

Abstract
Identification of the signaling pathways that control the proliferation of stem cells (SCs), and whether they act in a cell or non-cell autonomous manner, is key to our understanding of tissue homeostasis and cancer. In the adult Drosophila midgut, the Jun N-Terminal Kinase (JNK) pathway is activated in damaged enterocyte cells (ECs) following injury. This leads to the production of Upd cytokines from ECs, which in turn activate the Janus kinase (JAK)/Signal transducer and activator of transcription (STAT) pathway in Intestinal SCs (ISCs), stimulating their proliferation. In addition, the Hippo pathway has been recently implicated in the regulation of Upd production from the ECs. Here, we show that the Hippo pathway target, Yorkie (Yki), also plays a crucial and cell-autonomous role in ISCs. Activation of Yki in ISCs is sufficient to increase ISC proliferation, a process involving Yki target genes that promote division, survival and the Upd cytokines. We further show that prior to injury, Yki activity is constitutively repressed by the upstream Hippo pathway members Fat and Dachsous (Ds). These findings demonstrate a cell-autonomous role for the Hippo pathway in SCs, and have implications for understanding the role of this pathway in tumorigenesis and cancer stem cells.
Keywords: Hippo pathway, Intestinal stem cell, Regeneration, Drosophila
INTRODUCTION
SCs underlie the turnover of differentiated cells in many adult tissues (Blanpain and Fuchs, 2009; Reya et al., 2001). In the absence of damage, the baseline level at which SCs replace differentiated cells can be thought of as `normal homeostasis'. After an acute injury when many cells are simultaneously lost, these are replaced expediently by an increase in SC or transit-amplifying progenitor cell division (Blanpain and Fuchs, 2009; Ito et al., 2005; Jiang et al., 2009). This accelerated cell production, which can be thought of as `acute regeneration', allows the damaged tissue to return to its original size and to its normal homeostatic state. The transition from normal homeostasis to acute regeneration occurs quickly and often during the lifetime of an animal, as tissues can be damaged by exposure to pathogens, chemical and mechanical injuries, and disease conditions. Yet, the cell biology and molecular mechanisms that mediate this switch are poorly understood.
Understanding how SCs stop dividing during tissue regeneration is of fundamental importance, not only to regenerative medicine, but also in understanding the evasion of growth control during oncogenesis (Hanahan and Weinberg, 2000). In recent years, the Hippo signaling pathway has emerged as an evolutionarily conserved pathway intimately connected to the regulation of organ growth and cancer (Saucedo and Edgar, 2007). Genetic studies in Drosophila have identified a number of its signaling components and provided a framework of the structure of this pathway. Activation of the Serine/Threonine kinase Hippo (Hpo) leads to the suppression of the transcriptional co-activator Yorkie (Yki) (Dong et al., 2007; Huang et al., 2005). Overexpression of Yki or mutations in any one of the Hippo pathway members, such as the Serine/Threonine kinase Warts (Wts), lead to cellular proliferation coupled with resistance to cell death (Hamaratoglu et al., 2006; Saucedo and Edgar, 2007; Willecke et al., 2006; Wu et al., 2003b). Hence, the current model is that Hippo signaling acts to prevent Yki from causing tissue hyperplasia. The most upstream components of the pathway, Fat and Ds, are atypical cadherins that share limited similarities with classic Type 1 cadherins (Bennett and Harvey, 2006; Halbleib and Nelson, 2006; Willecke et al., 2006). Fat is thought to be a receptor for Ds, and one model of Hippo signaling pathway is that their binding initiates signaling in the cells expressing Fat (Matakatsu and Blair, 2004; Rogulja et al., 2008; Willecke et al., 2008). This model provides insight into how a cell might sense its surrounding neighbors and simultaneously regulate its growth and division.
Insensitivity to growth control itself is a defining feature of cancer (Hanahan and Weinberg, 2000), thus placing the Hippo pathway as an important candidate in tumorigenesis. Indeed, a number of recent studies support this notion. The human orthologs of Drosophila Yki, YAP1/2 are oncogenes (Zender et al., 2006), the overexpression of which has been linked to hyperplasia in the liver and small intestine of mice (Camargo et al., 2007; Dong et al., 2007). Mammalian orthologs of the Hippo pathway have conserved roles and their disruption also induces growth (Zeng and Hong, 2008). For example, mice mutant for Lats, the ortholog of Drosophila Wts, develop ovarian tumors and soft-tissue sarcomas (St John et al., 1999) and mutations in Mst1/2, the ortholog of Drosophila Hpo, lead to liver tumors (Zhou et al., 2009). These studies underscore the importance of this pathway in controlling growth in cancer, but it is unclear how Hippo growth control occurs constitutively in adult tissues and what its role might be therein. Furthermore, whereas Type 1 cadherins have been found to play important roles in SC biology (Song et al., 2002), the roles of atypical cadherin components of the Hippo pathway in SCs have not yet been investigated in detail.
To explore the role of the Hippo pathway in SCs specifically, we decided to examine its role in the Drosophila midgut epithelium, which contains a self-renewing population of ISCs that maintain homeostasis in the adult (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006). The lineage of ISCs is simple, as these cells divide to produce enteroblasts (Eb) that differentiate directly into an enterocyte (EC) or enteroendocrine cell (ee) without further division (Fig. 1A). Two signaling pathways, Wnt (Lee et al., 2009; Lin et al., 2008) and Notch (Ohlstein and Spradling, 2007), ensure the division of ISCs and differentiation of ISC progeny, respectively. Two studies on acute regeneration have so far identified ECs as sensors of damage, and ISCs as responders to cues originating from ECs (Jiang et al., 2009; Staley and Irvine, 2010). For example, in the event of injury, Jun N-Terminal Kinase (JNK) signaling is activated in dying ECs, which are thought to subsequently secrete Unpaired (Upd) cytokines – Upd/Os, Upd2 and Upd3 – to activate JAK/STAT signaling in ISCs (Jiang et al., 2009). In response, ISCs increase their rate of division and the regenerative response of ISCs ceases once their progeny replace the cells lost due to injury. In addition, Staley and Irvine (Staley and Irvine, 2010) recently reported that injury-induced JNK activity results in the activation of Yki in ECs (Staley and Irvine, 2010), placing Hippo signaling between JNK and JAK/STAT signaling in the EC response to acute injury. Importantly, this study failed to demonstrate a role for the Hippo pathway in the ISCs themselves.
Fig. 1.

