

Abstract
The ductus arteriosus is an arterial vessel that shunts blood flow away from the lungs during fetal life, but normally occludes after birth to establish the adult circulation pattern. Failure of the ductus arteriosus to close after birth is termed patent ductus arteriosus and is one of the most common congenital heart defects. Mice with smooth muscle cell-specific deletion of Jag1, which encodes a Notch ligand, die postnatally from patent ductus arteriosus. These mice exhibit defects in contractile smooth muscle cell differentiation in the vascular wall of the ductus arteriosus and adjacent descending aorta. These defects arise through an inability to propagate the JAG1-Notch signal via lateral induction throughout the width of the vascular wall. Both heterotypic endothelial smooth muscle cell interactions and homotypic vascular smooth muscle cell interactions are required for normal patterning and differentiation of the ductus arteriosus and adjacent descending aorta. This new model for a common congenital heart defect provides novel insights into the genetic programs that underlie ductus arteriosus development and closure.
Keywords: Congenital heart defects, Notch signaling pathway, Vascular smooth muscle differentiation, Mouse
INTRODUCTION
The evolutionarily conserved Notch signaling pathway plays a major role in vascular development in mammals and other vertebrates (Gridley, 2010; Hofmann and Iruela-Arispe, 2007; Phng and Gerhardt, 2009; Roca and Adams, 2007). Among the developmental decisions regulated by the Notch pathway during embryonic vascular development are arterial-venous specification, endothelial tip cell differentiation, and vascular smooth muscle cell differentiation. Whereas most Notch signaling pathway components are expressed in endothelial cells, Jag1 (which encodes a ligand for Notch pathway receptors) is unusual in that it is expressed in both endothelial cells and vascular smooth muscle cells (Villa et al., 2001). Mice homozygous for a Jag1 null mutation die from hemorrhage early during embryogenesis, exhibiting defects in remodeling of the embryonic and yolk sac vasculature (Xue et al., 1999). Endothelial cell-specific deletion of Jag1 with a Cre deleter that is highly expressed during embryogenesis also results in early embryonic lethality owing to vascular defects resulting from the defective development of vascular smooth muscle (High et al., 2008). Similarly, endothelial cell-specific deletion of Jag1 using an inducible Cre deleter line results in reduced coverage of retinal arteries by vascular smooth muscle cells (Benedito et al., 2009). However, it has not been established whether Jag1 plays an essential, cell-autonomous role in the vascular smooth muscle cell lineage.
We report here the phenotype of embryos and mice with smooth muscle-specific Jag1 deletion. These mice die in the early postnatal period from patent ductus arteriosus (PDA), a defect of the outflow tract of the heart. The ductus arteriosus is an arterial blood vessel that connects the pulmonary artery and the descending aorta during fetal life. After birth, the ductus arteriosus is normally rapidly and permanently occluded, separating the pulmonary and systemic circulations to establish the normal adult circulatory pattern. Failure of the ductus arteriosus to close after birth is termed PDA and is one of the most common human congenital heart defects. PDA patients are at increased risk of pulmonary and cardiac problems such as pulmonary hemorrhage, congestive heart failure, chronic lung disease, sepsis and necrotizing enterocolitis (Clyman, 2006; Forsey et al., 2009; Schneider and Moore, 2006). Mice with smooth muscle-specific deletion of Jag1 exhibit defects in contractile smooth muscle cell differentiation in the vascular wall of the ductus arteriosus and adjacent descending aorta. These defects appear to arise through an inability to propagate the JAG1-Notch signal by lateral induction throughout the vascular wall of these vessels. Our novel model for this common congenital heart defect provides new insights into the genetic programs that underlie ductus arteriosus development and closure.
MATERIALS AND METHODS
Mice
We described previously the Jag1 null allele (Xue et al., 1999) and Jag1 conditional allele (Jag1flox) (Kiernan et al., 2006). SM22a-Cre (official name Tagln-Cre) (Holtwick et al., 2002) and Tek-Cre (also known as Tie2-Cre) (Koni et al., 2001) mice were obtained from the Jackson Laboratory. Animal protocols were approved by the Jackson Laboratory Animal Care and Use Committee.
Intracardiac injections
To better visualize the outflow tract and ductus arteriosus, hearts of SM22a-Cre/+; Jag1flox/– mutant and control littermate mice were injected with Microfil silicone rubber injection compound (MV-122; Flow Tech). Pups were isolated at E18.5 by caesarean section, and were euthanized 7 hours post-surgery. The chest cavities were opened and fixed in 10% neutral buffered formalin. After fixing for 1 hour, neonates were rinsed and Microfil compound was injected into the left ventricle using a 27 gauge needle.
Indomethacin treatment
To determine whether postnatal indomethacin administration could rescue closure of the ductus arteriosus of SM22a-Cre/+; Jag1flox/– neonatal mice, newly born pups were injected subcutaneously with indomethacin (6 mg/kg body weight) within 12 hours of birth. Pups were euthanized 6 hours after injection, the chest cavities were opened and closure of the ductus arteriosus was scored visually. In some treated mice, outflow tracts were visualized by Microfil injection.
Histology and immunofluorescence
Embryos were fixed in Dent's (20% DMSO, 80% methanol) and/or 4% paraformaldehyde. Chest cavities were embedded in paraffin, sectioned and stained with Hematoxylin and Eosin. For immunohistochemistry, the sections were de-waxed in a standard xylene and ethanol series, then rehydrated with phosphate-buffered saline (PBS). An antigen-retrieval step was performed in boiling 10 mM sodium citrate (pH 6.0) for 10 minutes for antibodies except anti-PECAM1 (0.01% trypsin for 15 minutes at 37°C) and anti-phospho-histone H3 (10 mg/ml proteinase K for 5 minutes). Slides were then blocked in 5% goat serum and 2% BSA in PBST (PBS + 0.1% Tween-20) for 2 hours at room temperature before being incubated with diluted primary antibodies at 4°C overnight. Tyramide-amplified immunofluorescent staining for the NOTCH1 Val1744 epitope was performed as described (Del Monte et al., 2007). Primary antibodies used in this study include: anti-PECAM1 (BD Biosciences Pharmingen; 1:100; only works for Dent's-fixed embryos); polyclonal anti-smooth muscle 22α protein (Abcam; 1:400); polyclonal anti-smooth muscle alpha actin (Abcam; 1:200); polyclonal anti-smooth muscle myosin heavy chain (Biomedical Technologies; 1:200); anti-phospho-histone H3 (Upstate Biotechnology; 1:100); polyclonal anti-JAG1 (Santa Cruz, H-114; 1:50); polyclonal anti-cleaved NOTCH1 (Val1744; Cell Signaling); and polyclonal anti-NOTCH3 (Abcam ab23426; 1:250). Alexa Fluor fluorescent secondary antibodies (Molecular Probes), which included goat anti-rabbit Alexa Fluor 488 and 555, goat anti-rat Alexa Fluor 488 and 555, were applied for 2 hours at room temperature or at 4°C overnight. All slides were mounted using Vectashield mounting medium with DAPI (Vector Laboratories).
