodynamics (PD), on the other hand, examines what the drug does to the body (Holford and Sheiner, 1982). One of its aims is to study the time course of the drug's pharmacological effect after dosing (Derendorf et al., 2000). Yet, the often observed time lag between the drug's response and its plasma concentration (as seen by a counterclockwise hysteresis loop of the corresponding plot, Figure 1) made it initially difficult to reconcile PK to PD. Accordingly, they were long considered as separate disciplines.
Figure 1.
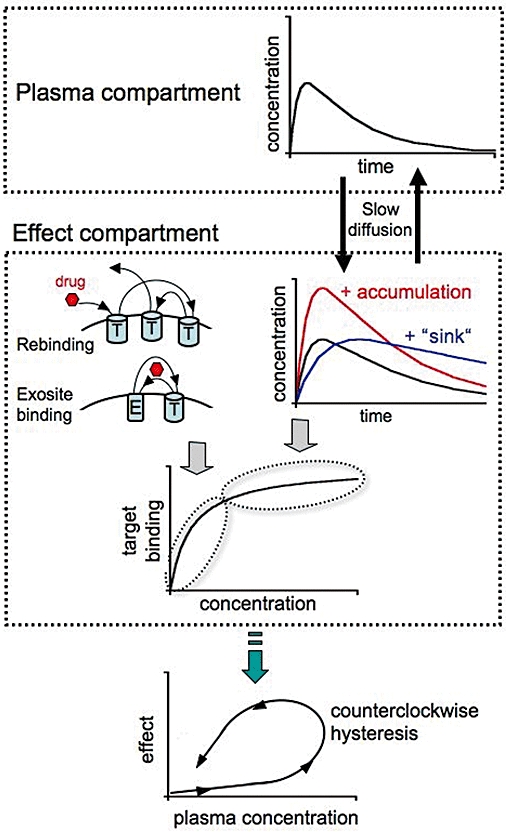
Factors contributing to long-lasting drug action. Classical pharmacokinetics (PK) contribution relates to a slow decline in free drug concentration in the plasma – and effect compartments (due to its slow metabolism and/or elimination) as well as to slow equilibration of the drug between both compartments. Local PK phenomena at the effect compartment that contribute to prolong high target occupancy include (i) increased drug concentration near the target by its uptake in the cell membrane from where it reaches the target (red curve); and (ii) prolonged presence of the drug near the target by its uptake in, and subsequent release from ‘sinks’ like cell membranes (i.e. the ‘diffusion microkinetic’ model, blue curve). When the target occupancy starts to decline, additional phenomena may contribute to prolong the occupancy. They include (i) slow dissociation of the drug-target complexes; (ii) ability of the drug to bind to multiple target (T) molecules before drifting away (i.e. rebinding); and (iii) ability of the drug to shuffle between the target and a (still hypothetical) high affinity ‘exosite’ (E). Combined, these phenomena may lead to a substantial delay between the drug's concentration in the plasma and the observed clinical effect (often presented by a counterclockwise hysteresis plot).
This came to an end by the introduction of a hypothetical compartment, the ‘effect compartment’, wherein the drug's target was defined to reside (Sheiner et al., 1979; Holford and Sheiner, 1982). The time needed for the drug to equilibrate between the plasma and effect compartments could then be held responsible for the delayed response (Figure 1). This paved the way to a wide range of combined PK/PD models to link the temporal variations in both the drug concentration in different body compartments and its clinical effect (Meibohm and Derendorf, 1997; Csajka and Verotta, 2006). The hysteresis loop could be forced to collapse by linking the effect to the effect compartment concentration instead of the plasma concentration at least in some cases (Della Paschoa et al., 1998), but not in all. Until a decade ago, the prevalent strategy for explaining the remaining hysteresis was to invoke a delay between the drug-target interaction and the generation of the response (Meibohm and Derendorf, 1997; Derendorf and Meibohm, 1999). Except for irreversibly acting drugs, those PK/PD models essentially regarded the amount of target-bound drug molecules to be in constant equilibrium with their free concentration at the effect site, so that attention could be focused on the potency/affinity and efficacy of the drug. It was thus sufficient to describe the drug-target interaction (and in most cases the thereby generated response) by equilibrium equations originating from the law of mass action and receptor theory (Hill, 1909; Ariens et al., 1956; Black and Leff, 1983).
Newer, mechanism-based PK/PD models originated from a better understanding of the molecular mechanism of the drug-target interaction and the thereby triggered biochemical and physiological events. The investigation of such mechanisms is now also considered to be part of the PD domain (Derendorf et al., 2000). Indeed, several in vitro studies have shed light on the hitherto overlooked importance of the rate of drug-target association and dissociation in linking the time-related changes in the concentration and the effect of a number of drugs (Shimada et al., 1996; Äbelöet al., 2001, 2006; Yassen et al., 2005; Yun et al., 2005). The importance of this kinetic concept is now gaining general recognition (Mager et al., 2003; Ploeger et al., 2009) and it has even led to the new ‘residence time’ terminology to describe the stability of the drug–target complexes (Copeland et al., 2006; Tummino and Copeland, 2008; Zhang and Monsma, 2009). The residence time (τ) of the drug is inversely proportional to the first-order dissociation rate constant (koff) of a binary drug-target complex. The term ‘residence time’ does not really epitomize a novel descriptive element in face of the more traditionally used dissociation half-life (t1/2 = 0.69/koff) but, as it is intuitively more appealing, it is likely to gain ground.
Terminology and focus
Here, we will still use the traditional ‘dissociation t1/2’ terminology and refer to ‘receptor occupancy t1/2’ to describe the average time needed to liberate half of the initially occupied receptors under conditions in where drug association (i.e. in open systems, in where the free drug concentration is allowed to vary with time, Figure 2) or rebinding (i.e. when a drug can bind to several target sites before drifting away) can take place. Also, as targets may have diverse functionalities and locations, for simplicity we will mainly focus on ligand-membrane-associated receptor interactions in the ensuing sections.
Figure 2.
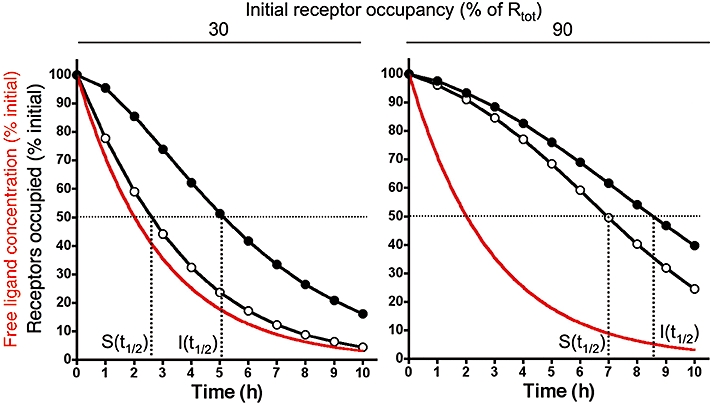
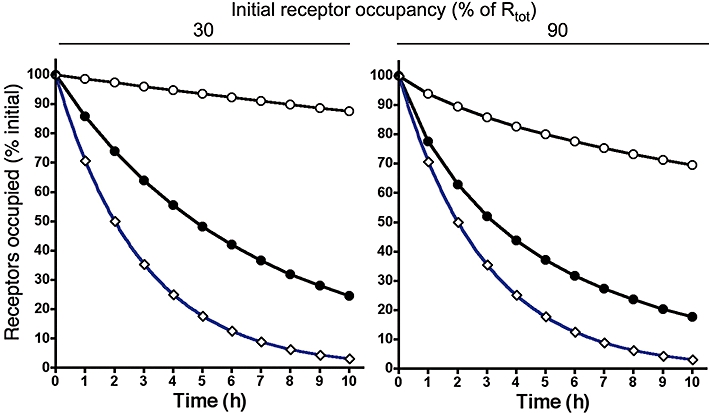
Receptor occupancy by a fast reversible (surmountable, S) ligand (○, koff = 5.2 min−1, kon = 1 × 108 M−1× min−1) and a slow dissociating (insurmountable, I) ligand (•, koff = 5.7 × 10−3 min−1, kon = 1 × 108 M−1× min−1) when the free ligand concentration decreases exponentially with time (with t1/2 = 2 h, red curve). At the start, free and receptor bound ligands are at equilibrium and either 30% (left panel) or 90% (right panel) of the receptors (Rtot) are occupied. Data points were obtained by integrating the differential equations describing a first-order decrease in free ligand concentration and a bimolecular ligand-receptor interaction as previously described (Vauquelin et al., 2001; Vauquelin and Van Liefde, 2006). Receptor occupancy half-life of the insurmountable ligand, I(t1/2), is 2.0-fold the corresponding half-life by the surmountable ligand, S(t1/2), at 30% initial occupancy. This value drops to 1.2-fold at 90% initial occupancy.
Contribution of drug-binding kinetics to clinical therapy
In drug-screening studies, ligand-receptor interactions are traditionally quantified in terms of affinity and efficacy only. Until recently, and besides a few exceptions (Leysen and Gommeren, 1986), only little attention has been paid to the kinetic properties of drug-target complexes. A likely reason for this is that the dissociation rate of the drug-target has historically been assumed to be too rapid to play a significant role. However, an increasing number of studies reveal that this is not always the case. Table 1 enumerates a few examples of slow-dissociating drugs, more can be found in Swinney (2004, 2006a) and Copeland et al. (2006). Among them, it was recently advanced that the slow dissociation of the muscarinic antagonist tiotropium bromide and of NK1 Neurokinin receptor antagonists from their cognate receptors was the key to explain their prolonged duration of action (Disse et al., 1999; Lindström et al., 2007). These and other examples made it clear that the dissociation rates of the complexes may have far-reaching repercussions with respect to the drug discovery process. A number of interesting principles have also been advanced in the emanating review papers (Swinney, 2004, 2006a,b, 2008, 2009; Copeland et al., 2006; Vauquelin and Van Liefde, 2006; Tummino and Copeland, 2008; Van Liefde and Vauquelin, 2009; Zhang and Monsma, 2009). Among them, a distinction should be made about whether the drug-target complex is a source of adverse events or not. In the affirmative case, the complexes should be preferred to dissociate swiftly (i.e. to be readily surmountable) so that the occupancy of the target by its endogenous ligands is only dictated by their ability to compete with free drug molecules. This is the case for neuroleptics/antipsychotics used in clinical therapy to attenuate psychotic episodes (Creese et al., 1976). Neuroleptics of the first generation, the typical ones, displayed high potency and long-lasting D2 dopamine receptor blockade. Yet, the incidental refractoriness of the striatal receptors in responding to fast fluctuations in the local dopamine concentration generated extrapyramidal side effects, including a Parkinsonian-like syndrome and tardive dyskinesia (Deutch, 1993). These adverse events are less prominent with second-generation, atypical neuroleptics because of their swifter dissociation from the D2 dopamine receptors so that those in the striatum remain responsive to peaks in the dopamine concentration (Kapur and Seeman, 2000, 2001). Without intrinsic adverse events, also denoted as ‘mechanism-based toxicity’, long-lasting target-binding drugs are expected to be the most suitable agents for therapies that require continuing, high levels of target occupancy (Copeland et al., 2006; Swinney, 2006b; Zhang and Monsma, 2009). Compared with solely relying on the pharmacokinetic properties of the drug, such dissociation characteristics are likely to offer greater control over the issue of selectivity and toxicity. Indeed, slow dissociation is often associated with high binding affinity, and such drugs are likely to display pronounced target selectivity and a broad therapeutic window. Conversely, because of their lower affinity for collateral targets, such drugs are also likely to dissociate faster from undesirable, adverse event-mediating ones. In such case, the drug-primary target complexes will last longer than the undesirable complexes so that the beneficial effects of the drug are likely to outlast the adverse events. Yet, there are also examples where the dissociation rate and equilibrium affinity are not directly related (Leysen and Gommeren, 2004). Tiotropium exhibits similar binding affinity at both the M3 muscrinic receptor, which provides efficacy, and the M2 subtype that is responsible for cardiovascular side effects. However, it dissociates 10 times more rapidly from the M2 receptor, displaying a beneficial profile that has been coined as ‘kinetic selectivity’ (Disse et al., 1999). This clearly illustrates that it can be advantageous to not only rely on affinity measurements at equilibrium, but also to investigate the kinetic properties of new drugs (Dowling and Charlton, 2006). This may also be the case for allosteric ligands (which bind an alternative site to the endogenous agonist) that may, by their nature, have slow offset kinetics (Kenakin, 2008). Taken together, these considerations clearly illustrate the general concept that parameters that describe the in vivo selectivity of a drug are not necessarily static but could depend on the post-administration time.