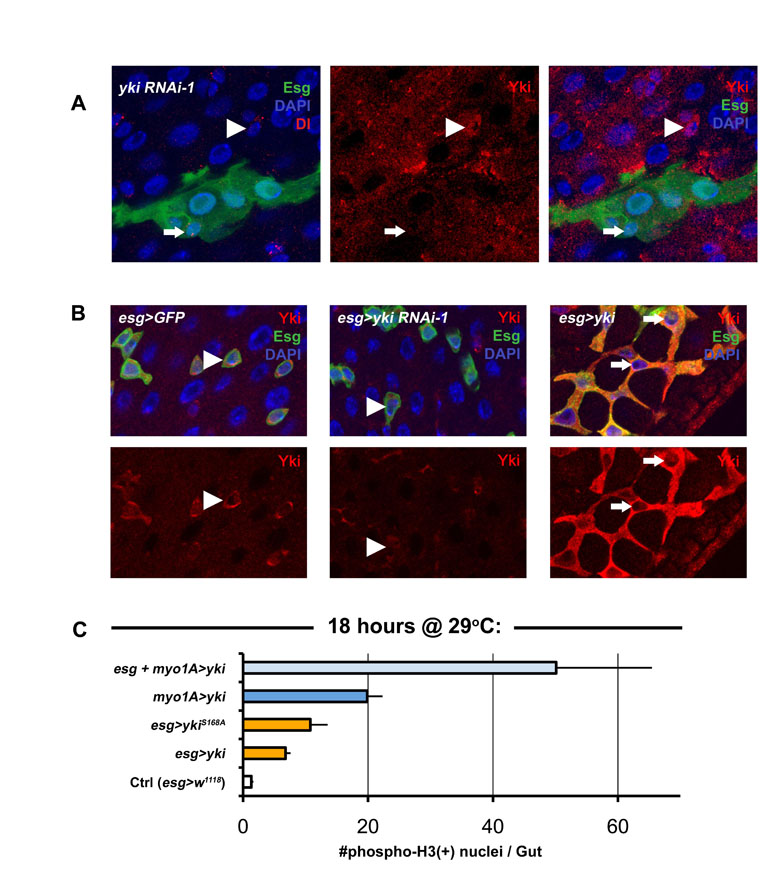
Yorkie induces ISC/Eb overproliferation in the gut epithelium. (A) The ISC lineage. (B) Esg(+) ISC/Eb cells in the wild-type posterior midgut epithelium stained with anti-Yki antibody. (C) Shown are: (i) wild type (w1118/+; MST1096-Gal4/+) versus (ii) Yorkie RNAi (MST1096-Gal4/+; yki RNAi-1/+) adult wings. (D) Confocal micrographs of wild type (first panel is esg>GFP: w1118/+; esg-Gal4, UAS-GFP, Tub-Gal80TS/+), RNAi against yki (middle panel is esg>yki RNAi-1: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; yki RNAi-1/+) and overexpression of yki (left panel is esg>yki: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; UAS-yki/+). The same hairpin that reduces wing size in C has no effect on ISC proliferation, whereas yki overexpression causes overproliferation. (E) Quantification of total mitoses in the entire midgut (left), and the frequency of Esg(+) cells in one field of view (FOV) in the posterior region of the midgut (right). All constructs were expressed in ISC/Eb cells. Both Yki and mutated Yki (YkiS168A) accelerated cell division, whereas RNAi constructs against yki produced no apparent changes when compared with wild type (Ctrl). Error bars indicate s.e.m. (P<0.05). (F) Confocal images showing Yki-overexpressing clones (shown on right, yki is hsFlp/+; UAS-GFP, act>CD2>Gal4/+; UAS-yki/+) and wild-type clones (left, w1118 is hsFlp/+; UAS-GFP, act>CD2>Gal4/+). Clones mutant for yki (ykiB5 is hsFlp, Tub-Gal4, UAS-nlsGFP/+; FRT42D, Tub-Gal80TS/FRT42D ykiB5) are no different from wild-type (arm-LacZ is hsFlp, Tub-Gal4, UAS-nlsGFP/+; FRT42D, Tub-Gal80TS/FRT42D arm-LacZ).
We sought to examine a role for Hippo signaling in ISCs, and, in contrast to Staley and Irvine (Staley and Irvine, 2010), we demonstrate a cell autonomous role for the Hippo pathway in these cells. We show that ISC proliferation is constitutively controlled by the Hippo pathway, in part through the upstream components Fat and Ds, and that this control is perturbed by damage. Using both ISC-specific overexpression and loss of function experiments, we demonstrate that Yki activation is crucial in ISCs during acute regeneration. Specifically, we show that yki mutant clones are unable to participate in acute regeneration, and find an important role for the Hippo pathway in promoting ISC survival. Furthermore, we demonstrate that the ability of Yki to promote proliferation is also mediated through the autocrine activation of the JAK/STAT pathway. Thus, similar to its role in ECs, Yki regulates the expression of the Upd cytokines in ISCs, activating JAK/STAT signaling in ISCs themselves. These findings reveal a redundancy in both autocrine and paracrine JAK/STAT activity during regeneration and, altogether, demonstrate a novel function for the Hippo pathway in ISCs that occurs in parallel to the indirect Hippo pathway activity in ECs.
MATERIALS AND METHODS
Transgenic and mutant lines
Drosophila stocks were as follows:
w1118
hsFlp; act>CD2>Gal4, UAS-nlsGFP/CyO
MST1096-Gal4/CyO
esg-Gal4, UAS-GFP, Tub-Gal80TS
Tub-Gal80TS/FM7; esg-Gal4/CyO
Tub-Gal80TS/FM7; myo1A-Gal4, UAS-GFP/CyO
yw; UAS-srcGFP; pros-Gal4
hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT40A/CyO
hsFlp, Tub-Gal4, UAS-GFP/FM7; Tub-Gal80, FRT82B/TM6 B
hsFlp, Tub-Gal4, UAS-GFP/FM7; FRT42D, Tub-Gal80/CyO
w; arm-LacZ, FRT40A/CyO
w; FRT42D arm-LacZ
w; FRT42D ykiB5/CyO
w; FRT42D hpo42-47/CyO
IF/CyO; wtsX1, FRT82B/TM6B
w; exe1, FRT40A/CyO
UAS-hpoKD(II)
UAS-hpo (II)
UAS-yki/TM3 Sb
yw; UAS-ykiS168A::GFP (II)
ds-LacZ; cn1/CyO
w; ex-LacZ
DIAP1-LacZ (2BC2 on X)
DIAP1-LacZ/TM6B
CycE-LacZ (III)
bantam GFP sensor/CyO
Upd-LacZ
w; 10xSTAT-GFP (II)
UAS-Upd/CyO
UAS-hopTUML/CyO
UAS-p35
The following RNAi stocks were obtained from the TRiP (Harvard Medical School, Boston, MA, USA):
SH00018.0 (white #1); JF01545 (white #2); JF03245 (fat); JF02842 (dachsous); JF03120 (expanded); JF02841 (merlin); SH00079.N (hippo #1); JF02740 (hippo #2); SH00083.N (warts #1); JF02741 (warts #2); SH00081.N (yorkie #1); JF01666 (yorkie #2); SH00045.N (Stat92E) and JF01268 (hopscotch).
Information on these lines can be found at http://www.flyrnai.org/TRiP-HOME.html.
Temperature-controlled expression
Crosses were carried out at 18°C using the temperature-sensitive Esg (esg-Gal4, UAS-GFP, Tub-Gal80TS) or NP1 drivers (Tub-Gal80TS/FM7; myo1A-Gal4, UAS-GFP/CyO). Offspring were maintained at 18°C to 2-3 days post-eclosion, then shifted to 29°C to suppress Gal80. Flies were transferred onto fresh media every 1-2 days. Guts were examined at the timepoints indicated in the figures. RNAi against the white gene was carried out to control for RNAi effects in this system, these were identical to w1118 controls (data not shown).
Mosaic analysis
Crosses and progeny were maintained at 25°C to the indicated times, and transferred onto fresh media every 1-2 days. For flip-out clones (Basler and Struhl, 1994) (hsFlp; act>CD2>Gal4, UAS-nlsGFP/CyO), a single 10-, 30- or 45-minute 37°C heat shock was applied to 2- to 3-day-old flies. MARCM clones (hsFlp, Tub-Gal4, UAS-nlsGFP/+; arm-LacZ, FRT40A/Gal80TS FRT40A and hsFlp, Tub-Gal4, UAS-nlsGFP/+; wtsX1, FRT82B/Gal80TS, FRT82B and hsFlp, Tub-Gal4, UAS-nlsGFP/+; exe1, FRT40A/Gal80TS, FRT40A), were exposed to either a single 45-minute or two 1-hour 37°C heat shocks at 2-3 days following eclosion. In both flip out and MARCM clones, the minimal heat shock time was applied for clonal analysis. One or two MARCM clones per five guts were noted in non-heat shocked flies, indicating some leakiness in the MARCM system. This does not affect our analysis because >10 clones were scored per midgut, with ∼10 midguts scored per condition. A region of leaky GFP expression was noted in the anterior region of the midgut using the flip out system (hsFlp; act>CD2>Gal4, UAS-nlsGFP/CyO); thus, this region was omitted from these analyses. Both MARCM and flip out clones were scored as clusters of two or more adjacent cells.