Microscopy
For transmission electron microscopy, chest cavities of E18.5 and P0 mice were fixed in 2% glutaraldehyde with 0.1 M sodium cacodylate (pH 7.2) overnight at 4°C. The ductus arteriosus was carefully dissected out and embedded in resin at 70°C for 48 hours. Sections (90 nm) were collected on a 300-mesh copper grid and stained with 2% uranyl acetate and Reynold's lead, then viewed on a JEOL 1230 JM transmission electron microscope. For light microscopy, histological and immunofluorescent images were taken with a Zeiss Axioplan microscope or a Leica SP5 confocal microscope.
Quantitative (q) RT-PCR
The ductus arteriosus and ascending aorta from P0 neonates and E18.5 embryos were microdissected and immersed in RNAlater (Ambion). Genotypes were identified by allele-specific PCR. We combined two to five ductus arteriosi or ascending aortas of the same genotype to increase the RNA extraction yield. RNA was prepared using the Qiagen Mini mRNA Extraction Kit. RNA (100 ng of each sample) was reverse transcribed with random hexamers (Ambion). qRT-PCR was performed with the Super SYBR Master Mix Kit (Applied Biosystems). For each gene tested we performed three experimental repeats and three biological repeats. Expression levels were normalized to Gapdh expression. The study was performed on the ABI 7500 platform using SDS software. The primers used were designed by Primer Bank (Spandidos et al., 2008) (forward and reverse; 5′ to 3′): smooth muscle 22α, CAACAAGGGTCCATCCTACGG and ATCTGGGCGGCCTACATCA; smoothelin, ATGGCAGACGAGGCTTTAGC and AGTGTAGCCAGTTCTCCTTGTT; smooth muscle actin α2, GTCCCAGACATCAGGGAGTAA and TCGGATACTTCAGCGTCAGGA; smooth muscle actin γ2, CCGCCCTAGACATCAGGGT and TCTTCTGGTGCTACTCGAAGC; smooth muscle myosin heavy chain, AAGCTGCGGCTAGAGGTCA and CCCTCCCTTTGATGGCTGAG; calponin, AGAACCGGCTCCTGTCCAA and GTGTGCATAGGATAACCCCATC; Gapdh, AGGTCGGTGTGAACGGATTTG and TGTAGACCATGTAGTTGAGGTCA.
RESULTS
Mouse embryos with endothelial cell-specific deletion of the Jag1 gene phenocopy Jag1 null mutants
To assess whether Jag1 function is required in both endothelial and vascular smooth muscle cells, we performed tissue-specific Jag1 gene deletions. Endothelial-specific Jag1 deletion has been reported previously (High et al., 2008) using an independently generated Jag1 conditional allele. Endothelial cell-specific deletion of our Jag1 conditional allele (Kiernan et al., 2006) yielded results identical to those published previously (High et al., 2008). Endothelial cell-specific Jag1 mutant embryos phenocopied constitutive Jag1 null mutant embryos (Xue et al., 1999). Endothelial cell-specific Jag1 mutant embryos died at approximately embryonic day (E) 10.5, exhibiting yolk sac vascular remodeling defects (see Fig. S1 in the supplementary material), hemorrhages (see Fig. S1 in the supplementary material) and non-cell-autonomous defects in vascular smooth muscle cell differentiation (see Fig. S2 in the supplementary material). These results demonstrate that the early lethality of Jag1 null embryos is due to Jag1 function in endothelial cells, and that Jag1 expression in endothelial cells is required for the differentiation of adjacent mural cells into vascular smooth muscle.
Mice with smooth muscle-specific deletion of Jag1 die postnatally due to patent ductus arteriosus
To assess whether Jag1 function is also required in vascular smooth muscle cells, we performed smooth muscle-specific deletion using SM22a-Cre (also known as Tagln-Cre) mice (Holtwick et al., 2002). SM22a-Cre/+; Jag1flox/– (hereafter referred to as Jag1-SMcko, for Jag1 smooth muscle conditional knockout) mice were generated by crossing SM22a-Cre/+; Jag1+/– males with Jag1flox/flox female mice. Unlike embryos with endothelial cell-specific Jag1 deletion, Jag1-SMcko mice were found alive and in the expected Mendelian ratios at postnatal day (P) 0, the day of birth. However, ∼50% of the Jag1-SMcko neonates died by P1, and no Jag1-SMcko mice survived past P2 (Table 1). Jag1-SMcko mice in the early postnatal period could be identified by their cyanotic appearance, which suggested the possibility of cardiovascular defects. We therefore assessed the morphology of the heart and cardiac outflow tract in Jag1-SMcko and control littermate mice from E18.5 through P2. This analysis revealed that 95% (53/56) of Jag1-SMcko mice born exhibited PDA (Fig. 1B,C,G; Table 2; see Fig. S3 in the supplementary material). In addition to PDA, a subset of Jag1-SMcko mice also exhibited another outflow tract defect termed retroesophageal right subclavian artery (Fig. 1C; Table 2).
Table 1.
Survival of Jag1-SMcko embryos and mice

Fig. 1.