Table 1.
Example dissociation half-lifes of several G protein-coupled receptor ligands
| Receptor | Ligand | Dissociation half-life (t1/2) | References |
|---|---|---|---|
| M3 muscarinic receptor | Tiotropium | 7.7 h | Dowling and Charlton, 2006 |
| Ipratropium | 0.16 h | ||
| CCR5 chemokine receptor | Maraviroc | 136 h | Watson et al., 2005 |
| 873140 | >136 h | ||
| µ opioid receptor | Alvimopan | 30–44 min | Cassel et al., 2005 |
| N-methylnaloxone | 0.46 min | ||
| Histamine H1 receptor | Desloratidine | >8.7 h | Anthes et al., 2002 |
| GSK1004723 | 5.8 h | Slack et al., 2009 | |
| Angiotensin type 1 receptor | Candesartan | 2 h | Vauquelin and Van Liefde, 2006 |
| Olmesartan | 70 min | ||
| Neurokinin 1 receptor | aprepitant | 3.6 h | Hale et al., 1998 |
Physiological relevance of drug dissociation rates
The stability of drug-target complexes varies a lot: from being irreversible in the case of covalently bound ligands such as β-haloalkylamines (Furchgott, 1966) and gastric proton pump inhibitors like omeprazole (Wallmark et al., 1984) to very fast dissociating ones like atypical neuroleptics. Yet, most of the drugs will experience intermediate, more-or-less slow dissociation from their target (Swinney, 2004; Copeland et al., 2006). Whether or not the dissociation rate of the drug has clinical relevance needs, first of all, to be regarded in light of the half-life of the free drug in the effect compartment. Moreover, in the case of receptor antagonists, attention should also be paid to the duration of receptor exposure to endogenous messengers like hormones and neurotransmitters (Kapur and Seeman, 2000, 2001; Vauquelin and Van Liefde, 2006).
This latter issue is clearly illustrated in classical organ bath experiments (and related intact cell experiments) in where the methodology consists of an initial exposure (for a reasonably long time period) of a tissue to a fixed concentration of the antagonist of interest, after which (still in the presence of the antagonist) the tissue is further incubated with increasing concentrations of an agonist (Leff and Martin, 1986). The response of the tissue is recorded at each agonist concentration and, compared with the control agonist concentration-response curve for a naïve tissue, the maximal response will obviously decline (in the absence of ‘spare receptors’) in case of an irreversibly binding antagonist. This type of antagonism is truly ‘insurmountable’ because the occupied receptors are forever refractive to agonist stimulation (Vauquelin et al., 2002; Kenakin et al., 2006). At the other extreme, antagonists may dissociate so swiftly that subsequently added agonist molecules can already access all the receptor molecules before the response is recorded. Such antagonists will only produce parallel rightward shifts of the agonist concentration-response curve. They are denoted as ‘surmountable’ because all the receptor molecules can get stimulated if the agonist concentration is high enough. In between resides a broad range of more-or-less slowly dissociating antagonists. In organ bath experiments, such antagonists will produce a mixed type (i.e. partially insurmountable) inhibition if the ensuing challenge with the agonist is to short to allow a new mass-action equilibrium to be fully operational before the response is measured (Lew et al., 2000, 2001). The insurmountable behaviour of such antagonists only reflects a temporal hemi-equilibrium as it can be overcome by prolonging the agonist exposure. In practice, however, this may be impossible to achieve because of methodological constraints of the experiment. A final note of caution with respect to interpreting these experiments is that insurmountable antagonism could also result from allosteric/non-competitive interactions and that partial insurmountability could originate from the co-occurrence of two antagonist-bound receptor conformations/states with only one of them being surmountable (Vauquelin et al., 2001; Swinney, 2004; Copeland et al., 2006; Vauquelin and Van Liefde, 2006; Tummino and Copeland, 2008).
In contrast to intact and fractionated cell-based experiments and the usual organ bath experiments, administered drugs are gradually eliminated from a living organism. This implies that the concentration of free drug (or its active metabolite) in the effect compartment will also decline with time. If the drug in question dissociates swiftly from its receptor (i.e. when bound and free drug concentrations are in quasi-permanent equilibrium), the receptor occupancy t1/2 will depend both on the rate of drug elimination from the effect compartment and the initial level of receptor occupancy (Vauquelin and Van Liefde, 2006; Szczuka et al., 2009). Indeed, when the free drug concentration is at or below its equilibrium dissociation constant (KD) for the receptor, the initial occupancy will only be modest and its decline will closely follow the decline in free drug concentration. On the other hand, when the free drug concentration is in large excess of its KD, the initial receptor occupancy will be nearly maximal and, because of the hyperbolic shape of Emax model-based saturation binding curve (Figure 1), it will decline much slower than the free drug concentration. Hence, high local drug concentrations in effect compartment will already lead to a long lasting effect by itself. However, being surmountable, such drugs will also continuously compete with other receptor ligands like metabolites and endogenous messengers for binding to the target.
On the other hand, the example of proton pump inhibitors (Wallmark et al., 1984) clearly illustrates that the therapeutic action of irreversibly binding drugs might persist even after the free drug was fully eliminated from the effect compartment. In fact, their action can only be abolished by de novo synthesis of target molecules by the organism. Here again, the situation is more complex for the reversible but slow dissociating drugs. For such drugs, a first series of simulations already suggested that their binding could appreciably outlast that of a fast reversible/surmountable drug provided that their dissociation is slower than their elimination from the relevant compartment (Vauquelin and Van Liefde, 2006; Tummino and Copeland, 2008). The present simulations constitute an extension thereof by focusing on the potential impact of the free drug elimination t1/2 (from 15 min to 12 h) and of the initial fraction of receptor occupancy (30, 80 and 95%) on the receptor occupancy t1/2. Three drugs were compared, a surmountable/fast reversible one, a slow dissociating one (dissociation t1/2 = 2 h) and a quasi-irreversible one (dissociation t1/2 = 12 h). An example of the results is illustrated in Figure 2 and an example of derived receptor occupancy t1/2 versus free drug elimination t1/2 plots is illustrated in Figure 3 for initial 80% receptor occupancy. An interesting observation is that the latter plots are quasi-linear and parallel for the three drugs examined and that their intercept with the ordinate closely amounts their dissociation t1/2-values (hence, the vertical separation between the plots corresponds to the difference in their dissociation t1/2).
Figure 3.
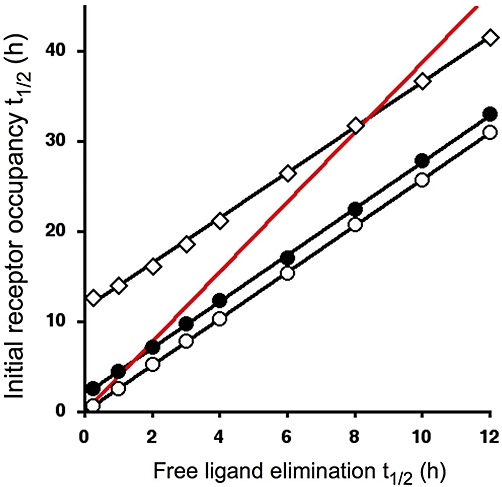
Receptor occupancy t1/2 as a function of the free ligand's elimination t1/2. Experimental set-up and data generation and analysis are the same as in Figure 2. Initial receptor occupancy amounts 80% of Rtot. Three ligands were compared: a fast reversible (surmountable, S) one (○, koff = 5.2 min−1), a slow dissociating one (•, koff = 5.7 × 10−3 min−1) and a quasi-irreversible one (◊, koff = 4.8 × 10−4 min−1) and kon = 1 × 108 M−1× min−1 for all. Binding of the latter ligands was arbitrarily considered to outlast that of the surmountable one when their receptor occupancy t1/2 exceed 1.5 times S(t1/2) (this rule applies to the sections of the plots that are located above the red line representing this threshold value). Table 2 lists such threshold free ligand elimination t1/2-values for the different fractions of initial receptor occupancy.
We here arbitrarily opted for a ≥50% increase in receptor occupancy t1/2 to establish a threshold above which the binding of an insurmountable drug appreciably outlast that of a fast reversible one. In Figure 3, this rule applies to the sections of the plots that are located above the red line. Upon inspection, these and additional (not shown) simulations suggest that it is not the elimination rate itself but rather the ratio between the dissociation and elimination rates that determines whether the binding of an insurmountable drug will appreciably outlast that of the fast reversible one. Moreover, the threshold is not constant as it depends on the initial degree of receptor occupancy (Table 2). At 30% initial occupancy, it is already sufficient for the dissociation to take place at half the elimination rate; at 80%, the dissociation already needs to be a little slower; while at 95%, the dissociation already needs to be about three times slower. The reason for this dependence may be found in the hyperbolic shape of a saturation binding curve at equilibrium and the resulting impact of the extent of initial receptor occupancy upon the rate by which it declines in case of a surmountable agonist, as discussed previously (Figure 1). Taken together, the present, extended simulations confirm that the terms ‘slow dissociation’ and ‘long-lasting receptor blockade’ by antagonists should not be used in strict synonymy.
Table 2.
Threshold t1/2-values (in h) for insurmountable ligand elimination below which their receptor occupancy t1/2 outlasts that of the surmountable ligand by ≥50%: influence of the initial fraction of occupied receptors and the ligand's dissociation t1/2
| Initial receptor occupancy (% of Rtot) | |||
|---|---|---|---|
| Ligand dissociation t1/2 (h) | 30 | 80 | 95 |
| ◊, 2 | 4 | 1.5 | 0.7 |
| •, 12 | 26 | 8.3 | 4.5 |
Nevertheless, even when the elimination of a free antagonist drug is rate limiting, slow dissociation/insurmountablility may still improve its efficacy in vivo, particularly in systems with rapid and transient signalling characteristics (Vauquelin and Van Liefde, 2006). The most extreme example of such a system is neurotransmission, where neurotransmitter is released locally within a synapse at very high concentrations, but which is very rapidly removed by reuptake and metabolic routes. In the synapse, the concentration of agonist is so high that if left to reach equilibrium, it would compete off the antagonist and bind to all the receptors. The extremely transient nature of high neurotransmitter concentration, however, means that most antagonist ligands will have a long-enough duration at the receptor to effectively antagonize the agonist over the very short period of high agonist concentrations, completely blocking transmission even in situations of very high agonist concentrations.