Feeding
Flies were maintained on 5% w/v sucrose + H2O for the timepoints indicated. Flies were fed 5% w/v DSS (MP Biomedicals) dissolved in sucrose (Amcheslavsky et al., 2009), or Pseudomonas aeruginosa bacteria (Apidianakis et al., 2009). Pseudomonas was grown in LB media with 100 μg/μl Rifampicin (Sigma) overnight at 32°C, 50 ml aliquots of these bacterial cultures were spun down and one pellet was reconstituted in 500 μl sucrose. For the chase, flies were exposed to DSS as above, then maintained on normal media at 18°C up to the timepoint indicated.
Dissection and staining
Female guts were dissected in 1×PBS (Gibco) and fixed in either: (1) 4% paraformaldehyde (Electron Microscopy Sciences) diluted with 1×PBS or (2) 5% paraformaldehyde diluted in 100 mM sodium phosphate buffer (Sigma) for 40 minutes. Samples were washed three times with PBS, then blocked for 30 minutes in 1×PBS, 1% BSA (Sigma) and 0.2% Triton X-100 (Sigma). Samples were stained for primary antibodies overnight at 4°C using the following antibodies: mouse anti-Armadillo, mouse anti-Delta, mouse anti-Prospero (Developmental Studies Hybridoma Bank), rat anti-Yki (a gift from Helen McNeill, Mount Sinai Hospital, Toronto, Canada), rat anti-Fat (a gift from Michael Simon, Stanford University School of Medicine, Stanford, CA, USA), rat anti-Ds (a gift from Michael Simon), rabbit anti-Yki (a gift from Ken Irvine, Rutgers University, Piscataway, NJ, USA) and rabbit anti-β-galactosidase (Cappel). Samples were washed three times in PBS, and stained at 4°C for 1.5 hours with secondary antibodies as follows: donkey anti-mouse Alexa 555, donkey anti-mouse Alexa 594, donkey anti-mouse Alexa 647, goat anti-rat Alexa 555, donkey anti-rabbit Alexa 594, goat anti-rabbit Alexa 647 (Molecular Probes) and goat anti-rat HRP (Millipore). Following secondary staining with HRP-conjugated antibody, Fat protein signal was amplified using the Tyramide TSA Kit (Perkin Elmer, #NEL700A) as per the manufacturer's instructions. TUNEL labeling (Roche, #12156792910 In situ Cell Death Kit, TMR Red) was performed according to the manufacturer's instructions. All samples were counterstained with DAPI (Molecular Probes) for 10 minutes, washed three times with PBS, and mounted using Vectashield (Vector).
RT-qPCR
Twelve midguts from each genotype were dissected and collected on ice (n=3 biological replicates). RNA was isolated using the RNEasy Mini Kit (Qiagen) and cDNA was transcribed using the iScript cDNA Synthesis Kit (Biorad). qPCR was then performed using iQ SYBR Green Supermix on a CFX96 Real-Time System/C1000 Thermal Cycler (Biorad). All samples were treated according to the manufacturer's instructions, gene expression was normalized to the control transcript Rp49, then normalized relative to the w1118 control. qPRC primers used were: Rp49-F, ATCGGTTACGGATCGAACAA; Rp49-R, GACAATCTCCTTGCG CTTCT; Upd-F, TCAGCTCAGCATCCCAATCAG; Upd-R, ATAGTCGATCCAGTTGCTGTTCCG; Upd2-F, TGCTATCGCTGAGGCTCTCG; Upd2-R, GACTCTTCTCCGGCAAATCAGA; Upd3-F, AAATTGAATGCCAGCAGTACG; Upd3-R, CCTTGCTGTGCGTTTC GTTC; Ex-F, GCACCGCACCATTGTTCATC; Ex-R, CAACTGA TGGCTGCAAACCG.
Microscopy
Fluorescent microscopy was performed on a Zeiss Axioskop 2motplus upright, confocal images were obtained using the Leica TCS SP2 AOBS system.
Data analysis
Images were processed in Photoshop CS2. Statistics (t-test or ANOVA as appropriate) were carried out using Graphpad Prism 5.0. The frequency of Esg(+) cell per field of view (FOV) was determined as the ratio of Esg(+) cells/total number of DAPI(+) nuclei (including ISCs, Ebs, ECs and ee cells) taken in one region of the posterior midgut using a 40× objective. Using this method, ∼300-400 cells were scored per midgut, and at least six midguts were analyzed per genotype.
RESULTS
To address whether the Hippo pathway is involved in ISC growth control following regeneration, we first examined whether the target of the Hippo pathway is expressed by ISCs. The Drosophila midgut ISC and Eb cells express the transcription factor Escargot (Esg) (Micchelli and Perrimon, 2006). We found that Esg(+) cells in the posterior midgut were enriched for Yki, but that its localization was predominantly cytoplasmic (Fig. 1B). This cytoplasmic localization of Yki suggests that it is inactivated in these cells, as Yki phosphorylation and exclusion from the nucleus is regulated by Hippo signaling (Dong et al., 2007). Only following translocation into the nucleus, can Yki activate transcription factors involved in growth during development (Dong et al., 2007; Oh and Irvine, 2008; Saucedo and Edgar, 2007). The cytoplasmic localization of Yki in ISCs suggests that removal of Yki should not lead to any obvious ISC phenotypes, which we tested by knockdown of Yki by RNAi in the midgut using the temperature-controlled Esg-Gal4 system. No obvious undergrowth phenotype was noticed when Yki RNAi is expressed in ISC/Eb cells (Fig. 1D). This result is probably not due to the RNAi reagent that we used, as RNAi against Yki in the Drosophila wing, where Yki is required for normal organ growth, produces a severe undergrowth phenotype (Fig. 1C) and reduces Yki protein levels (see Fig. S1A in the supplementary material).
Next, we tested whether overexpression of either wild-type Yki, or the constitutively active YkiS168A, in ISCs could affect their proliferative abilities. Strikingly, although removal of Yki showed no phenotype in the midgut, overexpression of Yki in ISCs increased division, as judged by an increase in mitotic phosphorylated-Histone3 positive nuclei, and also increased ISC/Eb frequency, as shown by an increase in esg>GFP signal (Fig. 1D,E). Our results contrast with the recent report of Staley and Irvine (Staley and Irvine, 2010), who failed to detect a phenotype in ISC following overexpression of Yki in ISCs. This may be due to a difference in the strength of the expressed Yki in these cells, as we used a wild-type UAS-yki transgene (Huang et al., 2005) and the UAS-ykiS168A:GFP (Oh and Irvine, 2008), both of which are random transgene insertions. The UAS-ykiS168A:V5 used in by Staley and Irvine is a targeted insertion (attP2 at 68A) (Oh and Irvine, 2009). Indeed, division was induced only 18 hours after Yki overexpression in ISC/Ebs using the esg-Gal4 driver, suggesting that Yki plays a cell-autonomous role when it is activated in these cells (see Fig. S1C in the supplementary material).