Jag1-SMcko mice exhibit patent ductus arteriosus. (A-C) Outflow tracts of P0 mice. In the control littermate (CT) outflow tract (A), the ductus arteriosus (arrowhead) has closed. The ductus arteriosus in Jag1-SMcko mice (B,C, arrowhead) is not closed and blood can be observed flowing through the still-patent ductus. The Jag1-SMcko mouse shown in B represents the most common Jag1-SMcko mutant phenotype. These mice have a normally patterned outflow tract, but exhibit patent ductus arteriosus (PDA). The mouse shown in C represents a subset of Jag1-SMcko mice that displays both PDA and retroesophageal right subclavian artery (arrow). (D,G) Histological analysis confirmed PDA in P0 Jag1-SMcko (G) versus control (D) mice. (E,F,H,I) Fluorescent immunostaining using markers for smooth muscle cells (orange, anti-SM22α antibody) and endothelial cells (green, anti-PECAM1 antibody) revealed reduced vascular smooth muscle cell differentiation in the ductus arteriosus and adjacent descending aorta of Jag1-SMcko (H,I) versus control (E,F) mice. F and I are higher magnification views of the descending aorta vessel wall shown in E and H, respectively. All sections are counterstained with DAPI to highlight cell nuclei (blue). aAo, ascending aorta; dAo, descending aorta; DA, ductus arteriosus; PT, pulmonary artery trunk; RCA, right common carotid artery; LCA, left common carotid artery; RSA, right subclavian artery. Scale bars: 1 mm in A-C; 200 μm in D,E,G,H.
Table 2.
Phenotypes of Jag1-SMcko embryos and mice

Jag1-SMcko mice exhibit defects in vascular smooth muscle differentiation in the ductus arteriosus and the adjacent descending aorta
Ductus arteriosus closure postnatally requires differentiated vascular smooth muscle cells to contract in response to a drop in circulating prostaglandin levels and a rise in oxygen tension that occur after birth. We assessed vascular smooth muscle cell differentiation in the walls of the outflow tract vessels by immunofluorescent staining for several smooth muscle contractile proteins. Using an antibody against smooth muscle 22α protein (SM22α) at P0 we found that, in contrast to the control littermate ductus arteriosus, which has multiple layers of differentiated smooth muscle cells occupying almost the entire width of the ductus arteriosus wall (Fig. 1E,F), the Jag1-SMcko ductus arteriosus has a much thinner region of SM22α-expressing cells adjacent to the endothelial cell layer (Fig. 1H,I). Reduced numbers of SM22α-expressing cells were also observed in the medial wall of the connecting descending aorta (Fig. 1H,I). Analyzing additional contractile smooth muscle cell markers, such as smooth muscle α-actin (SMA; also known as ACTA2) and smooth muscle myosin heavy chain (SMMHC; also known as MYH11), defects in vascular smooth muscle cell differentiation in the ductus arteriosus and descending aorta of Jag1-SMcko embryos could be observed at E17.5 (Fig. 2A-C versus 2A′-C′), when the ductus arteriosus in both the Jag1-SMcko and littermate control embryos is still patent. Surprisingly, we found that vascular smooth muscle cell differentiation defects in Jag1-SMcko mice were restricted to the region of the ductus arteriosus and the adjacent descending aorta. Both the ascending limb of the aorta (Fig. 2D′-F′) and the pulmonary artery trunk (Fig. 2G′-I′) of the mutant embryos exhibited normal differentiation of contractile vascular smooth muscle cells throughout the medial wall.
Fig. 2.

Jag1-SMcko mouse embryos exhibit regionally restricted vascular smooth muscle cell differentiation defects. (A-I′) Immunofluorescent staining of outflow tract vessels in E17.5 control littermate (A-I) and Jag1-SMcko (A′-I′) embryos stained with antibodies (red) against smooth muscle 22α (SM22), smooth muscle myosin heavy chain (SMMHC) and smooth muscle actin (SMA). All sections were counterstained with DAPI (blue). In the control (A-C), expression of these contractile smooth muscle cell proteins was observed throughout the width of the medial wall of the ductus arteriosus (DA) and descending aorta (dAo). However, in the Jag1-SMcko ductus arteriosus and descending aorta (A′-C′), expression of all three contractile smooth muscle cell proteins was observed only in the region adjacent to the endothelial cell layer lining the lumens of the ductus arteriosus and descending aorta. By contrast, in both the ascending aorta (aAo) (D-F versus D′-F′) and pulmonary artery (PA) (G-I versus G′-I′), no differences in expression of these contractile smooth muscle cell proteins were observed between the control littermates and Jag1-SMcko embryos. Asterisks (A-C′) mark the entry point of the ductus arteriosus (left side) with the descending aorta (right side). Scale bars: 100 μm for A-F′; 50 μm for G-I′.
Additional assays support our finding of defects in vascular smooth muscle cell differentiation in the outer layers of the medial wall of the ductus arteriosus and descending aorta of Jag1-SMcko mice. We assessed expression levels of genes involved in contractile smooth muscle cell differentiation by quantitative real-time RT-PCR of RNA isolated at P0 from the ductus arteriosus (which displays the differentiation defect) and ascending aorta (which does not display the defect) of Jag1-SMcko and control littermate mice. The expression of all these contractile smooth muscle cell marker genes was downregulated in the ductus arteriosus of Jag1-SMcko compared with wild-type littermates (Fig. 3). However, in RNA isolated from the ascending aorta, only the SMMHC gene (Myh11) exhibited substantial downregulation in Jag1-SMcko mutants. Ultrastructural analysis further revealed that whereas the control ductus arteriosus medial wall is composed of compact, well-organized smooth muscle cell layers throughout the width of the vessel wall (Fig. 4A,C), the Jag1-SMcko ductus arteriosus is composed of more loosely organized cells surrounded by thicker layers of extracellular matrix (Fig. 4B,D). Taken together, these data demonstrate that PDA in Jag1-SMcko mice is caused by defects in vascular smooth muscle cell differentiation in the ductus arteriosus and descending aorta.
Fig. 3.

Transcription of contractile smooth muscle cell marker genes is reduced in Jag1-SMcko mice. Relative gene expression levels were examined in ductus arteriosus (top) and ascending aorta (bottom) in P0 wild-type (WT) and Jag1-SMcko (CKO) mice. Genes included encode smooth muscle 22α (SM22), smooth muscle actin α2 (SMA2a), smooth muscle actin γ2 (SMA2g), smooth muscle myosin heavy chain (SMMHC), smoothelin (SML) and calponin 2 (CALP). P<0.05 for control versus mutant DA comparisons (Student's t-test); aAo comparisons (except the SMMHC comparison) were not statistically significant. Error bars indicate standard deviation of the mean.
Fig. 4.

