

Abstract
It has become clear in recent decades that the post-translational modification of protein cysteine residues is a crucial regulatory event in biology. Evidence supports the reversible oxidation of cysteine thiol groups as a mechanism of redox-based signal transduction while the accumulation of proteins with irreversible thiol oxidations is a hallmark of stress-induced cellular damage. The initial formation of cysteine sulfenic acid (SOH) derivatives, along with the reactive properties of this functional group, serves as a crossroads whereby the local redox environment may dictate the progression of either regulatory or pathological outcomes. Protein-SOH are established as transient intermediates in the formation of more stable cysteine oxidation products both under basal conditions and in response to several redox-active extrinsic compounds. This review details both direct and multi-step chemical routes proposed to generate protein-SOH, the spectrum of secondary reactions that may follow their initial formation and the arsenal of experimental tools available for their detection. Both the pioneering studies that have provided a framework for our current understanding of protein-SOH as well as state-of-the-art proteomic strategies designed for global assessments of this post-translational modification are highlighted.
1. Introduction
A correlation has been established between disruptions of cellular redox status and a number of age-related diseases including cancer, atherosclerosis and neurodegeneration (1). The elevated levels of oxidants associated with these pathologies are thought to mediate the chemical modification of proteins, lipids and nucleic acids. At the same time, organisms have evolved protein cysteine-based regulatory switches designed to sense and respond to oxidative stress and environmental toxicants (2). For example, molecular mechanisms whereby the initial stress-induced oxidation of cysteine residues is transmitted into alterations in gene expression are conserved from microbes to humans (3–5). The thiol functional group is exclusive to cysteine amongst other amino acids and therefore engenders cysteine residues with unique structural, catalytic and regulatory features within proteins. The nucleophilicity of the thiol group facilitates roles for cysteine residues in electron donation, disulfide bonding and metal ion coordination while leaving them acutely sensitive to post-translational modification by oxidants and electrophiles (6). Reversible oxidation of cysteine residues can trigger structural and functional changes in proteins and has been established as a means through which signal transduction pathways can be altered (7–8). On the other hand, irreversible cysteine oxidations may serve as biomarkers of diseases linked to oxidative stress (9). Since a variety of reactive metabolites are capable of generating protein-SOH intermediates, and the subsequent biochemistry of this functional group supports the progression to either reversible or irreversible products, protein-SOH are positioned at a critical point from which both regulatory and toxic outcomes may arise. Protein-SOH are implicated as transient derivatives in peroxide metabolizing enzymes, oxidant-responsive transcription factors and a diverse set of proteins that utilize reactive cysteine residues in their active site. Investigations performed in recent decades have cultivated a growing appreciation for the involvement of protein-SOH and their secondary reaction products in the modulation of signal transduction pathways. This article aims to review scientific evidence regarding 1) the various biochemical pathways proposed to produce protein-SOH, 2) the myriad of additional reactions that may follow the initial formation of protein-SOH and 3) the current state of methodologies designed to detect protein-SOH both in simple isolated preparations and complex biological samples.
2. Chemical Pathways to Protein-SOH Formation
Historical Perspective
Examinations of the mechanism of oxidation of low molecular mass thiol compounds initiated by various oxidants offered the first glimpses into the possible existence of the SOH moiety. A report on the oxidation of thiol compounds by H2O2 presented polarimetric and colorimetric data that suggested the oxidation of cysteine and glutathione to their corresponding disulfides by dithioformamidine proceeded through two consecutive bimolecular reactions that involved an SOH intermediate (10). The proposed mechanism consisted of the initial oxidation of the thiol to the SOH oxidation state followed by disulfide formation upon reduction of the SOH group by a second thiol compound. This reaction mechanism is still considered to be the major pathway for oxidant-induced disulfide formation. Shortly thereafter, cysteine-SOH was identified as the primary oxidation product upon the reaction of cysteine with permonosulfuric acid (11). Analysis of the precipitate formed following the oxidation of cysteine perchlorate in isoamyl alcohol established that cysteine-SOH sulfate was the chief constituent. Other early evidence for SOH as an oxidation product of low molecular mass thiols arises from observations of reaction stoichiometry. For example, a study on the oxidation of various thiol compounds by the dye 2,6-dichlorophenol indophenol described a reaction stoichiometry of 1 mole of dye reduced per mole of thiol oxidized and led the authors to conclude that an SOH functional group was formed (12). This dye to thiol ratio was indicative of the SOH product as opposed to the disulfide in which the reaction stoichiometry would be expected to be 2 moles thiol oxidized per mole of dye. Other critical observations based on studies of small thiol compounds were that oxidation rates increased as a function of increasing pH, while at constant pH, those thiols with the lowest pKa values were oxidized most rapidly (13). These observations of lipid peroxide-mediated thiol oxidation led the authors to conclude that the thiolate anion (S−) is the reactive species rather than the unionized thiol group.