To further analyze the effect of Yki expression, we compared the size of clones of cells overexpressing Yki with wild type. When examined at 14 days after induction, wild-type clones typically contain fewer than 10 cells (see Fig. 2F), most of which are ECs, distinguished by their large polyploid nuclear size (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006); only one or two are ISCs, distinguished by the expression of the ISC marker Delta (Dl) (Ohlstein and Spradling, 2007) (Fig. 1F). Yki-overexpressing clones were larger than in wild type and sometimes contained more than two Dl(+) cells, suggesting either an expansion of the ISC number or stress, which has been shown to prevent the differentiation of Dl(+) Eb cells (Biteau et al., 2008) (Fig. 1F; see Fig. S2 in the supplementary material). However, the presence of large Dl(–), polyploid EC nuclei and staining for the ee marker Prospero (Ohlstein and Spradling, 2007) (Pros) (see Fig. S2A in the supplementary material) suggests that differentiation of Ebs is not impaired following Yki overexpression. Loss-of-function yki clones were also examined and, consistent with our RNAi depletion (Fig. 1D), no difference was noted between these and their wild-type counterparts under normal conditions (Fig. 1F, Fig. 2E,F). Clonal analysis was then applied using yki RNAi to further confirm these findings (see Fig. S2 in the supplementary material).
Fig. 2.

Perturbation of Hippo signaling causes hyperplasia in the gut epithelium. (A) RNAi against hpo leads to overproliferation (bottom panel is esg>hpo RNAi-1: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; hpo RNAi-1/+) when compared with wild type (top panel is esg>GFP: w1118/+; esg-Gal4, UAS-GFP, Tub-Gal80TS/+). (B) Quantification of Esg(+) cell frequency (top) and mitoses (bottom). ds RNAi shows no difference compared with wild type (Ctrl), whereas every other Hippo pathway member tested shows overproliferation when expressed in ISC/Eb cells. Error bars indicate s.e.m. (P<0.05). (C) When ds is knocked down in ECs (Tub-Gal80TS/+; myo1A-Gal4, UAS-GFP/+; ds RNAi/+), it causes strong overproliferation. Error bars indicate s.e.m. (P<0.05). (D) Mosaic analysis shows that hpo mutation (hpo42-47 is hsFlp, Tub-Gal4, UAS-nlsGFP/+; FRT42D, Tub-Gal80TS/FRT42D hpo42-47) phenocopies yki overexpression (compare with Fig. 1F). (E) Quantification of clonal frequency reveals an expansion in clone number in hpo mutant clones, but not in yki mutant clones, which are similar to wild type (arm-LacZ). Genotypes are same as above, frequencies are shown as percentage normalized to the 4-day timepoint when each gut contained 5-15 clones in total (n=10-12 guts examined at each timepoint). Error bars indicate s.e.m. (P<0.05). (F) Histograms show that large clones are more frequent among hpo mutant clones than in wild type (arm-LacZ) or yki mutants. Brackets show total number of clones analyzed at this timepoint (14 days after clone induction).
As Yki cytoplasmic retention is caused by Hippo signaling, we carried out RNAi experiments against components of the Hippo pathway to determine whether these repress Yki in ISCs. Knockdown of Hpo, Wts, Mer, Expanded (Ex) and Fat in ISCs produced an increase in both ISC/Eb frequency and ISC division in the midgut (Fig. 2A,B). For example, by 11 days, knockdown of Hpo produced a strong phenotype that closely resembled the overexpression of Yki in these cells (Fig. 2A). Some variability in the frequency of Esg(+) cells was observed at different time points, most likely reflecting variability in the level of knockdowns associated with RNAi (see Fig. S4A,B in the supplementary material). To corroborate the finding that Hippo signaling deactivation in ISCs leads to overproliferation, the overexpression of a dominant-negative kinase dead Hpo transgene (Wu et al., 2003a) was found to elicit a similar phenotype as RNAi (Fig. 2B).
To further investigate the effect of removal of Hpo, we compared the size of hpo mutant clones (Fig. 2D,F) and clones expressing Hpo RNAi (see Fig. S2 in the supplementary material) with wild type. The loss of Hpo also increased the proportion of large clones (Fig. 2F), and slightly increased the proportion of Dl(+) cells per clone (see Fig. S3C in the supplementary material). An expansion in clone number was observed. Whereas the frequency of at least two-cell wild-type clones is constant over time, the frequency of hpo mutant and RNAi-depleted clones increases over the same period (Fig. 2E; see Fig. S2 in the supplementary material). These data suggest either an expansion in ISC number and their dispersal from the initial clones into the midgut epithelium, or the expansion of the surrounding tissue which separates ISC clones. It has been suggested that ISCs may mobilize in the gut epithelium (Ohlstein and Spradling, 2006), and our results support this notion. Similar clones were obtained using yki overexpression and the Hippo pathway mutant alleles wtsX1 and exe1 (see Figs S2, S3 in the supplementary material). We stained clones for Pros and found that ee production was not affected by the loss of hpo (see Fig. S3C in the supplementary material). Similarly ECs are present in hpo clones (see Figs S2, S3 in the supplementary material), indicating that differentiation is not impaired in the absence of Hpo pathway activity. Quantification of Dl(+) and Pros(+) cell number using the Esg-Gal4 driver, further confirmed the ability of Ebs to differentiate with either Yki overexpression or Hippo pathway knockdown (see Fig. S4C in the supplementary material).
An important question is whether the proliferation observed in response to Yki activity or Hippo signaling disruption was due to the division of ISCs or whether Ebs, which normally do not divide, could be induced to proliferate. We noted little overlap in Esg(+) and markers of differentiation, Pros (not shown) and Pdm1 (see Fig. S4D in the supplementary material). Staining for the ISC marker Dl and phosphorylated-Histone3 revealed that nearly all of the mitotic events occurred in Dl(+) cells (Fig. 3A) suggesting that it is the ISC division and not the division of transit amplifying progenitors that is regulated by Hippo signaling. However, we cannot exclude the possibility that Ebs dedifferentiate into ISCs under the control of the Hippo pathway.
Fig. 3.

Hippo signaling occurs in different cell types within the midgut epithelium. (A) Confocal images show wild type (left panel is esg>GFP: w1118/+; esg-Gal4, UAS-GFP, Tub-Gal80TS/+), overexpression of yki (center panel is esg>yki: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; UAS-yki/+) and RNAi against hpo (right panel is esg>hpo RNAi-1: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; hpo RNAi-1/+) stained for Dl (green) and phosphorylated Histone 3 (red). Nearly all mitotic cells are also Dl(+), as revealed by quantification of percentages (number of phospho-H3 cells counted are in brackets). (B) Fat staining is located in the basal region of wild-type posterior gut epithelium. Top panels show confocal sections through the epithelial wall, with an ISC labeled for Esg (green) located basally. The outside of the gut is just below the ISC. A crescent of Fat protein, coincident with Armadillo (Arm) which marks the cell cortex, is located between the ISC and its neighboring EC cells. Lower panels show confocal sections taken through the basal region of the epithelium, where ISC/Eb cells are located. Fat staining is present around the cortex of Esg(+) cells. (C) Confocal images show that the Ds protein, also coincident with Arm, is located at the edges of wild-type ECs – in some cases where the ECs appose Esg(+) cells. (D) Confocal images of a Tub-Gal80TS/ds-LacZ; myo1A-Gal4, UAS-GFP/+ posterior midgut. ds-LacZ is expressed in the EC cells which are GFP-labeled using the NP1 Gal4 driver. (E) Confocal images of a ds-LacZ/+; esg-Gal4, UAS-GFP, Tub-Gal80TS/+ midgut. Strong ds-LacZ expression is also present in Pros(+) ee cells.