Related situations may also occur in other systems, for example, chemotaxis of leukocytes into areas of inflammation. Ligands that competitively antagonize chemotactic receptors equilibrate with the receptor in the periphery and only come into contact with high concentrations of chemoattractant for brief periods as they pass through an inflammatory focus. The concentrations of chemoattractant can be very high in those foci, up to 1000-fold their EC50 for the CXCR2 chemokine receptors on human neutrophils (Nocker et al., 1996; Traves et al., 2002). A rapidly dissociating compound will re-equilibrate with the chemoattractant in the time it takes for the leukocyte to pass the area of inflammation, possibly resulting in an incomplete blockade of infiltration into tissue. In these situations, receptor blockade will depend upon the relative concentrations of each ligand. A slowly dissociating ligand will, however, remain largely bound to the receptor during the time taken to pass the inflammatory focus, effectively antagonizing the chemoattractant, thereby demonstrating higher efficacy. Recently, the kinetic characteristics of Sch527123, a novel CXCR2 antagonist developed by Schering Plough, have been directly determined using a tritiated version of the molecule. The binding of [3H]Sch527123 to CXCR2 receptors in CHO cells was found to be slowly reversible with a dissociation t1/2 of approximately 22 h (Gonsiorek et al., 2007). It is possible that this slow dissociation contributes to its highly efficacious inhibition of pulmonary inflammation in animal models (Chapman et al., 2007).
Evidence for rebinding in radioligand dissociation experiments
Several experimental strategies can be deployed to determine the dissociation rate of drug-receptor complexes (Table 3) (Vauquelin and Szczuka, 2007). When a drug is available in a radiolabelled form and if it displays sufficiently high affinity and specificity for its cognate receptor, its dissociation can be directly measured by following the time-course of its specific/receptor binding under wash-out conditions. All these experiments require a preliminary incubation of the receptor-containing preparation with the radioligand but, for the subsequent wash-out, four major experimental paradigms can be discerned depending on whether or not the radioligand-containing medium is replaced by fresh medium and whether or not an excess of unlabelled competitive ligand is supplied (Table 3). Exposure to new medium containing an excess of unlabelled ligand (Method 1) ensures its fast occupancy of all the free receptor sites so that the binding of any remaining free radioligand or even receptor-dissociated radioligand molecules is effectively prevented (Limbird, 1996). This is presumably the safest method determining the radioligand's genuine dissociation rate. Even without replacing the medium, the mere addition of unlabelled ligand (Method 2) will reduce radioligand association. Yet, for this to be effective, one needs to ensure that the concentration of the unlabelled ligand is high enough. One way to deal with this issue is dilute the medium before adding the unlabelled ligand. With this in mind, it is clearly less satisfactory for radioligand dissociation to be monitored in fresh medium alone (i.e. without unlabelled ligand, Method 3) and even worse if the initial incubation medium is not replaced by, but simply diluted with an excess of fresh medium (Method 4).
Table 3.
Methods for measuring radioligand dissociation rates: advantages and limitations
| Method | Removal of free radioligand after pre-incubation with receptor | Excess unlabelled ligand in wash- out medium | Advantages and limitations |
|---|---|---|---|
| 1 | Yes | Yes | Correct koff* |
| 2 | No | Yes | Correct koff* |
| 3 | Yes | No | Rebinding possible |
| 4 | Only dilution | No | Rebinding possible and approximate koff# |
Only if both radioligand and unlabelled ligand interact competitively, that is, when they bind to overlapping sites at the receptor.
Due to the establishment of a new mass-action equilibrium with diluted free radioligand in the wash-out medium.
Because dissociated radioligand molecules are capable of binding again to their cognate receptor molecules with Method 3 but not with Method 1, a comparison of the time-wise decline of the specific radioligand binding under both wash-out conditions offers an elegant approach to find out whether such ‘rebinding’ or ‘reassociation’ can effectively take place. By applying this approach on intact plated recombinant cell lines expressing the human G protein coupled receptors of interest, we have already detected such rebinding behaviour for a number of radiolabelled antagonist molecules. They include the AT1-type angiotensin II receptor antagonists [3H]-candesartan, [3H]-olmesartan and [3H]-telmisartan; the D2 dopamine receptor antagonists [3H]-spiperone and [3H]-raclopride (but to a much lower extent); and the CB1 cannabinoid receptor antagonists [3H]-rimonabant and [3H]-taranabant (Fierens et al., 1999; Le et al., 2007; Packeu et al., 2008; Szczuka et al., 2009; Wennerberg et al., 2010). Compared with the mono-exponential decline in radioligand binding and the involvement of the total receptor population when the wash-out takes place in an excess of unlabelled ligand, dissociation seems to be greatly delayed if not halted entirely. In this case, dissociation only appears to occur from a fraction of the labelled receptor molecules when the wash-out takes place in fresh medium only. Upon surveying the literature, it appears that such differences in dissociation behaviour have also been observed for other receptor systems (Table 4). Yet, rather than evoking the rebinding hypothesis, authors tend to focus on the faster dissociation of the radioligand in the presence of the unlabelled ligand (Vauquelin and Szczuka, 2007). Indeed, this phenomenon is generally regarded as a hallmark for negative cooperativity between the radioligand and the unlabelled ligand and, in turn, could point to the occurrence of allosteric interactions (Park et al., 1990; Limbird, 1996; Gill et al., 1999; Urizar et al., 2005). To be more precise, by binding to a remote/‘allosteric’ site, the unlabelled ligand is able to trigger/favour a conformational change of the receptor molecule (or multimeric complex therof) and this could enhance the dissociation of the radioligand from the receptor's ‘orthosteric’ site (Christopoulos and Kenakin, 2003).
Table 4.
Selected situations in where rebinding is likely to prolong drug/ligand-target binding
| Ligand | Target | System |
|---|---|---|
| (1)Thyrotropin | Thyrotropin receptors | Human thyroid membranes |
| (2)Naloxone | µ opioid receptor | Recombinant CHO cells |
| (3)Sartans | AT1 angiotensin II receptors | Recombinant CHO cells |
| (4)Spipeone | D2 dopamine receptors | Recombinant CHO cells |
| (5)Rimonabant, taranabant | CB1 cannabinoid receptors | Recombinant CHO cells |
| (6)Epidermal growth factor | EGF receptors | A431 cells |
| (7)Dinitrophenyl ligands | Cell-surface IgE | Rat basophilic leukemia cells |
| (8)Fibroblast growth factor-2 | Heparan sulphate proteoglycans | Vascular smooth muscle cells |
| (9)Diprenorphine, naloxone | Opioid receptors | Rat brain, in vivo |
| (10)CH 23390 | D1 dopamine receptors | Rat brain, in vivo |
| (11)Fasciculin 2 | Acetylcholinesterase | Neuromuscular junction |
| (12)Lectins | Selectin | Cell-cell interactions |
| (13)Peptide-MHC class II ligands | T cell receptors | Cell-cell interactions |
| (14)Antibodies (also monomeric) | Immobilized ampicillin | BIAcore biosensor |
Many other experimental observations in where target binding is shortened by the presence of an excess of other drugs/ligands have been published but they were, rightfully or not, merely commented in view of non-competitive interactions. References: (1) Powell-Jones et al., 1979; (2) Spivak et al., 2006; (3) Fierens et al., 1999; Le et al., 2007; (4) Packeu et al., 2008 (5) Szczuka et al., 2009; Wennerberg et al., 2010; (6) Wiley (1988); (7) Goldstein et al., 1989; Posner et al., 1992; (8) Chu et al., 2004; (9) Perry et al., 1980; (10) Gifford et al., 1998; (11) Krejci et al., 2006; (12) Lou et al., 2006; Thomas, 2006; (13) Germain, 1997; (14) Nieba et al., 1996.
Fortunately, at least two criteria offer the opportunity to discriminate between the ‘rebinding’ and the ‘allosteric interaction’ models. First is to compare the dissociation behaviour of the radioligand when the wash-out is carried out in the presence of increasing concentrations of different unlabelled ligands of the examined receptor. If those unlabelled ligands produce a concentration-dependent increase in the radioligand's (apparent) dissociation rate with the same order of potencies as in competition binding experiments and if their maximal effect is the same, it is already very likely that the ‘rebinding’ model accounts for the observations. Indeed, it should already be quite unlikely for distinct allosterically acting ligands to trigger/favour the same conformational change of the receptor and, hence, to increase the dissociation rate of the orthostetic ligand (i.e. the radioligand) to the same extent (Christopoulos and Kenakin, 2003). Second, the likelihood of this interpretation is further supported if the unlabelled equivalent of the radioligand increases the radioligand's dissociation rate to the same extent as the other unlabelled ligands and if the radioligand binding experiments show no evidence of negative cooperativity (i.e. when the Hill coefficient of the saturation binding plot is close to 1). The radiolabelled antagonists [3H]-candesartan and [3H]-spiperone have already been inspected according to those criteria and, at least for those there is ample evidence for rebinding to take place if the wash-out takes place in fresh medium only (i.e. Method 3) (Fierens et al., 1999; Packeu et al., 2008).
Finally, although the most widely used procedure to prevent radioligand rebinding is to include an excess of unlabelled ligand in the wash-out medium, Goldstein et al. (1989) proposed that the same result can also be obtained by capturing free radioligand molecules with high affinity binding proteins such as specific antibodies. As bovine serum albumin (BSA) also avidly binds certain classes of drugs (Simard et al., 2006); it could also act as an external ‘sink’ in such cases. In agreement, rebinding of [3H]-candesartan and [3H]-taranabant to their receptors was indeed partly prevented by addition of 1% w/v BSA in the wash-out medium (Fierens et al., 1999; Szczuka et al., 2009). The effect of these soluble proteins also points at rebinding as it can only take place if the radioligand is free in solution (and, hence, physically separated from its receptor) for some time. An alternative interpretation of such phenomena in terms of an unfavourable conformational change of the receptor seems less probable because it requires the soluble protein to interact with an allosteric site at the receptor.
Functional organ bath ‘wash-out’ experiments are often also carried out in compliance with Method 3. In such experiments, pre-incubation of the target tissue with the drug is followed by brief wash steps and a final, long-term incubation of the tissue in the wash-out medium. If the drug is an agonist, the remaining response can be recorded at distinct time intervals (Austin et al., 2003). If the drug is an antagonist, the tissue needs to be challenged with and excess of agonist at distinct time intervals and the response monitored soon thereafter. By this method, the time-wise loss of receptor occupancy by the antagonist can be indirectly deduced from the restoration in receptor's responsiveness. Using this strategy, and in agreement with the [3H]-candesartan dissociation experiments in medium only, there was only a very slow recovery of the contractile response of candesartan-pretreated rabbit aortic strips and rat portal veins to angiotensin II (Ojima et al., 1997; Morsing et al., 1999). Similar functional wash-out experiments, but this time with AT1 receptor expressing recombinant Chinese Hamster Ovary cells, also yielded a very slow recovery of the response (here measured in terms of inositol phosphates accumulation) (Vanderheyden et al., 1999). Together with the radioligand dissociation experiments, these functional wash-out experiments are indicative for the physiological relevance of rebinding phenomena.
Current mechanistic interpretations of rebinding
According to a widespread pharmacologist's view, radioligand molecules that are freshly dissociated from their receptor distribute rapidly all over the wash-out medium so that its rebinding is only a matter of restoring a new mass-action equilibrium between free and bound radioligand (Sutter et al., 1979; Limbird, 1996). This implies that rebinding should only be preeminent when the radioligand (or antagonist in functional wash-out experiments) displays very high affinity for its receptor. Although [3H]-candesartan complied with this requisite, [3H]-telmisartan also experienced considerable rebinding under the same experimental conditions and this despite its much lower affinity for the AT1 receptor (Fierens et al., 1999; Le et al., 2007). Moreover, computer-assisted simulations revealed that, for simply restoring a new mass-action equilibrium between homogenously distributed free and bound [3H]candesartan, the free concentration had to be eight times higher than the total amount of radioligand that what was left in the wells (Fierens et al., 1999). As alternative explanation, it was suggested that the released radioligand molecules are subjected to local accumulation in the vicinity of their receptors. This interpretation joins the already ancient ‘unstirred layer’ concept (Silhavy et al., 1975). Although earlier dismissed in the context of ligand-membrane associated receptor interactions (Limbird and Lefkowitz, 1976; De Meyts et al., 1977), this concept has recently been restored in honour (Copeland et al., 2006; Spivak et al., 2006).