Ultrastructural changes in the ductus arteriosus wall of Jag1-SMcko mouse embryos. (A-D) The tunica media of E18.5 control littermate ductus arteriosus (A) is more compactly organized than that of the Jag1-SMcko ductus arteriosus (B), particularly in layers distal to the vessel lumen (Lu). The ductus arteriosus medial wall of the control littermate (A) is composed of vascular smooth muscle cells with spindle-shaped nuclei (asterisks in high-magnification view C) forming compact, well-organized layers throughout the width of the vessel wall. These cells are surrounded by thin layers of extracellular matrix (arrows in C). The Jag1-SMcko ductus arteriosus is composed of more loosely organized cells (B), with irregularly shaped nuclei that are surrounded by thicker layers of extracellular matrix (arrows in high-magnification view D) than in the control (C). Asterisks mark smooth muscle nuclei. Scale bars: 20 μm in A,B; 2 μm in C,D.
A possible explanation for the regional restriction of the vascular smooth muscle cell differentiation defect in Jag1-SMcko mice is that SM22a-Cre-mediated recombination is more efficient in the ductus arteriosus and its adjacent descending aorta than it is in the pulmonary trunk and ascending aorta. Cre-mediated deletion of our Jag1flox allele generates a nonfunctional, but still immunoreactive, JAG1 protein that exhibits altered cellular trafficking (Kiernan et al., 2006) (Fig. 5). Detection of this nonfunctional JAG1 protein by immunofluorescence enabled us to assess Cre-mediated Jag1 deletion in the outflow tract of Jag1-SMcko embryos (Fig. 5). Whereas the wild-type JAG1 protein localized primarily around the cell surface (Fig. 5A), the nonfunctional JAG1 protein generated after deletion of the Jag1flox allele appeared to localize to the cytoplasm (Fig. 5B). Using this assay, we compared Jag1 gene deletion in Jag1-SMcko and control littermate embryos at E13.5. This analysis revealed equivalent Jag1 gene deletion in the embryonic ascending aorta (Fig. 5H), which does not exhibit the differentiation defect, and embryonic ductus arteriosus and descending aorta (Fig. 5F). These data demonstrate that regional restriction of the vascular smooth muscle cell differentiation defect in Jag1-SMcko embryos is not caused by regional differences in Jag1 gene deletion.
Fig. 5.
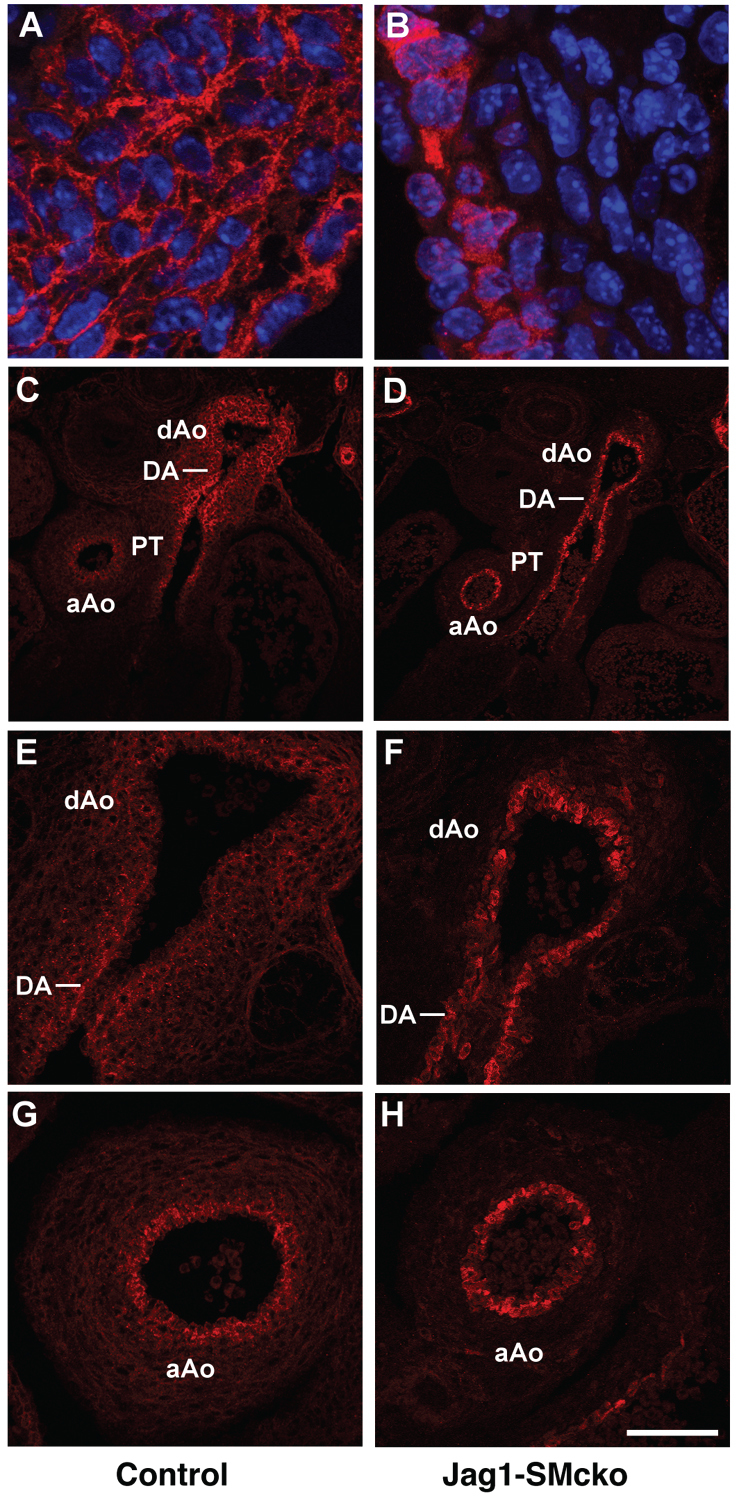
Regional restriction of vascular smooth muscle cell differentiation defects in Jag1-SMcko mouse embryos is not caused by regional differences in Jag1 gene deletion. (A) In the control littermate embryo, wild-type JAG1 protein (red) localizes primarily at the cell surface. Nuclei are stained with DAPI (blue). (B) However, in the Jag1-SMcko embryo, the nonfunctional JAG1 protein generated after deletion of the Jag1flox allele is localized to the cytoplasm. (C-G) The differences in JAG1 protein localization are apparent from immunostaining of outflow tract vessels of E13.5 control littermate (C,E,G) and Jag1-SMcko (D,F,H) embryos. In the mutant embryo, the same JAG1 staining pattern was observed in the ascending aorta (aAo) (H), which does not exhibit the vascular smooth muscle differentiation defect, as was observed in the embryonic ductus arteriosus (DA) and descending aorta (dAo) (F), which does exhibit the differentiation defect. PT, pulmonary artery trunk. Scale bars: 200 μm in C,D; 100 μm in E-H.