Interestingly, knockdown of the Hippo pathway ligand Ds in ISCs did not increase ISCs/Eb frequency or ISC division (Fig. 2B). Ds is thought to act as the ligand of Fat through which cell-cell contact signaling is achieved (Matakatsu and Blair, 2004; Rogulja et al., 2008; Willecke et al., 2008), and the depletion of Fat in ISCs leads to ISC overproliferation. Thus, the nests of ISCs/Ebs, which are surrounded by their EC progeny, might be signaling through the Fat/Ds cadherins. We tested the effect of removing Ds in ECs using the temperature-sensitive NP1 Myo1A-Gal4 driver that is expressed in EC cells (Jiang et al., 2009) (see Fig. S5B in the supplementary material). Strikingly, knockdown of Ds produced overproliferation if carried out in ECs (Fig. 2C) but did not seem to affect the proportions of ISC or ee cells present (see Fig. S4C in the supplementary material). To provide further support to our functional analyses, we examined the expression of Hippo pathway components in the midgut epithelium. The receptor of the Hippo signaling pathway, Fat, was found to be expressed in the Esg(+) cells (Fig. 3B), including Dl(+) ISCs (see Fig. S5A in the supplementary material). Its ligand, Ds, was also present in the midgut epithelial cells, in particular along the borders of EC cells (Fig. 3C), as well as along the borders where diploid Esg(+) cells attached to their EC neighbors. Ds staining was not uniform throughout the epithelium, suggesting that either the expression of the ligand is dynamically regulated, or that gut epithelial cells are heterogenous for the expression Ds. To determine which cells express the Ds ligand, a ds-LacZ reporter was used that carries a lacZ insertion in the ds gene. Staining for β-galactosidase revealed that ds expression is present in EC cells but that not all ECs expressed ds strongly (Fig. 3D). We found that ee cells also express ds, whereas Esg(+) ISC/Eb cells did not show high ds expression (Fig. 3E). Altogether, our analysis suggests that ISCs and Eb cells express Fat, while their differentiated progeny express Ds, most likely to inhibit tissue expansion. The observation that Ds expression appears dynamic suggests that EC and ee cells might only express Ds transiently, e.g. depending on their maturity during differentiation. Alternatively, it is possible that only some subtypes of EC and ee cells express Ds.
Our data demonstrate that the Hippo signaling pathway limits the proliferation of ISCs but does not affect the differentiation of Eb progeny. The complete inhibition of Yki in ISCs raises the issue of what role this transcriptional co-activator plays in this system. The regeneration of damaged tissue provides a context where ISCs must divide quickly to replace lost cells in the midgut epithelium. Therefore, we tested whether the loss of Yki activity would have consequences on the proliferation of ISCs in a gut regeneration model. Midguts in which Yki was knocked down by RNAi were exposed to dextran sodium sulfate (DSS) (Fig. 4A) and Pseudomonas bacteria, both of which have previously been shown to induce tissue injury (Amcheslavsky et al., 2009; Apidianakis et al., 2009; Jiang et al., 2009). Yki depletion strongly suppressed the DSS-induced division of ISCs (Fig. 4A,B) whereas Pseudomonas showed a lesser effect (Fig. 4B). Under both DSS and Pseudomonas damage conditions, the division of ISCs and frequency of ISCs/Ebs in the posterior midgut was decreased (Fig. 4B,C), confirming that the damage response is reduced when Yki is knocked down. We re-examined yki mutant clones under injury conditions (Fig. 4D,4E) and found that the size of the clones was significantly reduced from their former levels, in the absence of damage, where they were equivalent to controls (Fig. 1F, Fig. 2F). Yki mutant clones were also less frequent than wild types during DSS damage: whereas wild-type clones increased in frequency to 7.8±1.7 times their original numbers, yki clones increased to only 2.8±0.6 times theirs. The expansion of clones during damage again suggests the dispersal of ISCs during tissue regeneration. The presence of dividing yki mutant clones itself suggests that Yki is not needed for the survival of all ISCs during damage, but their small size and reduced frequency suggests that Yki is needed for either the survival of their differentiated progeny or for the proliferation of ISCs following injury. Cell death was examined using TUNEL labeling, but no increases in apoptosis were observed in ykiB5 clones (see Fig. S7 in the supplementary material), suggesting that Yki primarily functions to increase division. The survival requirement was further tested by the simultaneous depletion of yki and overexpression of the cell death inhibitor p35 (Fig. 4B). P35 was able to partially rescue the loss of dividing cells but not the decrease in Esg(+) under DSS injury conditions. Thus, we find some evidence that supports a role for Yki in preventing the cell death of ISCs during damage. Because under normal homeostasis, Yki overexpression induces ISC division (Fig. 1, Fig. 3A), it seems likely that Yki functions in ISCs to promote both proliferation and survival.
Fig. 4.

Yki accelerates ISC division during regeneration. (A) The Yki-depleted midgut (bottom panel is esg>yki RNAi-1: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; yki RNAi-1/+) contains fewer Esg(+) cells in response to DSS-induced damage, compared with the wild-type gut (top panel is esg>GFP: w1118/+; esg-Gal4, UAS-GFP, Tub-Gal80TS/+). (B) Quantification of mitoses shows that DSS (top) and Pseudomonas (bottom) exposure reduces the regenerative response of ISCs expressing yki RNAi constructs. Error bars indicate s.e.m. (P<0.05). (C) The frequency of Esg(+) cells in the posterior midgut is also reduced, when flies are fed either DSS or Pseudomonas, and Yki is concomitantly depleted. Error bars indicate s.e.m. (P<0.05). (D) Confocal images show a reduction in the size of yki mutant clones (ykiB5) when compared with wild type (arm-LacZ) when the midgut is injured, although both contain ISCs marked by Dl (arrows). (E) After injury, yki mutant clones are smaller than wild type (arm-LacZ). However, under baseline conditions, these are similar in size to wild type. Brackets show total number of clones analyzed at this timepoint (14 days after clone induction), error bars indicate s.e.m. (P<0.05). (F) Confocal images showing the esg>Gal4/ex-LacZ reporter line (esg-Gal4, UAS-GFP, Tub-Gal80TS/ex-LacZ) under normal conditions and injury conditions. Samples were prepared identically and images were taken using the same parameters.