Brownian motion refers to the random walks of molecules in solution resulting from their continuous collisions with molecules of the solvent and a large set of such random walks can be formulated by the classical equations of macroscopic diffusion. In this respect, bimolecular ligand-receptor interactions are customarily described by equations that apply to solutes, that is, molecules that are homogeneously distributed in a solvent with unrestricted 3D diffusion therein. Classical PK models also assume that compartments are homogenous and well stirred (Gifford et al., 1998; Pang et al., 2007). Even in this situation, diffusion already plays a role in the ligand's association and dissociation characteristics. Indeed, according to the prevalent biophysical model (DeLisi, 1981; DeLisi and Wiegel, 1981), binding between the ligand (L) and its receptor (R) proceeds according to a two-step process (Figure 4). Diffusion of both molecules will first bring them sufficiently close together to form a so-called ‘encounter complex’[(L… R)] and it is only then that the reversible binding process (formation of L.R) can take place. This model separates the reversible ligand-receptor binding in two components: one depends on the transport characteristics of both partners (designated by the diffusive rate constants k+ and k-), while the other one depends on the interaction mechanism (designated by the reaction rate constants k1 and k−1). It can also be represented by a single-step process with the same mathematical form as the classical one describing such interaction between soluble molecules in a well-mixed system except that the ‘fundamental’ association and dissociation rate constants (kon and koff) are replaced by the ‘effective’ forward rate coefficient kf and the ‘effective’ reverse rate coefficient kr (Figure 4) (Goldstein and Dembo, 1995). However, kf and kr are not constant in other circumstances (please see next) and this is why the term ‘coefficient’ is to be preferred.
Figure 4.
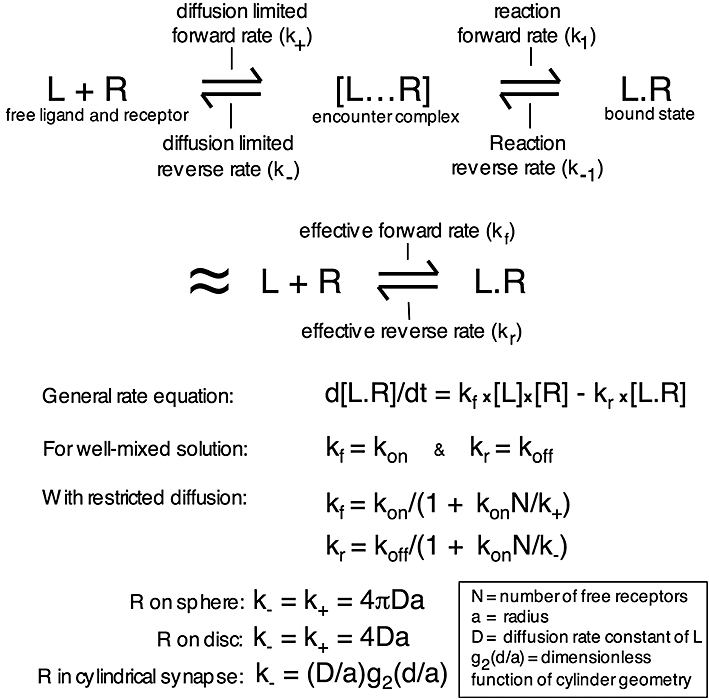
Schematic representation of drug (L) – receptor (R) binding with the intermediate formation of an encounter complex [L… R] (DeLisi, 1981; DeLisi and Wiegel, 1981). This binding will also be represented by a single-step process with the ‘effective’ forward rate cefficient kf = k+k1/(k-+ k1) and the ‘effective’ reverse rate coefficient kr = k-k−1/(k-+ k1). In a well-mixed system, these ‘effective’ rate coefficients correspond to the ‘fundamental’ association and dissociation rate constants (kon and koff). As kf and kr are not always constant, such as in the following situations, the term ‘coefficient’ is to be preferred. When receptors are present at the cell surface, kf and kr are related to kon and koff (respectively) by a factor that depends on kon, the diffusion rate of the ligand (D), the geometry of the system and the amount of free receptors available (N) (Goldstein et al., 1989; Goldstein and Dembo, 1995; Coombs and Goldstein, 2004).
Yet, although essentially overlooked in pharmacology, receptors have only a very restricted diffusion capability when they are confined to cell membranes and even the very presence of the membrane itself already constitutes a hindrance/wall for free 3D diffusion of the ligand. Because of these topological characteristics of the receptor environment, calculations according to biophysical models indicate that ligands are more prone to experience rebinding phenomena than in a situation where the same receptors are uniformly distributed in solution (Berg and Purcell, 1977; DeLisi, 1981; Shoup and Szabo, 1982; Goldstein et al., 1989; Goldstein and Dembo, 1995). Indeed, based on the premises that the membrane constitutes a reflective surface and that the receptors only constitute a minor portion of this surface, most ligand molecules should first hit the membrane at a non-receptor locus. Calculations by Berg and Purcell (1977) according to this model further revealed that, after that initial collision, the ligands are most likely to make several more encounters with non-receptor loci at the membrane before they interact with an associated receptor molecule or drift away. Because of this initial tendency of the ligand to remain in close proximity of the membrane surface, it has a higher probability to hit a receptor within a given time-span as compared with the situation where ligand and receptor molecules are homogeneously dispersed in solution. The same reasoning is obviously also applicable to ligand molecules that freshly dissociated form their cognate receptor and it was calculated that this could already substantially increase the probability of rebinding when the cell surface carried 3500 receptor molecules or more (DeLisi, 1981). In this respect, it is important to realize that the initial binding to and release of a ligand from the receptor (as well as its adsorption into and release from the plasma membrane, see further) is in most cases not a necessary condition for rebinding to take place. Indeed, compared with simply bouncing off the cell wall, binding to a receptor only delays the ligand but it does not affect the subsequent spatial trajectory of the dissociated ligand (Andrews, 2005). It is only if a bi/multidentate ligand can carry out caterpillar-like crawling between adjacent receptors/targets that this initial interaction may count (Levin et al., 2002; Gopalakrishnan et al., 2005).
Based on these and other biophysical considerations, Goldstein and colleagues could equate kf and kr as a function of the ‘fundamental’ rate constants kon and koff for membrane-associated receptor molecules in different spatial contexts, including a whole surface of a spherical cell, a cluster-mimicking disc or the bottom of a synapse-mimicking cylinder (Goldstein and Wiegel, 1983; Goldstein et al., 1989; Posner et al., 1992; Goldstein and Dembo, 1995; Coombs and Goldstein, 2004). In these equations, kf and kr are still proportional to kon and koff respectively (Figure 4). An important property of the proportionality factor (F) is that it is not constant but that it depends on the amount of free receptor molecules (N) at the surface in question. This allows F to account for rebinding phenomena when radioligand dissociation is measured in fresh medium only. Indeed, it is only at full initial receptor occupancy (i.e. when n = 0) that F is equal to one (Figure 4). Then, as more receptors become free with time, F (and, hence, kr) starts to decrease as well. As illustrated by the simulations in Figure 5, this may give rise to (at first sight) only partial radioligand dissociation and lead to interpretations in terms of receptor heterogeneity. Moreover, F depends on kon but not on KD. This distinction is important as it implies that a ligand's aptness to undergo rebinding will depend on its reaction forward rate constant rather than on its affinity for the receptor. Finally, F also depends on the diffusive reverse rate constant (k-, characterizing the flux of ligands away from the receptor-bearing surface), which not only depends on the translational diffusion coefficient (D) of the ligand (receptor diffusion is to slow to be accounted for) but also on geometric characteristics of the receptor environment (Figure 4). In synapses or similar flat cavities, rebinding is promoted by the propensity of dissociated ligand molecules to return to the receptor-bearing surface many times before they manage to diffuse out of the cavity (Coombs and Goldstein, 2004).
Figure 5.
Effect of rebinding on the time-wise decline in receptor occupancy by a slow dissociating ligand (koff = 5.7 × 10−3 min−1, kon = 1 × 108 M−1× min−1) after wash-out of the free ligand. At the start, free and receptor bound ligand is at equilibrium and either 30% (left panel) or 90% (right panel) of the receptors (Rtot) are occupied. Data points were obtained by integrating the differential equations describing a bimolecular ligand-receptor interaction without rebinding (◊ and curve in blue, ‘effective’ reverse rate coefficient kr = koff) or with rebinding (kr = koff/(1 + kon.[R].k-) at low total surface receptor density (•, 3.89 × 1012 receptors·cm−2 and kon.[R].k- = 1.22 and 0.174 at 30 and 90% initial occupancy, respectively) and 20-fold higher surface receptor density (○, kon.[R].k- = 24.3 and 3.47 at 30 and 90% initial occupancy respectively). Note that because of the greater availability of free receptors for ligand rebinding, this process is more outspoken at high total receptor density and low initial receptor occupancy.
Restricted ligand diffusion plays an important role in biology and it is therefore not surprising that alternative mathematical descriptions have also been proposed to model reactions that take place under dimensional constraints such as those at the surface of membranes, in unstirred media and when a large fraction of the volume is taken up by macromolecules (i.e. ‘macromolecular crowding’) (Pang et al., 2007). Among them, the ‘fractal reaction kinetics’ model (Kopelman, 1988) only necessitates a simple adaptation of the classical formalism to describe reversible bimolecular reactions based on unrestricted 3D reactant diffusion. In this model, the association rate is not constant throughout the reaction course but it decreases in an exponential fashion with time: that is, k(t) = ko.t-h, where k(t) and ko is the rate coefficient at time t and t = 0, respectively, and h is a measure of the ‘dimensionality’ of the system. This time-wise decrease in k reflects the tendency of the reactants to undergo spatial segregation when a reaction progresses in an unstirred medium with limited diffusion. Although the use of this equation has been well documented in case of association phenomena (Kopelman, 1988; Schnell and Turner, 2004), it is also likely to apply to rebinding phenomena. Indeed, despite the different form and the heuristic nature of the equation, the ‘fractal reaction kinetics’ approach can depict rebinding phenomena in closely the same way as the approach devised by Goldstein and coworkers (please see legend of Figure 5)
Taken together, ligand rebinding implies that a fraction of the receptor-dissociated ligand molecules choose to bind again rather than to drift away. This may result in consecutive binding of the same ligand molecule to neighbouring receptor molecules in the cell membrane. The said interpretation of the rebinding phenomenon differs from the still prevailing ‘novel mass action equilibrium’ model (Limbird, 1996) that it is a highly localized phenomenon and, hence, does not require the presence of ligand in the aqueous bulk phase.
Physiological relevance of, and traits that promote ligand rebinding
One of the most evocative studies supporting the in vivo relevance of ligand rebinding was presented by Gifford et al. (1998). Following a bolus injection of the D1 dopamine receptor antagonist [3H]-SCH 23390 in rats, the tracer accumulated far more in the striatum as in the cerebellum (the control region with very low D1 dopamine receptor content) and this striatal content was found to decline very slowly with time despite the fast breakdown of the tracer in plasma. Similarly, when the rats were sacrificed soon after the injection, a very slow decrease in the radioligand content of the ‘still living’ striatal slices was noticed in wash-out medium alone. This slow decrease was attributed to rebinding because it could be dramatically enhanced by the presence of an excess of unlabelled ligand. Related experiments, but in where brain was homogenized instead of sliced, failed to show accelerated radioligand release in the presence of unlabelled ligand (Sadée et al., 1982). This was imputed to the disruption of a diffusion boundary next to the receptor sites (receptor micro-compartment) during the homogenization process. Ligand rebinding has also been invoked by many more authors to explain unusually long in vivo binding properties of radioligands as well as positron emission tomography and single photon emission computed tomography radiotracers, but usually without providing further experimenal evidence (e.g. Perry et al., 1980; Frost and Wagner, 1984; Votaw et al., 1993; Frost, 2001).