JAG1 regulates its own expression via lateral induction
Previous studies have shown that Jag1 is highly expressed in both endothelial cells and smooth muscle cells of large arteries at early embryonic stages in mice (High et al., 2008; Loomes et al., 1999). However, Jag1 expression in the outflow tract at later embryonic stages has not been closely examined. We assessed JAG1 protein expression by immunofluorescence from E13.5 through E16.5. At E13.5, JAG1 was expressed at high levels across the full width of the vessel wall of the ductus arteriosus and descending aorta (Fig. 6A,J). By contrast, in the ascending aorta and pulmonary artery trunk, only the layer of smooth muscle cells adjacent to the endothelial layer displayed a similarly high level of JAG1 expression (Fig. 6A,J versus 6D,G). The expression pattern seen at E13.5 was maintained at stages as late as E15.5 (see Fig. S4 in the supplementary material), although the high levels of JAG1 expression observed in the ductus arteriosus and descending aorta at E13.5 gradually declined. By E16.5, JAG1 expression had decreased to a low level in all parts of the outflow tract.
Fig. 6.

JAG1 protein is highly expressed in the ductus arteriosus and descending aorta and is required for its own expression. Transverse sections of E13.5 mouse embryos through the level at which the ductus arteriosus (DA) connects to the descending aorta (dAo). Sections are immunostained with JAG1 antibody (red); A-L are also counterstained with DAPI (blue) to highlight nuclei. (A-C) Low-magnification views showing the JAG1 expression pattern in the ductus arteriosus and descending aorta, ascending aorta (aAo) and pulmonary trunk (PT). (D-L) High-magnification views of the medial walls of the ascending aorta (D-F), pulmonary trunk (G-I) and descending aorta (J-L). (M-O) JAG1 immunofluorescent staining reveals the superimposition of the wild-type JAG1 and mutant JAG1 expression patterns throughout the entire medial wall in the descending aorta of SM22a-Cre/+; Jag1flox/+ embryos (O). Scale bars: 200 μm in A-C; 40 μm in D-L; 25 μm in M-O.
We utilized the altered cellular localization of the mutant JAG1 protein to assess the consequences of inactivation of JAG1 function in Jag1-SMcko embryos. Expression of mutant JAG1 protein in Jag1-SMcko ductus arteriosus and descending aorta was restricted to vascular smooth muscle cells in the region of the medial wall adjacent to the endothelium (Fig. 6B,K). This suggests that expression of functional JAG1 protein at the surface of differentiated vascular smooth muscle cells adjacent to the endothelium is required for JAG1 expression in adjacent smooth muscle cells located more distally in the ductus arteriosus and descending aorta medial wall.
To confirm that the mutant JAG1 protein is capable of being expressed throughout the medial wall of the ductus arteriosus and descending aorta, we analyzed JAG1 expression in SM22a-Cre/+; Jag1flox/+ embryos. These embryos contain the SM22a-Cre and Jag1flox alleles, but also have the wild-type Jag1 allele. SM22a-Cre/+; Jag1flox/+ embryos and mice therefore express both the wild-type JAG1 protein and the mutant JAG1 protein that exhibits altered cellular localization. Since JAG1-mediated Notch signaling is active in SM22a-Cre/+; Jag1flox/+ embryos, which exhibit no mutant phenotype, we should observe the superimposition of the wild-type JAG1 and mutant JAG1 expression patterns. Analysis of E13.5 SM22a-Cre/+; Jag1flox/+ embryos revealed that both the wild-type and mutant JAG1 proteins were expressed throughout the entire medial wall in SM22a-Cre/+; Jag1flox/+ embryos (Fig. 6F,I,L,O). Taken together, these results indicate that expression of functional JAG1 protein at the cell surface of smooth muscle cells adjacent to the endothelial cell layer is required for expression of JAG1 protein in smooth muscle cell layers located more distally, which is a hallmark of Notch signaling via lateral induction (Eddison et al., 2000; Lewis, 1998; Saravanamuthu et al., 2009).
Cleaved NOTCH1 protein is reduced in Jag1-SMcko embryos at E13.5
We assessed expression of cleaved NOTCH1 by utilizing the anti-Val1744 antibody, which recognizes an epitope generated by gamma secretase cleavage of the NOTCH1 protein (Del Monte et al., 2007; Schroeter et al., 1998). At E13.5, at the level of the connection between the ductus arteriosus and descending branch of the dorsal aorta, cleaved NOTCH1 protein was observed throughout the developing medial wall in the control littermate (Fig. 7A), whereas in the Jag1-SMcko mutant cleaved NOTCH1 was observed in only a few cells adjacent to the vascular endothelium (Fig. 7B). By E17.5, levels of cleaved NOTCH1 were reduced in the medial wall of both control and mutant embryos (Fig. 7C,D). However, cleaved NOTCH1 was still observed in both controls and Jag1-SMcko mutants at E17.5 in other tissues, such as in the hair follicles in the skin (Fig. 7E,F). Analysis of NOTCH3 expression revealed that in both controls and Jag1-SMcko mutants, expression was high throughout the medial wall of the ductus arteriosus and descending dorsal aorta at both E13.5 and E17.5. (see Fig. S5 in the supplementary material).
Fig. 7.

Cleaved NOTCH1 protein expression is reduced in Jag1-SMcko mouse embryos at E13.5. (A-F) Immunofluorescent staining with anti-cleaved NOTCH1 antibody (Val1744 epitope; red) of a control littermate (A,C,E) and Jag1-SMcko mutant (B,D,F) at E13.5 (A,B) and E17.5 (C-F). At the level of the connection between the ductus arteriosus (left) and descending branch of the dorsal aorta (right) (A-D), cleaved NOTCH1 protein was observed throughout the developing medial wall in the control littermate at E13.5 (A), whereas in the Jag1-SMcko mutant (B) expression was only observed in a few cells adjacent to the vascular endothelium. By E17.5, cleaved NOTCH1 protein was downregulated in the medial wall of both control and mutant embryos (C,D), whereas nuclear cleaved NOTCH1 protein was still observed at this stage in hair follicles in the skin (E,F, arrowheads). Scale bars: 100 μm.