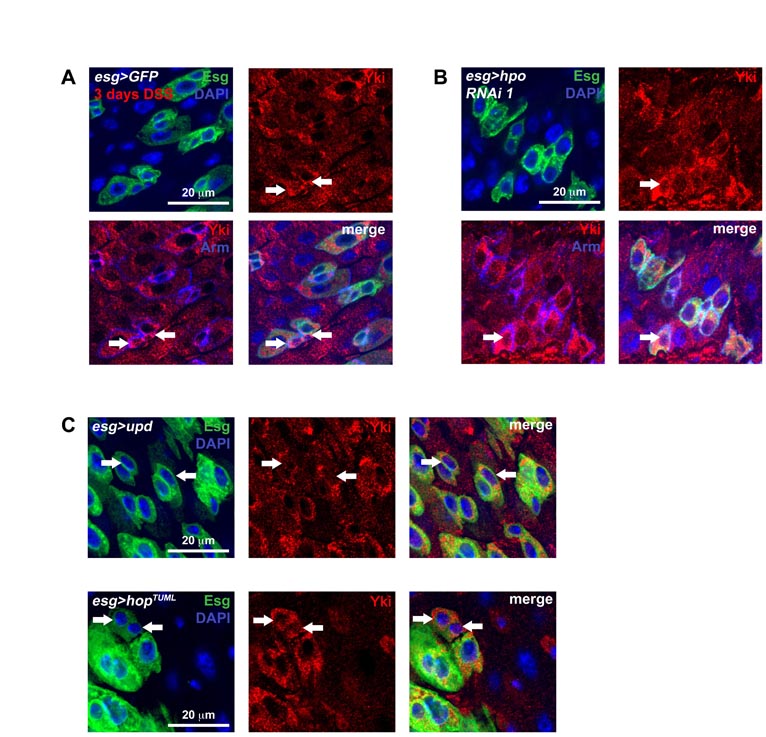
The activation of Yki and its targets (which are normally suppressed by Hippo signaling) were further investigated. Staining of endogenous Yki in damaged wild-type midguts revealed that the nuclear signal of Yki increases in Esg(+) ISCs/Ebs, similar to what is observed when Hpo is knocked down in these cells (see Fig. S6 in the supplementary material). The targets of Yki include the cell cycle regulator CyclinE (CycE), the apoptosis inhibitor DIAP1 and the microRNA bantam. Flies carrying reporters of these target genes were examined when injured by DSS and Pseudomonas bacteria (see Figs S8, S9 in the supplementary material). CycE, DIAP1 and bantam targets were elevated under both conditions of injury, although an increase in DIAP1 was also noted in EC cells. Altogether, these data demonstrate that, following injury, Yki functions to increase the regenerative capacity of ISCs. We also examined the expression of Ex, a component of the Hippo pathway itself regulated by the pathway (Hamaratoglu et al., 2006; Saucedo and Edgar, 2007) (see Figs S8, S9 in the supplementary material). Interestingly, ex reporter expression was transiently increased following injury but then returned to baseline levels when environmental insult was removed (Fig. 4F). This suggests that after acute regeneration is complete, Yki is deactivated to slow down accelerated ISC division.
The constitutive expression of Yki in ISCs reveals that these cells are primed to divide very rapidly. The baseline division of these cells is once per day (Ohlstein and Spradling, 2006) – even though their maximal cell cycle rate is much higher. This is evident under conditions of damage, where tissue loss induces much greater ISC output (Jiang et al., 2009). It has been shown that the response to damage involves the release of Upd cytokines, resulting in the activation of the JAK/STAT pathway (Jiang et al., 2009). To examine the relationship between JAK/STAT and Hippo signaling, we depleted both Hpo and the Janus kinase Hopscotch (Hop), or its target Stat92E, simultaneously in the ISCs. Concomitant Hpo and Yki knockdown (Fig. 5A,B) inhibited overproliferation induced by Hpo knockdown alone, consistent with Hpo acting through Yki. Similarly, Hop or Stat92E knockdown also suppressed Hpo depletion-dependant overproliferation, indicating that the JAK/STAT pathway acts either downstream or in parallel to Hpo (Fig. 5A,B). The same effects were observed when Yki was overexpressed and either Stat92E or Hop were depleted (see Fig. S10B in the supplementary material). Stat92E suppression of ISC division was somewhat less, but this was due to the infrequent presence of localized hyperplasias (see Fig. S10A in the supplementary material), which have been previously reported to occur in a subset of Stat92E mutant clones (Beebe et al., 2010).
Fig. 5.

JAK/STAT is integrated with Hippo signaling. (A) When hpo is depleted (top panel is esg>hpo RNAi-1: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; hpo RNAi-1/+), cell division is increased. Knockdown of Yki (middle panel is esg>hpo RNAi-1, yki RNAi-1: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; hpo RNAi-1/yki RNAi-1) and Stat92E (bottom panel is esg>hpo RNAi-1, stat92E RNAi: esg-Gal4, UAS-GFP, Tub-Gal80TS/+; hpo RNAi-1/stat92E RNAi) suppress this overproliferation. (B) Quantification of Esg(+) cell frequency (top) and mitosis (bottom). The depletion of yki, hop and stat92E suppress the overproliferation observed due to hpo depletion alone. Error bars indicate s.e.m. (P<0.05). (C) Quantification of Esg(+) cell frequency and mitosis, shows that when yki is depleted, JAK/STAT activation still results in increased proliferation. If both yki and hop, or yki and stat92E are depleted, no significant differences are observed from yki depletion alone. Error bars indicate s.e.m. (P<0.05). (D) Graphs show quantification of mitosis and Esg(+) cell frequency as above, genotypes examined are the same for both graph y-axes. The values for esg>yki and esg>hpo RNAi-1 are reproduced from Fig. 1E and Fig. 2B in order to compare the increase. Error bars indicate s.e.m. (P<0.05).
We next activated JAK/STAT signaling by overexpressing Upd/Os, or a constitutively active form of Hop and concomitantly depleted Yki (Fig. 5C). In contrast to the requirement of JAK/STAT activity for Yki-accelerated division, JAK/STAT activation resulted in overproliferation independent of Yki. Simultaneous knockdown of Yki and Hop, or Yki and Stat92E did not differ greatly from depletion of Yki alone (Fig. 5C). These interactions between the Hippo and JAK/STAT pathways were also recapitulated in the context of injury (see Fig. S11 in the supplementary material). To further test whether these pathways operate independently or interdependently, we activated Yki (by overexpression or by Hpo depletion) and simultaneously activated the JAK/STAT pathway (Fig. 5D). The proliferative effects of these two pathway activities together resulted in greatly increased division, suggesting a synergistic effect as division was greater than that observed with either pathway alone. Moreover, the activation of JAK/STAT pathway did not produce any obvious changes in Yki localization (see Fig. S6C in the supplementary material). These results support a model where the JAK/STAT and Hippo pathways operate independently to activate processes in ISCs; however, whereas JAK/STAT induces division alone, Yki induces division only when JAK/STAT is also active at some level.
We investigated whether Hippo signaling might upregulate JAK/STAT signaling. The activity of the JAK/STAT pathway was increased when either yki was overexpressed or when hpo was depleted in Esg(+) cells (Fig. 6A). Quantitative PCR revealed a slight increase in Upd/Os and Upd2 transcripts, and a larger increase in Upd3 transcript in midguts when hpo was depleted (Fig. 6B). Closer inspection of Esg(+) cells revealed that Upd/Os cytokine production was increased in these precursors when yki was overexpressed or when hpo was depleted (Fig. 6C). These increases are reminiscent to what is observed following injury (Jiang et al., 2009), suggesting that Upd(s) may be transcriptional targets of Yki in ISC/Eb cells. We further examined whether Yki is required for JAK/STAT activity in these cells when the midgut is undergoing damage. Neither JAK/STAT activity (Fig. 6A) nor Upd/Os production (Fig. 6C) was abolished when yki was depleted in ISCs/Ebs, indicating that Yki facilitates Upd/Os production but is not required to activate JAK/STAT activity during regeneration. Moreover, residual STAT activity, which functions independently of Yki, may account for the incomplete suppression of regeneration when yki is lost (Fig. 4).
Fig. 6.