Drug rebinding is likely to be favoured by the anatomical and physiological complexity of higher organisms. Indeed, biophysical models suggest that this phenomenon is favoured by a high local receptor density, a high reaction forward rate constant and obstacles that hinder the flux of the ligands from the receptor-bearing surface.
Of particular note is that the cell plasma membrane is not a homogenous structure but contains ordered cholesterol-rich microdomains such as lipid rafts with an average diameter in the 100–200 nm range and caveolae that manifest themselves as 50–100 nm ‘flask shaped’ invaginations and also contain cholesterol-binding protein caveolin-1 (Pike, 2003). Many receptors seem to be concentrated in such microdomains along with coupling factors, effector enzymes and substrates. Because of the spatial proximity of the interacting components, signal transduction would occur rapidly and efficiently. β2-adrenoceptors in cardiomyocytes constitute a typical example thereof (Okamoto et al., 1998; Ostrom et al., 2001; Steinberg, 2004). Because it is more likely for a dissociated drug to bind to a nearby receptor/target than to one that is far away, these and other clustering types have been proposed to promote rebinding phenomena (Andrews, 2005).
Rebinding phenomena may also take place in optical biosensors, such as the BIAcore, in where surface-immobilized target molecules are subjected to a flow of soluble target-binding analytes (Nieba et al., 1996). To minimize this effect, low levels of immobilized target as well as high flow rates have been recommended (Karlsson and Falt, 1997). In an elegant study with cell monolayers, Spivak et al. (2006) also noticed that the equilibration between free ligand molecules and their receptors deep within the clefts separating the cells is delayed when compared with the situation at the top surface and that this delay could be minimized by increasing the flow rate. In this respect, it should be noted that in vivo, most receptors are not expected to be subject to any convective stirring of significance. Indeed, they are likely to face rather flat liquid-filled cavities such as neuronal synapses, neuroendocrine and neuromuscular junctions and other interstitial spaces. Moreover, even free circulating immune cells are able to form immunological synapses among themselves as well as with endothelial cells (Taub et al., 1993; Grakoui et al., 1999). In complex tissues, ligand diffusion in and out of such extracellular spaces is also delayed by their need to traverse tortuous paths with eventually blind pockets within the tissue and the fractal networks of the microcirculatory system (Lovich et al., 2001; Dokoumetzidis et al., 2004; Hrabctová and Nicholson, 2004), so that rebinding may take place unabated. Additionally, the extracellular matrix (forming a structural net composed of different types of macromolecules) is also likely to constitute an obstacle to the escape of dissociated ligands by free 3D diffusion (Dityatev and Schacher, 2006; Vargová and Syková, 2008). This latter phenomenon is also referred to ‘macromolecular crowding’ and, in the same vein, the cytoskeleton and high protein content (occupying up to 40% of the cell volume) also constitute diffusion obstacles inside the cell (Hall and Minton, 2003; Rivas et al., 2004; Schnell and Turner, 2004). In this respect, free ligand diffusion in extracellular spaces may also be affected by a number of pathological conditions (Vargová and Syková, 2008). For example, ischaemia/anoxia has been observed to go along with cell swelling in the central nervous system (Sykováet al., 1999; Homola et al., 2006). This swelling causes a substantial reduction in the extracellular space volume and a concomitant increase in the concentration of existing free ligand diffusion barriers therein. Such conditions could also favour ligand rebinding.
Finally, small ligand molecules and peptides with adequate physicochemical properties are able to undergo electrostatic interactions with head groups of membrane phospholipids and reside close to the membrane- solution interface (Kane et al., 2008). If hydrophobic interactions with lipid hydrocarbon chains prevail, ligands may even become completely embedded within the lipid bilayer (Herbette et al., 1988; Sargent et al., 1988). Such ligands are able to undergo 2D diffusion at the cell surface and, because of this ‘reduction of dimensionality’, they are expected to find their receptor targets with greater ease than exclusively via 3D diffusion in solution (Adam and Delbrück, 1968). Moreover, while a molecule may never cross its starting point again if diffusing in dimension D > 2, it is likely to stay in the vicinity of the starting point when the diffusion is restricted to a lower dimension (de Gennes, 1982; Kopelman, 1988; Berry, 2002). Taken together, this restricted diffusion is already likely to favour ligand rebinding phenomena in its own right. Moreover, a number of such ligands have been proposed to gain access to their binding site at their receptor via lateral diffusion between the receptor's membrane-spanning α-helical domains, that is, without having to leave the membrane (reviewed in Vauquelin and Packeu, 2009). Here, two additional mechanisms could act hand in hand with the ‘reduction of dimensionality’ phenomenon to exacerbate the rebinding of such ligands. First, ligand–lipid interactions could exert translational, conformational and orientational constraints on such ligands (Sargent and Schwyzer, 1986; Rhodes et al., 1992; Schwyzer, 1995; Bader et al., 2001). While they have no unique 3D structure in solution, they could acquire well-defined conformations in the membrane that are favourable for receptor binding (Sargent et al., 1988; Contreras et al., 2001). Second, membrane-spanning proteins will also create macromolecular obstacles to the unrestricted 2D-diffusion of such ligands (Berry, 2002). Taken together, rebinding could become very pronounced in the case of membrane-incorporated ligands when the above-mentioned phenomena act together to reduce their diffusive reverse rate constant, k-, and to increase their reaction forward rate constant, k1, for binding to the receptor target.
Rebinding and other local pharmacokinetic mechanisms that prolong drug action
Importantly, by partitioning in the membrane, ligand molecules could acquire a high local concentration therein (Vauquelin and Packeu, 2009). Such ligands could also be released slowly back in the aqueous phase (Figure 1). This combination of properties was proposed to explain the comparatively gentle onset and long-lasting clinical action of the lipophilic dihydropyridine lacidipine despite its short plasma half-life (Herbette, 1994). It also constitutes the core of the ‘diffusion microkinetic’ model that was advanced to explain the long-lasting bronchodilatory effect of β2-adrenoceptor agonists like formoterol and salmeterol. This model stipulates that the plasma membrane can act as a depot/reservoir for the ligand rather than merely functioning as an inert substratum for the receptor (Anderson, 1993). Yet, the diffusion microkinetic model alone failed to explain why, in organ bath experiments, the long-lasting airway smooth muscle dilatory effect of salmeterol was independent of its initial concentration and the magnitude of its effect at the onset of the wash-out (Johnson et al., 1993). To overcome this limitation, it was proposed that this agonist is able to undergo specific and long-lasting binding to auxiliary sites, ‘exosites’, located either at the receptors themselves or in their immediate vicinity in the membrane (Johnson et al., 1993; Johnson and Coleman, 1995; Coleman et al., 1996). This theory was originally proposed by Rocha e Silva (1969) to explain the persistent antagonistic activity at histamine H1 receptors and, more recently, also for the muscarinic acetylcholine receptor agonist xanomeline (Christopoulos et al., 1998). The presence of an exosite implies that, once dissociated from the active site of a receptor, the ligand is not free to diffuse away (Figure 1). As such, the exosite and rebinding models are rather convergent. Indeed, the rebinding model could be regarded to represent a variant of the exosite model if one assumes that clustered receptors are exosites for one another (Szczuka et al., 2009). Alternatively, exosite binding could be regarded to represent an extreme form of rebinding as it calls a halt to the flux of free ligand molecules away from the receptor rather than merely reducing that flux.
If other pharmacokinetic mechanisms fail, the diffusion microkinetic model is certainly sufficient to prolong drug action as long as the plasma membrane (or any other depot close to the receptors) harbours a sufficiently high level of drug molecules. It is only when this mechanism exhausts (i.e. at low drug levels) that additional local pharmacokinetic phenomena like rebinding and/or exosite binding may start to play a preeminent role (Figure 1). To date, no solid experimental evidence has been presented in support of the participation of any of these latter mechanisms in the case of drugs like salmeterol and this may explain why this topic is still a matter of debate (Coleman, 2009; Szczuka et al., 2009). However, to settle this issue, there seems to be a consensus about the need for dedicated studies on simple experimental systems before moving to more complex ones like intact tissues.
Concluding remarks
Whereas pharmacokinetic modelling has traditionally been based on in vivo studies, newer mechanism-based PK/PD models also rely on in vitro observations dealing with drug transport, metabolism, distribution and target binding (Derendorf and Meibohm, 1999; Pang et al., 2007; Ploeger et al., 2009). In this respect, in vitro information about drug-target binding kinetics is hitherto most often based on observations with intact tissues (such as organ bath wash-out experiments) and/or on radioligand binding experiments on isolated cell membrane preparations (Figure 6). Despite the unquestionable physiological relevance of the former approach, intact tissues may be too complex to provide straightforward information about drug-target interaction kinetics as well as pharmacokinetic mechanisms that take place at the sub-cellular/molecular scale (Coleman, 2009; Szczuka et al., 2009). At first sight, experiments on membrane preparations seem to be more appropriate were it not that the so-obtained ligand-receptor dissociation rates sometimes markedly deviate from the rates observed in intact cells (Hara et al., 1998; Fierens et al., 2002; Vauquelin and Packeu, 2009). Little attention has been paid to this issue yet, but many more examples could be discovered if further dedicated comparative studies were to be performed. The rationale for this distinct behaviour is still unclear but a potential hint towards an explanation is that leaky cells already behave membrane-like (Verheijen et al., 2004; Vauquelin and Packeu, 2009). This highlights the fact that membrane receptors (and membrane-associated proteins in general) lose part of their natural environment when a cell is disrupted. This natural environment entails differences between the ionic composition and the redox potential at both sides of the plasma membrane as well as the organizing role of the cytoskeleton. Additionally, receptor molecules that are normally hidden from the cell surface due to their presence in intracellular compartments also become accessible when the cells get disrupted. Intact plated cells seem to offer a compromise between these two in vitro experimental systems (Figure 6). From the physiological point of view, they certainly offer a more relevant environment than the membrane preparations, not only because they are alive but also because, to some extent, they mimic the microanatomic complexity of intact tissues (Spivak et al., 2006; Grießner et al., 2009). Yet, compared with those tissues, they offer far greater experimental flexibility, including the ability to compare radioligand binding and functional data under the same experimental conditions, easy wash-out and the ability to parallel determinations on equivalent samples in each well rather than consecutive determinations on each tissue (Vauquelin et al., 2002). Recombinant cell systems offer additional advantages, including a better focus on the receptors of interest and the use of the naïve parent cells for the detection of receptor-unrelated phenomena. Therefore, they offer a good compromise between the information that can be obtained at the pharmacodynamic level (which requires a simple system) and the local pharmacokinetic level (which requires a tissue-like microanatomical complexity).
Figure 6.