Postnatal indomethacin injection rescues ductus arteriosus closure in a subset of Jag1-SMcko neonates
Prostaglandins are key regulators of ductus arteriosus patency. Cyclo-oxygenases 1 and 2 (COX1 and 2; PTGS1 and 2; formally known as prostaglandin H synthases 1 and 2) are rate-limiting enzymes involved in the conversion of arachidonic acid to prostaglandins. Non-steroidal anti-inflammatory drugs such as indomethacin and ibuprofen inhibit the formation of prostanoids by the COX enzymes (Mitchell and Warner, 2006). In premature infants exhibiting PDA, COX inhibitors are used clinically to induce ductus arteriosus closure by reducing prostaglandin levels, thereby inducing constriction of the ductus arteriosus and closure of its lumen.
We assessed whether indomethacin administration to naturally born neonatal mice could rescue the PDA of Jag1-SMcko neonates. Indomethacin administered by subcutaneous injection within 12 hours of birth rescued ductus arteriosus closure in ∼50% of the Jag1-SMcko mutants (Table 3; see Fig. S6 in the supplementary material). These data suggest that the ductus arteriosus of some Jag1-SMcko mutants retains sufficient smooth muscle to constrict in response to the drop in prostaglandin levels caused by postnatal indomethacin administration.
Table 3.
Indomethacin injection rescues ductus arteriosus closure in a subset of Jag1-SMcko mutant neonates

DISCUSSION
One of the primary physiological signals regulating patency of the ductus arteriosus is the level of circulating prostaglandins (Clyman, 2006; Forsey et al., 2009; Schneider and Moore, 2006). The most important of these is prostaglandin E2 (PGE2), which signals through the G protein-coupled receptor EP4 (also known as PTGER4). Targeted deletion of several genes encoding components of the prostaglandin signaling pathway, including the receptor EP4 (Nguyen et al., 1997; Segi et al., 1998), 15-hydroxy prostaglandin dehydrogenase (HPGD) (Coggins et al., 2002), the prostaglandin transporter PGT (also known as SLCO2A1) (Chang et al., 2010) and COX1 and 2 (Loftin et al., 2001), lead to PDA in neonatal mice. However, several recent studies have demonstrated that, in both mice and humans, defects in smooth muscle cell differentiation or function in the ductus arteriosus wall can cause PDA. Smooth muscle cells differ from most other differentiated cell lineages in adult mammals in that they are not irreversibly, terminally differentiated. Smooth muscle cells, unlike skeletal muscle cells and cardiomyocytes, retain the ability to reversibly alter their phenotype in response to various environmental, physiological and genetic cues (McDonald and Owens, 2007; Owens et al., 2004). This property has been termed phenotypic switching or smooth muscle cell plasticity. Contractile smooth muscle cells express high levels of contractile proteins involved in establishing and maintaining myofilament structure and function, including SM22α, SMA and SMMHC. By contrast, synthetic smooth muscle cells express lower levels of contractile muscle proteins and have higher rates of proliferation, migration and production of extracellular matrix components.
In humans, mutations in both the smooth muscle myosin heavy chain (MYH11) and the smooth muscle α-actin (ACTA2) genes are found in inherited forms of thoracic aortic aneurysm and dissection with PDA (Guo et al., 2007; Zhu et al., 2006). In mice, neural crest cell-specific deletion of the myocardin gene (Myocd) results in death of the mutant neonates from PDA (Huang et al., 2008). The Myocd conditional mutant mice exhibit reduced expression of smooth muscle-specific contractile proteins and display ultrastructural features indicating that vascular smooth muscle in the ductus arteriosus medial wall has acquired a synthetic, rather than a contractile, phenotype (Huang et al., 2008). Our analyses of Jag1-SMcko embryos and mice indicate that most smooth muscle cells in the medial wall of the ductus arteriosus and descending aorta fail to express contractile smooth muscle cell proteins. In the Jag1-SMcko mutants, the differentiation of contractile vascular smooth muscle cells is confined to the region adjacent to the endothelial cell layer.
Our studies of both Jag1-SMcko mutants and embryos with endothelial cell-specific Jag1 deletion indicate that Jag1-mediated Notch signaling induces Jag1 expression in the signal-receiving cells, which is a hallmark of Notch signaling via lateral induction (Eddison et al., 2000; Lewis, 1998; Saravanamuthu et al., 2009). Several other studies have also demonstrated that expression of Jag1 is induced by Notch signal reception (Ascano et al., 2003; Daudet et al., 2007; Eddison et al., 2000; Luo et al., 1997; Ross and Kadesch, 2004; Saravanamuthu et al., 2009), although some of these studies indicate that Jag1 might not be a direct Notch target gene (Ross and Kadesch, 2004). Significantly, a recent study demonstrated that both the JAG1 and NOTCH3 proteins were induced in vascular smooth muscle cells upon co-culture with endothelial cells (Liu et al., 2009).
Requirements for the NOTCH1 and NOTCH3 receptors have been studied previously in an in vivo vascular injury model (Li et al., 2009). Subsequent to vascular injury, smooth muscle cells in the vascular wall in the injured area proliferate and form a thickened layer of smooth muscle cells termed the neointima. Notch1+/– mice and mice with a heterozygous deletion of Notch1 in smooth muscle cells exhibit a 70% decrease in neointimal formation after carotid artery ligation. However, neointimal formation is unaffected after carotid artery ligation in Notch3–/– mice (Li et al., 2009). These data indicate that the NOTCH1 receptor plays a dominant role in vascular smooth muscle cells during the response to vascular injury. Whether NOTCH1 plays a similar dominant role during embryonic vascular smooth muscle development is not known.
Our analyses, along with those of others (Benedito et al., 2009; High et al., 2008), suggest a model for JAG1-mediated Notch signaling during development of the vascular wall of the ductus arteriosus and descending aorta (Fig. 8). In the ductus arteriosus and descending aorta of wild-type mice, endothelial cells expressing JAG1 protein undergo heterotypic interactions with adjacent vascular mural/smooth muscle cells expressing Notch receptors (most likely through the NOTCH1 receptor, although our data cannot exclude a role for the NOTCH3 receptor). Notch signal reception via lateral induction induces JAG1 expression in the receiving cells, leading to JAG1-mediated Notch signal transmission through homotypic interactions with the adjacent layer of smooth muscle cells. We further propose that Notch signal reception by vascular smooth muscle cells is required for their differentiation into contractile, rather than synthetic, smooth muscle cells. In embryos with endothelial cell-specific Jag1 deletion, the mutant endothelial cells are unable to signal to adjacent smooth muscle cells, which consequently differentiate into synthetic smooth muscle cells. In embryos with smooth muscle cell-specific Jag1 deletion, JAG1-expressing endothelial cells signal heterotypically to the adjacent layer of mutant vascular smooth muscle cells. These cells differentiate as contractile smooth muscle and upregulate expression of the mutant JAG1 protein. However, the Jag1-SMcko smooth muscle cells are unable to propagate the JAG1 signal through homotypic cellular interactions to more distal layers of smooth muscle, which therefore differentiate as synthetic smooth muscle cells.