Yki activates the release of JAK/STAT cytokines. (A) The activity of the JAK/STAT pathway, using the stat-GFP reporter (stat-GFP is Tub-Gal80TS/+; esg-Gal4/10xSTAT-GFP and where present the overexpression constructs yki RNAi-1, yki or hpo RNAi-1 are heterozygous on the third chromosome). When either yki is overexpressed or hpo is depleted by RNAi, an increase in STAT activity is observed. DSS damage induces a similar increase even when yki is depleted. (B) qPCR of tested transcripts following hpo knockdown using the esg-Gal4 driver. Signal is normalized to the control gene rp49 and levels are shown normalized to esg>GFP controls; error bars are s.e.m. (C) Upd/Os expression is elevated in Esg(+) cells when either hpo is knoced down or when yki is overexpressed (esg>GFP, upd-LacZ refers to w1118/upd-LacZ; esg-Gal4, UAS-GFP, Tub-Gal80TS/+ and where present the overexpression constructs yki RNAi-1, yki or hpo RNAi-1 are heterozygous on the third chromosome). Normally signal is weak or non-existent in wild type Esg(+) cells (esg>GFP, upd-LacZ), but Esg(+) cells (arrows) become positive for upd-LacZ (red) during Yki-induced overgrowth (esg>yki, upd-LacZ) or Hpo knockdown (esg>hpo RNAi-1, upd-LacZ). Similar to the results above, Upd/Os signal still shows an increase (arrows) during DSS damage even when yki is depleted (esg>yki RNAi-1, upd-LacZ).
DISCUSSION
We have analyzed the role of the Hippo pathway in the adult midgut and find several lines of evidence supporting a direct role for the Hippo pathway in ISCs. First, we show that Yki protein is enriched in ISCs (Fig. 1B), and that depletion of the Hippo pathway components that repress Yki in ISCs, leads to ISC overproliferation (Fig. 2, Fig. 3A). Second, we find that the Yki transcriptional targets CycE, Ex, and DIAP1 are upregulated in ISCs following injury (see Fig. S9 in the supplementary material), and that after such injury yki mutant clones are decreased in size and frequency relative to wild type (Fig. 4E). These data are consistent with a cell-autonomous survival and proliferation function for Yki in ISCs during damage. Furthermore, we have analyzed the interaction between the Hippo pathway and the JAK/STAT pathway, which has been previously implicated in ISC regeneration (Jiang et al., 2009). Our analysis reveals that JAK/STAT activity is required in ISCs for Yki-driven overproliferation (Fig. 5), and suggests that Yki acts, in part, by upregulating the Upd cytokines (Fig. 6). At least one of them, Upd/Os, acts in an autocrine fashion (Fig. 6C) to activate STAT (Fig. 6A).
Based on these findings, we propose that under normal homeostasis Yki is inactive in ISCs, but becomes activated when Hippo signal transduction is disrupted by cell contact cues resulting from injury (Fig. 7). Yki activity then leads to the transcription of known targets involved in proliferation (CycE), growth (bantam) and the inhibition of cell death (DIAP1). The expression of Upd cytokines is also transcribed following Yki activation (Fig. 6B) to promote the division of ISCs.
Fig. 7.

Hippo signaling restrains Yki-accelerated cell division in response to injury. Schematic shows the transition of ISCs between normal homeostasis, which occurs in the absence of injury, and acute regeneration following injury. Upon damage, the Hippo pathway ligand, Ds, no longer activates Hippo signaling in ISCs, allowing Yki to activate downstream targets. One of these targets is the Upd/Os cytokine, which stimulates proliferation of ISCs/Ebs through the JAK/STAT pathway. When ee and EC progeny are regenerated, Hippo signaling acts as a brake, cytokine production slows and the system returns to the homeostatic state.
Furthermore, we find that co-activation of Yki and STAT increase division over either pathway alone (Fig. 5D). These results are consistent with at least three hypothetical models of Hippo and JAK/STAT integration in ISCs: (1) Yki directly activates Upd(s) expression, further increasing the production of cytokines that occurs following damage, in order to boost STAT activity; (2) Yki may enhance STAT activity by increasing the expression of intermediates, such as CycE which have been proposed to stabilize STAT (Chen et al., 2003); (3) Yki activates both Upd/Os and DIAP1 in ISCs, which promotes survival and enhances the proliferative capacity of the ISC population during injury. These models are not mutually exclusive. As mentioned above, we observe the upregulation of Upd/Os, CycE and DIAP1, suggesting that the activity of Yki may promote a complex transcriptional response in ISCs to quickly heal tissues after damage.
We note that others have reported that injury activates the JNK pathway in ECs, which subsequently triggers the secretion of Upd cytokines to act in a paracrine fashion on ISCs (Jiang et al., 2009). Although we observed autocrine Upd/Os activity on ISCs, it is likely that there are multiple sources of Upd(s) in this tissue: from the surrounding muscle (Lin et al., 2010), the ECs (Jiang et al., 2009) or other midgut epithelial cells (Beebe et al., 2010; Liu et al., 2010). The respective roles and origin of the three Upd cytokines (Upd/Os, Upd2 and Upd3) in this tissue will need to be further clarified.
When Yki is activated in ISCs using the esg-Gal4 driver, we find that all three cytokines are increased in the midgut (Fig. 6B), reminiscent to what is observed during injury (Jiang et al., 2009). Importantly, Yki activation in ISCs using the esg-Gal4 driver increases Upd/Os production in ISCs themselves (Fig. 6C). Recently, it has been reported that the kinase Wts regulates the activity of Yki in ECs, and that ECs also provide a source of this cytokine when Yki activation is carried out in ECs using the myo1A-Gal4 driver (Staley and Irvine, 2010). Thus, in addition to the role of the Hippo pathway in ISCs that we describe, inhibition of Hippo signaling in damaged ECs also leads to production of Upd cytokines. We note an increase in ISC division 18 hours after Yki overexpression in ISCs using the esg-Gal4 driver, or in ECs using the myo1A-Gal4 driver, and a synergistic effect when overexpression was carried out in both simultaneously (see Fig. S1C in the supplementary material). This suggests that Yki activation in ISCs or ECs has autonomous and non-autonomous effects on ISCs. Because we also observe an increase in DIAP1 expression in ECs during injury (see Fig. S8 in the supplementary material), our findings are consistent with this report, and point to a role for Hippo in regulating the regenerative response in both ISCs and ECs in the midgut epithelium. The acute regeneration of this tissue appears to be controlled by both an ISC autonomous and non-autonomous response – both of which involve Hippo and JAK/STAT signaling. It is interesting that, depending on the state of the tissue, the Hippo pathway is used either as a means to accelerate tissue repair by SCs or as a brake on SCs in order to suppress tissue regeneration.
Hippo pathway members are strongly implicated in cancers, as revealed by the findings that mutations in many pathway members are enriched in human cancers (Zeng and Hong, 2008) and that the human ortholog of Yki is an oncogene (Zender et al., 2006). The issue of whether Hippo signaling occurs in SCs or in differentiated cells is important in resolving the cell of origin of carcinomas resulting from mutations in upstream Hippo pathway components. In particular, the mammalian ortholog of Merlin (Mer), NF2, has been shown to produce either hepatocellular carcinoma (Zhang et al., 2010) or both hepatocellular carcinoma and cholangiocarcinoma (Benhamouche et al., 2010). These contrasting reports bear important conceptual implications in understanding Hippo-induced tumorigenesis, because hepatocellular carcinoma is thought to originate from hepatocytes, while mixtures of both hepatocellular carcinoma and cholangiocarcinoma are thought to originate from the oval cell precursors of the liver. Thus, it is crucial to identify the physiological role of the Hippo pathway in both precursors versus their progeny, and the consequences of perturbing this pathway in either (Breuhahn and Schirmacher, 2010). Finally, the connection between Yki and Stat transcriptional activation in ISCs of the gut epithelium suggest that STAT regulation in Hippo-driven cancers need to be carefully evaluated.