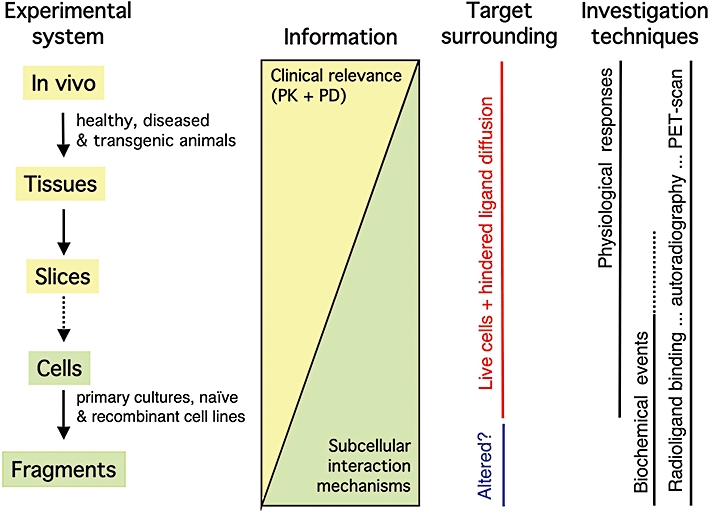
Pharmacological models and approaches: relevance of the obtained information. Nowadays, ligand-receptor/target interactions can be studied with a large variety of pre-clinical experimental systems (from cell fragments such as isolated membranes to in vivo animal models) and techniques (including the direct receptor labeling by radioligands and the measurement of receptor-mediated physiological responses and, by biochemical techniques, the intermediary intracellular signalling cascades). Of note is that the physiological/clinical relevance of the obtained information increases with the complexity of the experimental system. On the other hand, the observations on simple experimental systems are not only faster and cheaper but also easier to interpret in terms of molecular mechanisms. Yet, it is important to be aware that membrane-associated receptors/targets lose part of their natural environment when a cell is disrupted. This may alter their ligand interaction properties. PD, pharmacodynamics; PET, positron emission tomography; PK, pharmacokinetics.
Taken together, long-lasting clinical action of drugs not only depends on their macroscopic pharmacokinetic properties but also on their ability to bring about long-lasting occupancy of their target. Whereas slow dissociation starts to acquire general recognition, rebinding offers an additional but still little investigated option for enhancing the duration of drug action. By mimicking the complexity of tissues, intact cells offer the opportunity to investigate both mechanisms under the same, physiologically relevant conditions.
Acknowledgments
We are most obliged to the Queen Elisabeth Foundation Belgium, the Fonds voor Wetenschappelijk onderzoek Vlaanderen for their kind support.
Glossary
Abbreviations
- 2D and 3D
two-dimensional and three-dimensional
- BSA
bovine serum albumin
- Emax
maximal effect
- GPCR
G protein coupled receptor
- KD
equilibrium dissociation constant for bimolecular drug-target binding
- kf
effective forward rate coefficient
- koff
first-order dissociation rate constant of drug-target complex
- kon
second-order association rate constant of drug-target complex
- kr
effective reverse rate coefficient
- PD
pharmacodynamics
- PET
positron emission tomography
- PK
pharmacokinetics
- SPECT
Single photon emission computed tomography
- τ
residence time of drug-target complex
- t1/2
half-life
Conflict of interest
None.
Supporting Information
Supporting Information: Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Äbelö A, Andersson M, Holmberg AA, Karlsson MO. Application of a combined effect compartment and binding model for gastric acid inhibition of AR-HO47108: a potassium competitive acid blocker, and its active metabolite AR-HO47116 in the dog. Eur J Pharm Sci. 2006;29:91–101. doi: 10.1016/j.ejps.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Äbelö A, Gabrielsson J, Holstein B, Eriksson UG, Holmberg J, Karlsson MO. Pharmacodynamic modelling of reversible gastric acid pump inhibition in dog and man. Eur J Pharm Sci. 2001;14:339–346. doi: 10.1016/s0928-0987(01)00187-7. [DOI] [PubMed] [Google Scholar]
- Adam G, Delbrück M. Reduction of dimensionality in biological diffusion processes. In: Rich A, Davidson WH, editors. Structural Chemistry and Molecular Biology. San Francisco, CA: Freeman & Co.; 1968. pp. 198–215. [Google Scholar]
- Anderson GP. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–2160. doi: 10.1016/0024-3205(93)90729-m. [DOI] [PubMed] [Google Scholar]
- Andrews SS. Serial rebinding of ligands to clustered receptors as exemplified by bacterial chemotaxis. Phys Biol. 2005;2:111–122. doi: 10.1088/1478-3975/2/2/004. [DOI] [PubMed] [Google Scholar]
- Anthes JC, Gilchrest H, Richard C, Eckel S, Hesk D, West RE, et al. Biochemical characterization of desloratadine, a potent antagonist of the human histamine H(1) receptor. Eur J Pharmacol. 2002;449:229–237. doi: 10.1016/s0014-2999(02)02049-6. [DOI] [PubMed] [Google Scholar]
- Ariens EJ, Van Rossum JM, Simmonis AM. A theoretical basis of molecular pharmacology. Arzneimittel Forsch. 1956;6:611–621. [PubMed] [Google Scholar]
- Austin RP, Barton P, Bonnert RV, Brown RC, Cage PA, Cheshire DR, et al. QSAR and the rational design of long-acting dual D2-receptor/beta2-adrenoceptor agonists. J Med Chem. 2003;46:3210–3320. doi: 10.1021/jm020886c. [DOI] [PubMed] [Google Scholar]
- Bader R, Bettio A, Beck-Sickinger AG, Zerbe O. Structure and dynamics of micelle-bound neuropeptide Y: comparison with unligated NPY and implications for receptor selection. J Mol Biol. 2001;305:307–329. doi: 10.1006/jmbi.2000.4264. [DOI] [PubMed] [Google Scholar]
- Berg HC, Purcell EM. Physics of chemoreception. Biophys J. 1977;20:193–219. doi: 10.1016/S0006-3495(77)85544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry H. Monte Carlo simulations of enzyme reactions in two dimensions: fractal kinetics and spatial segregation. Biophys J. 2002;83:1891–1901. doi: 10.1016/S0006-3495(02)73953-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Cassel JA, Daubert JD, DeHaven RN. [(3)H]Alvimopan binding to the micro opioid receptor: comparative binding kinetics of opioid antagonists. Eur J Pharmacol. 2005;520:29–36. doi: 10.1016/j.ejphar.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Chapman RW, Minnicozzi M, Celly CS, Phillips JE, Kung TT, Hipkin RW, et al. A novel, orally active CXCR1/2 receptor antagonist, Sch527123, inhibits neutrophil recruitment, mucus production, and goblet cell hyperplasia in animal models of pulmonary inflammation. J Pharmacol Exp Ther. 2007;322:486–493. doi: 10.1124/jpet.106.119040. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2003;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Pierce TL, Sorman JL, El-Fakahany EE. On the unique binding and activating properties of xanomeline at the M1 muscarinic acetylcholine receptor. Mol Pharmacol. 1998;53:1120–1130. [PubMed] [Google Scholar]
- Chu CL, Buczek-Thomas JA, Nugent MA. Heparan sulfate proteoglycans modulate fibroblast growth factor-2 binding through a lipid-raft-mediated mechanism. Biochem J. 2004;379:331–341. doi: 10.1042/BJ20031082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA. On the mechanism of the persistent action of salmeterol: what is the current position? Brit J Pharmacol. 2009;158:180–182. doi: 10.1111/j.1476-5381.2009.00370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Johnson M, Nials AT, Vardey CJ. Exosites: their current status, and their relevance to the duration of action of long-acting beta2-adrenoceptor agonists. Trends Pharmacol Sci. 1996;17:324–330. [PubMed] [Google Scholar]
- Contreras LL, de Almeida RFM, Villalaín J, Fedorov A, Prieto M. Interaction of α-melanocyte stimulating hormone with binary phospholipid membranes: structural changes and relevance of phase behavior. Biophys J. 2001;80:2273–2283. doi: 10.1016/S0006-3495(01)76199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs D, Goldstein B. Effects of geometry of the immunological synapse on the delivery of effector molecules. Biophys J. 2004;87:2215–2220. doi: 10.1529/biophysj.104.045674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Disc. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Csajka C, Verotta D. Pharmacokinetic-pharmacodynamic modelling: history and perspectives. J Pharmacokinet Pharmacodyn. 2006;33:227–279. doi: 10.1007/s10928-005-9002-0. [DOI] [PubMed] [Google Scholar]
- de Gennes PG. Kinetics of diffusion-controlled processes in dense polymer systems. J Chem Phys. 1982;76:3316–3321. [Google Scholar]
- De Meyts P, Bianco R, Roth J. Site-site interactions among insulin receptors. Characterization of the negative cooperativity. J Biol Chem. 1977;251:1877–1888. [PubMed] [Google Scholar]
- DeLisi C. The effect of cell size and receptor density on ligand -receptor reaction rate constants. Mol Immunol. 1981;18:507–511. doi: 10.1016/0161-5890(81)90128-0. [DOI] [PubMed] [Google Scholar]
- DeLisi C, Wiegel FW. Effect of nonspecific forces and finite receptor number on rate constants of ligand-cell bound-receptor interactions. Proc Natl Acad Sci U S A. 1981;78:5569–5572. doi: 10.1073/pnas.78.9.5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Paschoa OE, Mandema JW, Voskuyl RA, Danhof M. Pharmacokinetic-pharmacodynamic modeling of the anticonvulsant and electroencephalogram effects of phenytoin in rats. J Pharmacol Exp Ther. 1998;284:460–466. [PubMed] [Google Scholar]
- Derendorf H, Lesko LJ, Chaikin P, Colburn WA, Lee P, Miller R, et al. Pharmacokinetoc/pharmacodynamic modeling in drug research and development. J Clin Pharmacol. 2000;40:1399–1418. [PubMed] [Google Scholar]
- Derendorf H, Meibohm B. Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm Res. 1999;16:176–185. doi: 10.1023/a:1011907920641. [DOI] [PubMed] [Google Scholar]
- Deutch AY. Prefrontal cortical dopamine systems and the elaboration of functional corticostriatal circuits: implications for schizophrenia and Parkinson's disease. J Neural Transm Gen Sect. 1993;91:197–221. doi: 10.1007/BF01245232. [DOI] [PubMed] [Google Scholar]
- Disse B, Speck GA, Rominger KL, Witek TJ, Jr, Hammer R. Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci. 1999;64:457–464. doi: 10.1016/s0024-3205(98)00588-8. [DOI] [PubMed] [Google Scholar]
- Dityatev A, Schacher M. The extracellular matrix and synapses. Cell Tissue Res. 2006;326:647–654. doi: 10.1007/s00441-006-0217-1. [DOI] [PubMed] [Google Scholar]
- Dokoumetzidis A, Karalis V, Iliadis A, Macheras P. The heterogeneous course of drug transit through the body. Trends Pharmacol Sci. 2004;25:140–146. doi: 10.1016/j.tips.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Dowling MR, Charlton SJ. Quantifying the association and dissociation rates of unlabelled antagonists at the muscarinic M3 receptor. Br J Pharmacol. 2006;148:1134–1142. doi: 10.1038/sj.bjp.0706819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierens FLP, Vanderheyden PML, De Backer J-P, Vauquelin G. Binding of the antagonist [3H]candesartan to angiotensin II AT1 receptor-transfected Chinese hamster ovary cells. Eur J Pharmacol. 1999;367:413–422. doi: 10.1016/s0014-2999(98)00965-0. [DOI] [PubMed] [Google Scholar]
- Fierens F, Vanderheyden PML, Roggeman C, Vande Gucht P, De Backer J-P, Vauquelin G. Distinct binding properties of the AT1 receptor antagonist [3H]candesartan to intact cells and membrane preparations. Biochem Pharmacol. 2002;63:1273–1279. doi: 10.1016/s0006-2952(02)00859-6. [DOI] [PubMed] [Google Scholar]
- Frost JJ. PET imaging of the opioid receptor: the early years. Nucl Med Biol. 2001;28:509–513. doi: 10.1016/s0969-8051(01)00221-9. [DOI] [PubMed] [Google Scholar]
- Frost JJ, Wagner HR., Jr Kinetics of binding to opiate receptors in vivo predicted from in vitro parameters. Brain Res. 1984;305:1–11. doi: 10.1016/0006-8993(84)91113-2. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res. 1966;3:21–55. [Google Scholar]
- Germain RN. T cell signaling: the importance of receptor clustering. Curr Biol. 1997;7:R640–R644. doi: 10.1016/s0960-9822(06)00323-x. [DOI] [PubMed] [Google Scholar]