Fig. 8.

Model for JAG1-Notch signaling in the ductus arteriosus and descending aorta. In the ductus arteriosus and descending aorta of wild-type mice, endothelial cells expressing JAG1 protein undergo heterotypic interactions with adjacent vascular mural/smooth muscle cells expressing Notch receptors (most likely through NOTCH1, although a role for NOTCH3 cannot be excluded). Notch signal reception via lateral induction induces JAG1 expression in the receiving cells, leading to JAG1-mediated Notch signal transmission through homotypic interactions with the adjacent layer of smooth muscle cells. We further propose that Notch signal reception by vascular smooth muscle cells is required for their differentiation into contractile, rather than synthetic, smooth muscle cells. In embryos with endothelial cell-specific Jag1 deletion, the mutant endothelial cells are unable to signal to adjacent smooth muscle cells, which consequently differentiate into synthetic smooth muscle cells. In embryos with smooth muscle cell-specific Jag1 deletion, JAG1-expressing endothelial cells signal heterotypically to the adjacent layer of mutant vascular smooth muscle cells. These cells differentiate as contractile smooth muscle and upregulate expression of the mutant JAG1 protein. However, these mutant smooth muscle cells are unable to propagate the JAG1 signal through homotypic cellular interactions to more distal layers of smooth muscle, which therefore differentiate as synthetic smooth muscle cells.
The mechanism that terminates the propagation of the JAG1-mediated signal in wild-type embryos is not understood. Our model proposes that the Jag1 gene is induced in vascular smooth muscle cell progenitors adjacent to the innermost layer of cells expressing the JAG1 ligand on their surface (Fig. 8). There is not an obvious signal to terminate propagation of this JAG1-mediated signal. Recent studies of NOTCH1-DLL1 signaling in mammalian cells have demonstrated that cis-interactions resulting in mutual inactivation of NOTCH1 and DLL1 in the same cell can generate a molecular switch between mutually exclusive signal-sending and signal-receiving states (Sprinzak et al., 2010). Whether such a mechanism contributes to termination of the JAG1-mediated signal during the development of vascular smooth muscle cells in the ductus arteriosus and descending aorta remains to be determined.
It is also unclear why the vascular smooth muscle cell differentiation defect in Jag1-SMcko mice is restricted to the ductus arteriosus and descending aorta. Our data indicate that regional restriction of the vascular smooth muscle cell differentiation defect is not caused by regional differences in Jag1 gene deletion. Jag1 deletion in the ascending aorta of the Jag1-SMcko embryos, which does not exhibit the differentiation defect, was equivalent to that in the embryonic ductus arteriosus and descending aorta, which do exhibit the defect (Fig. 5). The ductus arteriosus is a very specialized blood vessel, with a vascular wall composed of highly differentiated and contractile smooth muscle (Slomp et al., 1997). Smooth muscle of the ductus arteriosus activates a unique transcriptional program during its development (Ivey et al., 2008). This ductus arteriosus-specific transcriptional program might contribute to the regional restriction of the vascular smooth muscle differentiation defects exhibited by Jag1-SMcko mice.
The models for PDA exhibited by Jag1-SMcko mice and neural crest cell-specific Myocd gene deletion mice (Huang et al., 2008) represent a new paradigm for the etiology of PDA in mammals. In these mice, the differentiation of ductal smooth muscle cells along the synthetic, rather than the contractile, pathway leads to PDA. Further work will be required to determine whether this mechanism for the etiology of PDA occurs in humans.
Supplementary Material
Acknowledgments
We thank Greg Sousa and Chris Norton for helpful discussions. This work was supported by grants from the NIH (HD034883) and the March of Dimes Foundation (1-FY10-367) to T.G., and by a Center Grant from the National Cancer Institute (CA034196) to the Jackson Laboratory. Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.052043/-/DC1
References
- Ascano J. M., Beverly L. J., Capobianco A. J. (2003). The C-terminal PDZ-ligand of JAGGED1 is essential for cellular transformation. J. Biol. Chem. 278, 8771-8779 [DOI] [PubMed] [Google Scholar]
- Benedito R., Roca C., Sorensen I., Adams S., Gossler A., Fruttiger M., Adams R. H. (2009). The Notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124-1135 [DOI] [PubMed] [Google Scholar]
- Chang H. Y., Locker J., Lu R., Schuster V. L. (2010). Failure of postnatal ductus arteriosus closure in prostaglandin transporter-deficient mice. Circulation 121, 529-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyman R. I. (2006). Mechanisms regulating the ductus arteriosus. Biol. Neonate 89, 330-335 [DOI] [PubMed] [Google Scholar]
- Coggins K. G., Latour A., Nguyen M. S., Audoly L., Coffman T. M., Koller B. H. (2002). Metabolism of PGE2 by prostaglandin dehydrogenase is essential for remodeling the ductus arteriosus. Nat. Med. 8, 91-92 [DOI] [PubMed] [Google Scholar]
- Daudet N., Ariza-McNaughton L., Lewis J. (2007). Notch signalling is needed to maintain, but not to initiate, the formation of prosensory patches in the chick inner ear. Development 134, 2369-2378 [DOI] [PubMed] [Google Scholar]
- Del Monte G., Grego-Bessa J., Gonzalez-Rajal A., Bolos V., De La Pompa J. L. (2007). Monitoring Notch1 activity in development: evidence for a feedback regulatory loop. Dev. Dyn. 236, 2594-2614 [DOI] [PubMed] [Google Scholar]
- Eddison M., Le Roux I., Lewis J. (2000). Notch signaling in the development of the inner ear: lessons from Drosophila. Proc. Natl. Acad. Sci. USA 97, 11692-11699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsey J. T., Elmasry O. A., Martin R. P. (2009). Patent arterial duct. Orphanet. J. Rare Dis. 4, 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley T. (2010). Notch signaling in the vasculature. Curr. Top. Dev. Biol. 92, 277-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D. C., Pannu H., Tran-Fadulu V., Papke C. L., Yu R. K., Avidan N., Bourgeois S., Estrera A. L., Safi H. J., Sparks E., et al. (2007). Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488-1493 [DOI] [PubMed] [Google Scholar]