Supplementary Material
Acknowledgments
We thank all of the members of the TRiP and Perrimon Lab (in particular Chrysoula Pitsouli, Richelle Sopko and Richard Binari) for their help. We also thank Drs H. McNeill, D. Pan, B. Edgar, K. Irvine, M. Simon, X. Yang and S. Cohen for kindly sharing reagents. This work was supported in part by the Human Frontier in Science Program (P.K.). N.P. is an Investigator of the Howard Hughes Medical Institute. Deposited in PMC for release after 6 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.060483/-/DC1
References
- Amcheslavsky A., Jiang J., Ip Y. T. (2009). Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell 4, 49-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apidianakis Y., Pitsouli C., Perrimon N., Rahme L. (2009). Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. USA 106, 20883-20888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler K., Struhl G. (1994). Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature 368, 208-214 [DOI] [PubMed] [Google Scholar]
- Beebe K., Lee W. C., Micchelli C. A. (2010). JAK/STAT signaling coordinates stem cell proliferation and multilineage differentiation in the Drosophila intestinal stem cell lineage. Dev. Biol. 338, 28-37 [DOI] [PubMed] [Google Scholar]
- Benhamouche S., Curto M., Saotome I., Gladden A. B., Liu C. H., Giovannini M., McClatchey A. I. (2010). Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 24, 1718-1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett F. C., Harvey K. F. (2006). Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr. Biol. 16, 2101-2110 [DOI] [PubMed] [Google Scholar]
- Biteau B., Hochmuth C. E., Jasper H. (2008). JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell 3, 442-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C., Fuchs E. (2009). Epidermal homeostasis: a balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 10, 207-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuhahn K., Schirmacher P. (2010). A cellular view of Nf2 in liver homeostasis and tumorigenesis. Dev. Cell 19, 363-364 [DOI] [PubMed] [Google Scholar]
- Camargo F. D., Gokhale S., Johnnidis J. B., Fu D., Bell G. W., Jaenisch R., Brummelkamp T. R. (2007). YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054-2060 [DOI] [PubMed] [Google Scholar]
- Chen X., Oh S. W., Zheng Z., Chen H. W., Shin H. H., Hou S. X. (2003). Cyclin D-Cdk4 and cyclin E-Cdk2 regulate the Jak/STAT signal transduction pathway in Drosophila. Dev. Cell 4, 179-190 [DOI] [PubMed] [Google Scholar]
- Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S. A., Gayyed M. F., Anders R. A., Maitra A., Pan D. (2007). Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120-1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbleib J. M., Nelson W. J. (2006). Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 20, 3199-3214 [DOI] [PubMed] [Google Scholar]
- Hamaratoglu F., Willecke M., Kango-Singh M., Nolo R., Hyun E., Tao C., Jafar-Nejad H., Halder G. (2006). The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell. Biol. 8, 27-36 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2000). The hallmarks of cancer. Cell 100, 57-70 [DOI] [PubMed] [Google Scholar]
- Huang J., Wu S., Barrera J., Matthews K., Pan D. (2005). The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122, 421-434 [DOI] [PubMed] [Google Scholar]
- Ito M., Liu Y., Yang Z., Nguyen J., Liang F., Morris R. J., Cotsarelis G. (2005). Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 11, 1351-1354 [DOI] [PubMed] [Google Scholar]
- Jiang H., Patel P. H., Kohlmaier A., Grenley M. O., McEwen D. G., Edgar B. A. (2009). Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137, 1343-1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. C., Beebe K., Sudmeier L., Micchelli C. A. (2009). Adenomatous polyposis coli regulates Drosophila intestinal stem cell proliferation. Development 136, 2255-2264 [DOI] [PubMed] [Google Scholar]
- Lin G., Xu N., Xi R. (2008). Paracrine Wingless signalling controls self-renewal of Drosophila intestinal stem cells. Nature 455, 1119-1123 [DOI] [PubMed] [Google Scholar]
- Lin G., Xu N., Xi R. (2010). Paracrine unpaired signaling through the JAK/STAT pathway controls self-renewal and lineage differentiation of drosophila intestinal stem cells. J. Mol. Cell. Biol. 2, 37-49 [DOI] [PubMed] [Google Scholar]
- Liu W., Singh S. R., Hou S. X. (2010). JAK-STAT is restrained by Notch to control cell proliferation of the Drosophila intestinal stem cells. J. Cell Biochem. 109, 992-999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matakatsu H., Blair S. S. (2004). Interactions between Fat and Dachsous and the regulation of planar cell polarity in the Drosophila wing. Development 131, 3785-3794 [DOI] [PubMed] [Google Scholar]
- Micchelli C. A., Perrimon N. (2006). Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 439, 475-479 [DOI] [PubMed] [Google Scholar]
- Oh H., Irvine K. D. (2008). In vivo regulation of Yorkie phosphorylation and localization. Development 135, 1081-1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H., Irvine K. D. (2009). In vivo analysis of Yorkie phosphorylation sites. Oncogene 28, 1916-1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlstein B., Spradling A. (2006). The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature 439, 470-474 [DOI] [PubMed] [Google Scholar]
- Ohlstein B., Spradling A. (2007). Multipotent Drosophila intestinal stem cells specify daughter cell fates by differential notch signaling. Science 315, 988-992 [DOI] [PubMed] [Google Scholar]
- Reya T., Morrison S. J., Clarke M. F., Weissman I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105-111 [DOI] [PubMed] [Google Scholar]
- Rogulja D., Rauskolb C., Irvine K. D. (2008). Morphogen control of wing growth through the Fat signaling pathway. Dev. Cell 15, 309-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo L. J., Edgar B. A. (2007). Filling out the Hippo pathway. Nat. Rev. Mol. Cell Biol. 8, 613-621 [DOI] [PubMed] [Google Scholar]
- Song X., Zhu C. H., Doan C., Xie T. (2002). Germline stem cells anchored by adherens junctions in the Drosophila ovary niches. Science 296, 1855-1857 [DOI] [PubMed] [Google Scholar]
- St John M. A., Tao W., Fei X., Fukumoto R., Carcangiu M. L., Brownstein D. G., Parlow A. F., McGrath J., Xu T. (1999). Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 21, 182-186 [DOI] [PubMed] [Google Scholar]
- Staley B. K., Irvine K. D. (2010). Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr. Biol. 20, 1580-1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke M., Hamaratoglu F., Kango-Singh M., Udan R., Chen C. L., Tao C., Zhang X., Halder G. (2006). The fat cadherin acts through the hippo tumor-suppressor pathway to regulate tissue size. Curr. Biol. 16, 2090-2100 [DOI] [PubMed] [Google Scholar]
- Willecke M., Hamaratoglu F., Sansores-Garcia L., Tao C., Halder G. (2008). Boundaries of Dachsous Cadherin activity modulate the Hippo signaling pathway to induce cell proliferation. Proc. Natl. Acad. Sci. USA 105, 14897-14902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Huang J., Dong J., Pan D. (2003a). hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445-456 [DOI] [PubMed] [Google Scholar]
- Wu S., Huang J., Dong J., Pan D. (2003b). hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445-456 [DOI] [PubMed] [Google Scholar]
- Zender L., Spector M. S., Xue W., Flemming P., Cordon-Cardo C., Silke J., Fan S. T., Luk J. M., Wigler M., Hannon G. J., et al. (2006). Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253-1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q., Hong W. (2008). The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell 13, 188-192 [DOI] [PubMed] [Google Scholar]
- Zhang N., Bai H., David K. K., Dong J., Zheng Y., Cai J., Giovannini M., Liu P., Anders R. A., Pan D. (2010). The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 19, 27-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Conrad C., Xia F., Park J. S., Payer B., Yin Y., Lauwers G. Y., Thasler W., Lee J. T., Avruch J., et al. (2009). Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16, 425-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}