- Gifford AN, Gatley J, Volkow ND. Evaluation of the importance of rebinding to receptors in slowing the approach to equilibrium of high-affinity PET and SPECT radiotracers. Synapse. 1998;28:167–175. doi: 10.1002/(SICI)1098-2396(199802)28:2<167::AID-SYN7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Gill R, Verma C, Wallach B, Urso B, Pitts J, Wollmer A, et al. Modelling of the disulphide-swapped isomer of human insulin-like growth factor-1: implications for receptor binding. Protein Eng. 1999;12:297–303. doi: 10.1093/protein/12.4.297. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Dembo M. Approximating the effects of diffusion on reversible reactions at the cell surface: ligand-receptor kinetics. Biophys J. 1995;68:1222–1230. doi: 10.1016/S0006-3495(95)80298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein B, Posner RG, Torney DC, Erickson J, Holowka D, Baird B. Competition between solution and cell surface receptors for ligand. Dissociation of hapten bound to surface antibody in the presence of solution antibody. Biophys J. 1989;56:955–966. doi: 10.1016/S0006-3495(89)82741-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein B, Wiegel F. The effect of receptor clustering on diffusion-limited forward rate constants. Biophys J. 1983;43:121–125. doi: 10.1016/S0006-3495(83)84330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsiorek W, Fan X, Hesk D, Fossetta J, Qiu H, Jakway J, et al. Pharmacological characterization of Sch527123, a potent allosteric CXCR1/CXCR2 antagonist. J Pharmacol Exp Ther. 2007;322:477–485. doi: 10.1124/jpet.106.118927. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Forsten-Williams K, Nugent MA, Täuber UC. Effects of receptor clustering on ligand dissociation kinetics: theory and simulations. Biophys J. 2005;89:3686–3700. doi: 10.1529/biophysj.105.065300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- Grießner M, Bröker P, Lehmann A, Ehrentreich-Förster E, Bier F. Detection of angiotensin II type 1 receptor ligands by a cell-based assay. Anal Bioanal Chem. 2009;395:1937–1940. doi: 10.1007/s00216-009-3074-4. [DOI] [PubMed] [Google Scholar]
- Hale JJ, Mills SG, MacCoss M, Finke PE, Cascieri MA, Sadowski S, et al. Structural optimization affording 2-(R)-(1-(R)-3, 5-bis(trifluoromethyl)phenylethoxy)-3-(S)-(4-fluoro) phenyl-4-(3-oxo-1,2,4-triazol-5-yl)methylmorpholine, a potent, orally active, long-acting morpholine acetal human NK-1 receptor antagonist. J Med Chem. 1998;41:4607–4614. doi: 10.1021/jm980299k. [DOI] [PubMed] [Google Scholar]
- Hall D, Minton AP. Macromolecular crowding: qualitative and semiquantitative successes, quantitative challenges. BBA-Proteins Proteomics. 2003;1649:127–139. doi: 10.1016/s1570-9639(03)00167-5. [DOI] [PubMed] [Google Scholar]
- Hara M, Tozawa F, Itazaki K, Mihara S, Fujimoto M. Endothelin ET(B) receptors show different binding profiles in intact cells and cell membrane preparations. Eur J Pharmacol. 1998;345:339–342. doi: 10.1016/s0014-2999(98)00016-8. [DOI] [PubMed] [Google Scholar]
- Herbette LG. Membrane pathways for drug/ion channel interactions: molecular basis for pharmacokinetic properties. Drug Devel Res. 1994;33:214–222. [Google Scholar]
- Herbette LG, Trumbore M, Chester DW, Katz AM. Possible molecular basis for the pharmacokinetics and pharmacodynamics of three membrane-active drugs: propranolol, nimodipine and amiodarone. J Mol Cell Cardiol. 1988;20:373–378. doi: 10.1016/s0022-2828(88)80128-7. [DOI] [PubMed] [Google Scholar]
- Hill AV. The mode of action of nicotine and curare, determined by the form of the contraction curve and the method of temperature coefficients. J Physiol. 1909;39:361–373. doi: 10.1113/jphysiol.1909.sp001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holford NHG, Sheiner LB. Kinetics of pharmacological response. Pharmacol Ther. 1982;16:143–166. doi: 10.1016/0163-7258(82)90051-1. [DOI] [PubMed] [Google Scholar]
- Homola A, Zoremba N, Slais K, Kuhlen R, Syková E. Changes in diffusion parameters, energy-related metabolites and glutamate in the rat cortex after transient hypoxia/ischemia. Neurosci Lett. 2006;404:137–142. doi: 10.1016/j.neulet.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Hrabctová S, Nicholson C. Contribution of dead-space microdomains to tortuosity of brain extracellular space. Neurochem Int. 2004;45:467–477. doi: 10.1016/j.neuint.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Johnson M, Butchers PR, Coleman RA, Nials AT, Strong P, Sumner MJ, et al. The pharmacology of salmeterol. Life Sci. 1993;52:2131–2143. doi: 10.1016/0024-3205(93)90728-l. [DOI] [PubMed] [Google Scholar]
- Johnson M, Coleman RA. Mechanisms of action of β2-adrenoceptor agonists. In: Busse WW, Holgate ST, editors. Asthma & Rhinitis. Cambridge: Blackwell; 1995. pp. 1278–1295. [Google Scholar]
- Kane BE, Grant MKO, El-Fakahany EE, Ferguson DSM. Synthesis and evaluation of xanomeline analogs – probing the wash-resistant phenomenon at the M1 muscarinic acetylcholine receptor. Bioorg Med Chem. 2008;16:1376–1392. doi: 10.1016/j.bmc.2007.10.058. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J Psychiatry Neurosci. 2000;25:161–166. [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Seeman P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics? A new hypothesis. Am J Psychiatry. 2001;158:360–369. doi: 10.1176/appi.ajp.158.3.360. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Falt R. Experimental design for the kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J Immunol Meth. 1997;200:121–133. doi: 10.1016/s0022-1759(96)00195-0. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Seven transmembrane receptors as nature's prototype allosteric protein: de-emphasizing the geography of binding. Mol Pharmacol. 2008;74:541–543. doi: 10.1124/mol.108.050062. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Jenkinson S, Watson C. Determining the potency and molecular mechanism of action of insurmountable antagonists. J Pharmacol Exp Ther. 2006;319:710–723. doi: 10.1124/jpet.106.107375. [DOI] [PubMed] [Google Scholar]
- Kopelman R. Fractal reaction kinetics. Science. 1988;241:1620–1626. doi: 10.1126/science.241.4873.1620. [DOI] [PubMed] [Google Scholar]
- Krejci E, Martinez-Pena y Valenzuela I, Ameziane R, Akaaboune M. Acetylcholinesterase dynamics at the neuromuscular junction of live animals. J Biol Chem. 2006;281:10347–10354. doi: 10.1074/jbc.M507502200. [DOI] [PubMed] [Google Scholar]
- Le MT, Pugsley M, Vauquelin G, Van Liefde I. Angiotensin II AT1 antagonism and insurmountability: comparison of olmesartan and telmisartan. Br J Pharmacol. 2007;151:952–962. doi: 10.1038/sj.bjp.0707323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff P, Martin GR. Peripheral 5-HT2-like receptors. Can they be classified with the available antagonists? Br J Pharmacol. 1986;88:585–593. doi: 10.1111/j.1476-5381.1986.tb10239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin MD, Shimizu TS, Bray D. Binding and diffusion of CheR molecules within a cluster of membrane receptors. Biochem J. 2002;82:1809–1817. doi: 10.1016/S0006-3495(02)75531-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew MJ, Christtopoulos A, Ziogas J. Non-surmountable antagonism: transcending steady state. Trends Pharmacol Sci. 2001;22:65–66. [Google Scholar]
- Lew MJ, Ziogas J, Christopoulos A. Dynamic mechanisms of non-classical antagonism by competitive AT1 receptor antagonists. Trends Pharmacol Sci. 2000;21:376–381. doi: 10.1016/s0165-6147(00)01523-6. [DOI] [PubMed] [Google Scholar]
- Leysen JE, Gommeren W. Drug-receptor dissociation time, new tool for drug research: receptor binding affinity and drug-receptor dissociation profiles of serotonin-S2, dopamine-D2, histamine-H1 antagonists, and opiates. Drug Dev Res. 1986;8:119–131. [Google Scholar]
- Leysen JE, Gommeren W. Drug-receptor dissociation time, new tool for drug research: receptor binding affinity and drug-receptor dissociation profiles of serotonin-S2, Dopamine-D2, histamine-H1 antagonists, and opiates. Drug Devel Res. 2004;8:119–131. [Google Scholar]
- Limbird LE. Cell Surface Receptors: A Short Course in Theory and Methods. Boston, MA: Kluwer Academic Publishers; 1996. [Google Scholar]
- Limbird LE, Lefkowitz RJ. Negative cooperativity among β-adrenergic receptors in frog erythrocyte membrane. J Biol Chem. 1976;251:5007–5014. [PubMed] [Google Scholar]
- Lindström E, von Mentzer B, Påhlman I, Ahlstedt I, Uvebrant A, Kristensson E, et al. Neurokinin (NK)1 receptor antagonists: correlation between in vitro receptor interaction and in vivo efficacy. J Pharmacol Exp Ther. 2007;322:1286–1293. doi: 10.1124/jpet.107.124958. [DOI] [PubMed] [Google Scholar]
- Lou J, Yago T, Klopocki AG, Mehta P, Chen W, Zarnitsyna VI. Flow-enhanced adhesion regulated by a selectin interdomain hinge. J Cell Biol. 2006;174:1107–1117. doi: 10.1083/jcb.200606056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovich MA, Creel C, Hong K, Hwang CW, Edelman ER. Carrier proteins determine local pharmacokinetics and arterial distribution of paclitaxel. J Pharm Sci. 2001;90:1324–1335. doi: 10.1002/jps.1085. [DOI] [PubMed] [Google Scholar]
- Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Disp. 2003;31:510–519. doi: 10.1124/dmd.31.5.510. [DOI] [PubMed] [Google Scholar]
- Meibohm B, Derendorf H. Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modeling. Int J Clin Pharmacol Therap. 1997;35:401–413. [PubMed] [Google Scholar]
- Morsing P, Adler G, Brandt-Eliasson U, Karp L, Ohlson K, Renberg L, et al. Mechanistic differences of various AT1-receptor blockers in isolated vessels of different origin. Hypertension. 1999;33:1406–1413. doi: 10.1161/01.hyp.33.6.1406. [DOI] [PubMed] [Google Scholar]
- Nieba L, Krebber A, Plückthun A. Competition BIAcore for measuring true affinities: large differences from values determined from binding kinetics. Anal Biochem. 1996;234:155–165. doi: 10.1006/abio.1996.0067. [DOI] [PubMed] [Google Scholar]
- Nocker RE, Schoonbrood DF, van de Graaf EA, Hack CE, Lutter R, Jansen HM, et al. Interleukin-8 in airway inflammation in patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 1996;109:183–191. doi: 10.1159/000237218. [DOI] [PubMed] [Google Scholar]
- Ojima M, Inada Y, Shibouta Y, Wada T, Sanada T, Kubo K, et al. Candesartan (CV-11974) dissociates slowly from the angiotensin ATl receptor. Eur J Pharmacol. 1997;319:137–146. doi: 10.1016/s0014-2999(96)00837-0. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing ‘preassembled signaling complexes’ at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J Biol Chem. 2001;276:42063–42069. doi: 10.1074/jbc.M105348200. [DOI] [PubMed] [Google Scholar]