- High F. A., Lu M. M., Pear W. S., Loomes K. M., Kaestner K. H., Epstein J. A. (2008). Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 105, 1955-1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J. J., Iruela-Arispe M. L. (2007). Notch signaling in blood vessels: who is talking to whom about what? Circ. Res. 100, 1556-1568 [DOI] [PubMed] [Google Scholar]
- Holtwick R., Gotthardt M., Skryabin B., Steinmetz M., Potthast R., Zetsche B., Hammer R. E., Herz J., Kuhn M. (2002). Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 99, 7142-7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Cheng L., Li J., Chen M., Zhou D., Lu M. M., Proweller A., Epstein J. A., Parmacek M. S. (2008). Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J. Clin. Invest. 118, 515-525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivey K. N., Sutcliffe D., Richardson J., Clyman R. I., Garcia J. A., Srivastava D. (2008). Transcriptional regulation during development of the ductus arteriosus. Circ. Res. 103, 388-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan A. E., Xu J., Gridley T. (2006). The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet. 2, e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni P. A., Joshi S. K., Temann U. A., Olson D., Burkly L., Flavell R. A. (2001). Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193, 741-754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. (1998). Notch signalling and the control of cell fate choices in vertebrates. Semin. Cell Dev. Biol. 9, 583-589 [DOI] [PubMed] [Google Scholar]
- Li Y., Takeshita K., Liu P. Y., Satoh M., Oyama N., Mukai Y., Chin M. T., Krebs L., Kotlikoff M. I., Radtke F., et al. (2009). Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation 119, 2686-2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Kennard S., Lilly B. (2009). NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 104, 466-475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftin C. D., Trivedi D. B., Tiano H. F., Clark J. A., Lee C. A., Epstein J. A., Morham S. G., Breyer M. D., Nguyen M., Hawkins B. M., et al. (2001). Failure of ductus arteriosus closure and remodeling in neonatal mice deficient in cyclooxygenase-1 and cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 98, 1059-1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomes K. M., Underkoffler L. A., Morabito J., Gottlieb S., Piccoli D. A., Spinner N. B., Baldwin H. S., Oakey R. J. (1999). The expression of Jagged1 in the developing mammalian heart correlates with cardiovascular disease in Alagille syndrome. Hum. Mol. Genet. 8, 2443-2449 [DOI] [PubMed] [Google Scholar]
- Luo B., Aster J. C., Hasserjian R. P., Kuo F., Sklar J. (1997). Isolation and functional analysis of a cDNA for human Jagged2, a gene encoding a ligand for the Notch1 receptor. Mol. Cell. Biol. 17, 6057-6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald O. G., Owens G. K. (2007). Programming smooth muscle plasticity with chromatin dynamics. Circ. Res. 100, 1428-1441 [DOI] [PubMed] [Google Scholar]
- Mitchell J. A., Warner T. D. (2006). COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 5, 75-86 [DOI] [PubMed] [Google Scholar]
- Nguyen M., Camenisch T., Snouwaert J. N., Hicks E., Coffman T. M., Anderson P. A., Malouf N. N., Koller B. H. (1997). The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature 390, 78-81 [DOI] [PubMed] [Google Scholar]
- Owens G. K., Kumar M. S., Wamhoff B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767-801 [DOI] [PubMed] [Google Scholar]
- Phng L. K., Gerhardt H. (2009). Angiogenesis: a team effort coordinated by Notch. Dev. Cell 16, 196-208 [DOI] [PubMed] [Google Scholar]
- Roca C., Adams R. H. (2007). Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 21, 2511-2524 [DOI] [PubMed] [Google Scholar]
- Ross D. A., Kadesch T. (2004). Consequences of Notch-mediated induction of Jagged1. Exp. Cell Res. 296, 173-182 [DOI] [PubMed] [Google Scholar]
- Saravanamuthu S. S., Gao C. Y., Zelenka P. S. (2009). Notch signaling is required for lateral induction of Jagged1 during FGF-induced lens fiber differentiation. Dev. Biol. 332, 166-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D. J., Moore J. W. (2006). Patent ductus arteriosus. Circulation 114, 1873-1882 [DOI] [PubMed] [Google Scholar]
- Schroeter E. H., Kisslinger J. A., Kopan R. (1998). Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382-386 [DOI] [PubMed] [Google Scholar]
- Segi E., Sugimoto Y., Yamasaki A., Aze Y., Oida H., Nishimura T., Murata T., Matsuoka T., Ushikubi F., Hirose M., et al. (1998). Patent ductus arteriosus and neonatal death in prostaglandin receptor EP4-deficient mice. Biochem. Biophys. Res. Commun. 246, 7-12 [DOI] [PubMed] [Google Scholar]
- Slomp J., Gittenberger-de Groot A. C., Glukhova M. A., Conny van Munsteren J., Kockx M. M., Schwartz S. M., Koteliansky V. E. (1997). Differentiation, dedifferentiation, and apoptosis of smooth muscle cells during the development of the human ductus arteriosus. Arterioscler. Thromb. Vasc. Biol. 17, 1003-1009 [DOI] [PubMed] [Google Scholar]
- Spandidos A., Wang X., Wang H., Dragnev S., Thurber T., Seed B. (2008). A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics 9, 633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzak D., Lakhanpal A., Lebon L., Santat L. A., Fontes M. E., Anderson G. A., Garcia-Ojalvo J., Elowitz M. B. (2010). Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature 465, 86-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa N., Walker L., Lindsell C. E., Gasson J., Iruela-Arispe M. L., Weinmaster G. (2001). Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech. Dev. 108, 161-164 [DOI] [PubMed] [Google Scholar]
- Xue Y., Gao X., Lindsell C. E., Norton C. R., Chang B., Hicks C., Gendron-Maguire M., Rand E. B., Weinmaster G., Gridley T. (1999). Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8, 723-730 [DOI] [PubMed] [Google Scholar]
- Zhu L., Vranckx R., Khau Van Kien P., Lalande A., Boisset N., Mathieu F., Wegman M., Glancy L., Gasc J. M., Brunotte F., et al. (2006). Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 38, 343-349 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.