- Packeu A, De Backer J-P, Van Liefde I, Vanderheyden P, Vauquelin G. Antagonist-dopamine D2L-receptor interactions in intact cells. Biochem Pharmacol. 2008;75:2192–2203. doi: 10.1016/j.bcp.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Pang KS, Weiss M, Macheras P. Advanced pharmacokinetic models based on clearance, circulatory and fractal concepts. AAPS J. 2007;9:E268–E283. doi: 10.1208/aapsj0902030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park LS, Friend DJ, Schmierer AE, Dower SK, Namen AE. Murine interleukin 7 (IL-7) receptor. Characterization on an IL-7-dependent cell line. J Exp Med. 1990;171:1073–1089. doi: 10.1084/jem.171.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Mullis KB, Øie S, Sadée W. Opiate antagonist receptor binding in vivo: evidence for a new receptor binding model. Brain Res. 1980;199:49–61. doi: 10.1016/0006-8993(80)90229-2. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- Ploeger BA, van der Graaf PH, Danhof M. Incorporating receptor theory in mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling. Drug Metab Pharmacokinet. 2009;24:3–15. doi: 10.2133/dmpk.24.3. [DOI] [PubMed] [Google Scholar]
- Posner RG, Le BJ, Conrad DH, Holowka D, Baird B, Goldstein B. Aggregation of IgE-receptor complexes on rat basophilic leukemia cells does not change the intrinsic affinity but can alter the kinetics of the ligand-IgE interaction. Biochem. 1992;31:5350–5356. doi: 10.1021/bi00138a015. [DOI] [PubMed] [Google Scholar]
- Powell-Jones CHJ, Thomas CG, Jr, Nayfeh S. Thyrotropin receptors in normal human thyroid: nonclassical binding kinetics not explained by the negative cooperativity model. J Biol Chem. 1979;255:4001–4010. [PubMed] [Google Scholar]
- Rhodes DG, Newton R, Butler R, Herbette L. Equilibrium and kinetic studies of the interactions of salmeterol with membrane bilayers. Mol Pharmacol. 1992;42:596–602. [PubMed] [Google Scholar]
- Rivas G, Ferrone F, Herzfeld J. Life in a crowded world. EMBO Reports. 2004;5:23–27. doi: 10.1038/sj.embor.7400056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha e Silva M. A thermodynamic approach to problems of drug antagonism I. The ‘Charnière theory’. Eur J Pharmacol. 1969;6:294–302. doi: 10.1016/0014-2999(69)90188-5. [DOI] [PubMed] [Google Scholar]
- Sadée W, Perry DC, Rosenbaum JS, Herz A. [3H]-diprenorphine receptor binding in vivo and in vitro. Eur J Pharmacol. 1982;81:431–440. doi: 10.1016/0014-2999(82)90108-x. [DOI] [PubMed] [Google Scholar]
- Sargent DF, Bean JW, Schwyzer R. Conformation and orientation of regulatory peptides on lipid membranes. Key to the molecular mechanism of receptor selection. Biophys Chem. 1988;31:183–193. doi: 10.1016/0301-4622(88)80024-3. [DOI] [PubMed] [Google Scholar]
- Sargent DF, Schwyzer R. Membrane lipid phase as catalyst for peptide-receptor interactions. Proc Natl Acad Sci U S A. 1986;83:5774–5778. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell S, Turner TE. Reaction kinetics in intracellular environments with macromolecular crowding: simulations and rate laws. Progr Biophys Mol Biol. 2004;85:235–260. doi: 10.1016/j.pbiomolbio.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. 100 years lock-and-key concept: are peptide keys shaped and guided to their receptors by the target cell membrane? Biopolymers. 1995;37:5–16. doi: 10.1002/bip.360370104. [DOI] [PubMed] [Google Scholar]
- Sheiner LB, Stanski DR, Vozeh S, Miller RD, Ham J. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin Pharmacol Ther. 1979;25:358–371. doi: 10.1002/cpt1979253358. [DOI] [PubMed] [Google Scholar]
- Shimada S, Nakajima Y, Yamamoto K, Sawada Y, Iga T. Comparative pharmacodynamics of eight calcium channel blocking agents in Japanese essential hypertensive patients. Biol Pharm Bull. 1996;19:430–437. doi: 10.1248/bpb.19.430. [DOI] [PubMed] [Google Scholar]
- Shoup D, Szabo A. Role of diffusion in ligand binding to macromolecules and cell-bound receptors. Biophys J. 1982;40:33–39. doi: 10.1016/S0006-3495(82)84455-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy TJ, Szmelcman S, Boos W, Schwartz M. On the significance of the retention of ligand by protein. Proc Natl Acad Sci USA. 1975;72:2120–2124. doi: 10.1073/pnas.72.6.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JR, Zunszain PA, Hamilton JA, Curry S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J Mol Biol. 2006;361:336–351. doi: 10.1016/j.jmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Slack R, Browning C, Hall D, Begg D, Clark K. GSK1004723, a novel histamine H1/H3 receptor antagonist exhibiting slow H1 dissociation kinetics. 2009. Proceedings of the British Pharmacological Society. Available at: http://www.pA2online.org/abstracts/Vol7Issue4abst152P.pdf (accessed 8 July 2010.
- Spivak CE, Oz M, Beglan CL, Shrager RI. Diffusion delays and unstirred layer effects at monolayer cultures of Chinese hamster ovary cells: radioligand binding, confocal microscopy, and mathematical simulations. Cell Biochem Biophys. 2006;45:43–58. doi: 10.1385/CBB:45:1:43. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Beta(2)-adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol. 2004;37:407–415. doi: 10.1016/j.yjmcc.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Sutter A, Riopelle RG, Harris-Warrick RM, Shooter EM. Nerve growth factor receptors. Characterization of two distinct classes of binding sites on chick embryo sensory ganglia cells. J Biol Chem. 1979;254:5972–5982. [PubMed] [Google Scholar]
- Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004;3:801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- Swinney DC. Biochemical mechanisms of new molecular entities (NMEs) approved by United States FDA during 2001–2004: mechanisms leading to optimal efficacy and safety. Curr Top Med Chem. 2006a;6:461–478. doi: 10.2174/156802606776743093. [DOI] [PubMed] [Google Scholar]
- Swinney DC. Can binding kinetics translate to a clinically differentiated drug? From theory to practice. Lett Drug Des Disc. 2006b;3:569–574. [Google Scholar]
- Swinney DC. Applications of binding kinetics to drug discovery: translation of binding mechanisms to clinically differentiated therapeutic responses. Int J Pharm Med. 2008;22:23–34. [Google Scholar]
- Swinney DC. The role of binding kinetics in therapeutically useful drug action. Curr Opin Drug Discovery Dev. 2009;12:31–39. [PubMed] [Google Scholar]
- Syková E, Vargová L, Prokopová S, Simonová Z. Glial swelling and astrogliosis produce diffusion barriers in the rat spinal cord. Glia. 1999;25:56–70. doi: 10.1002/(sici)1098-1136(19990101)25:1<56::aid-glia6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Szczuka A, Packeu A, Wennerberg M, Vauquelin G. Molecular mechanism of the persistent bronchodilatory effect of the partial β2-adrenoceptor agonist salmeterol. Br J Pharmacol. 2009;158:183–194. doi: 10.1111/j.1476-5381.2009.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, et al. Recombinant human interferon–inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas W. Catch bonds, it all hinges on the interdomain region. J Cell Biol. 2006;174:911–913. doi: 10.1083/jcb.200609029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57:590–595. doi: 10.1136/thorax.57.7.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Urizar E, Montanelli L, Loy T, Bonomi M, Swillens S, Gales C, et al. Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 2005;24:1954–1964. doi: 10.1038/sj.emboj.7600686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Liefde I, Vauquelin G. Sartan-AT1 receptor interactions: in vitro evidence for insurmountable antagonism and inverse agonism. Mol Cell Endocrinol. 2009;302:237–243. doi: 10.1016/j.mce.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Vanderheyden PML, Fierens FLP, De Backer J-P, Fraeyman N, Vauquelin G. Distinction between surmountable and insurmountable selective AT1 receptor antagonists by use of CHO-K1 cells expressing human angiotensin II AT1 receptors. Br J Phamacol. 1999;126:1057–1065. doi: 10.1038/sj.bjp.0702398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargová L, Syková E. Extracellular space diffusion and extrasynaptic transmission. Physiol Res. 2008;57:S89–S99. doi: 10.33549/physiolres.931603. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Fierens F, Verheyen I, Vanderheyden P. Insurmountable AT1 receptor antagonism: the need for different antagonist binding states of the receptor. Trends Pharmacol Sci. 2001;22:343–344. doi: 10.1016/s0165-6147(00)01739-9. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Packeu A. Ligands, their receptors and … plasma membranes. Mol Cell Endocrinol. 2009;311:1–10. doi: 10.1016/j.mce.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Szczuka A. Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Neurochem Int. 2007;51:254–260. doi: 10.1016/j.neuint.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I. Slow antagonist dissociation and long-lasting in vivo receptor protection. Trends Pharmacol Sci. 2006;27:356–359. doi: 10.1016/j.tips.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I, Vanderheyden P. Models and methods for studying insurmountable antagonism. Trends Pharmacol Sci. 2002;23:514–518. doi: 10.1016/s0165-6147(02)02081-3. [DOI] [PubMed] [Google Scholar]
- Verheijen I, Tourlousse D, Vanderheyden PML, De Backer JP, Vauquelin G. Effect of saponin and filipin on antagonist binding to AT1 receptors in intact cells. Biochem Pharmacol. 2004;67:601–606. doi: 10.1016/j.bcp.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Votaw JR, Kessler RM, de Paulis T. Failure of the three compartment model to describe the pharmacokinetics in brain of a high affinity substituted benzamide. Synapse. 1993;15:177–190. doi: 10.1002/syn.890150303. [DOI] [PubMed] [Google Scholar]
- Wallmark B, Brändström A, Larsson H. Evidence for acid-induced transformation of omeprazole into an active inhibitor of (H++ K+)-ATPase within the parietal cell. Biochim Biophys Acta. 1984;778:549–558. doi: 10.1016/0005-2736(84)90406-1. [DOI] [PubMed] [Google Scholar]
- Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67:1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- Wennerberg M, Ballendran A, Clapham J, Vauquelin G. Unravelling the complex dissociation of [3H]-rimonabant from plated CB1 cannabinoid receptor- expressing cells. Fund Clin Pharmacol. 2010;24:181–187. doi: 10.1111/j.1472-8206.2009.00756.x. [DOI] [PubMed] [Google Scholar]
- Wiley HS. Anomalous binding of epidermal growth factor to A431 cells is due to the effect of high receptor densities and a saturable endocytic system. J Cell Biol. 1988;107:801–810. doi: 10.1083/jcb.107.2.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassen A, Olofsen E, Dahan A, Danhof M. Pharmacokinetic-pharmacodynamic modeling of the antinociceptive effect of buprenorphine and fentanyl in rats: role of receptor equilibration kinetics. J Pharmacol Exp Ther. 2005;313:1136–1140. doi: 10.1124/jpet.104.082560. [DOI] [PubMed] [Google Scholar]
- Yun H-Y, Yun M-H, Kang W, Kwon KI. Pharmacokinetics and pharmacodynamics of benedipine using a slow receptor binding model. J Clin Pharmacol Ther. 2005;30:541–547. doi: 10.1111/j.1365-2710.2005.00682.x. [DOI] [PubMed] [Google Scholar]
- Zhang R, Monsma F. The importance of drug-target residence time. Curr Opin Drug Discov Devel. 2009;12:488–496. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.