While these studies on the oxidation of small thiol compounds were certainly of interest in terms of mechanism, whether or not these same principles held true in the more complex environment of protein thiol groups remained an open question. Based on observations of a protein sulfenyl iodide (same oxidation state as SOH) intermediate, a general hypothesis of a two-step oxidation, with the intermediacy of SOH, in the formation of protein disulfides was proposed (14). This study presented evidence based on reaction stoichiometry, radioisotope incorporation and reversibility that the single thiol group of the tobacco mosaic virus coat protein could be converted to a sulfenyl iodide derivative that was sufficiently stable to allow detection. The general two-step oxidation hypothesis was then proposed as was the idea that certain protein environments may provide steric stabilization of cysteine derivatives of the SOH oxidation state. A similar observation of a protein thiol derivative of the same oxidation state as SOH was described upon treatment of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with tetrathionate (15). This article reported the formation of protein sulfenyl thiosulfate groups following the stoichiometric reaction of tetrathionate with thiol groups of the enzyme. Studies on the inhibition of the cysteine protease papain by H2O2 further established the idea that protein-SOH derivatives may be formed. Papain, like GAPDH, is an enzyme with a highly-reactive active site cysteine residue that is essential for activity. Evidence suggested that the reversible inactivation of papain by H2O2 does not involve intra- or inter-protein disulfide formation since sedimentation coefficient data eliminated the possibility of dimer formation and the presence of only one thiol group per protein precluded the formation of an intra-protein disulfide (16). Based on these findings, the authors concluded that the inactivation of papain by oxidizing agents like H2O2 may involve the conversion of the active site cysteine to the SOH form.
A series of reports published in 1969 provided experimental evidence that further established the existence of protein-SOH. The first substantial indication that stable protein-SOH derivatives can be formed upon thiol oxidation was demonstrated by the inhibition of GAPDH following exposure to o-iodosobenzoate (17). This inhibition was said to be due to the modification of the active site cysteine residue to an SOH since the enzyme could be reactivated by sodium arsenite treatment. This reductant is selective for SOH as compared to disulfides and thus, this reversibility suggested the formation of SOH. This study also advanced the idea that certain protein cysteine residues may exist in a steric environment that may support the stabilization of these oxidation products. A second observation that year on the 1:1 (oxidant:thiol) reaction stoichiometry of GAPDH inactivation by o-iodosobenzoate also led other investigators to conclude that the active site cysteine thiol may be modified to the SOH oxidation state (18). Another study of GAPDH inactivation also attributed the enzyme inhibition to the formation of an active site SOH derivative (19). This detailed report of H2O2-mediated inactivation of GAPDH provided several lines of evidence for SOH formation. Once again, oxidation stoichiometry (1 mole H2O2:1 mole thiol), sodium arsenite reversibility and lack of disulfide formation pointed towards the SOH functional group as the oxidation product. These authors also noted a pH-dependence that suggested the reaction with H2O2 involved the thiolate anion rather than the unionized parent thiol. Excess H2O2 or prolonged incubation times led to the formation of irreversible oxidation products. Finally, a report on the oxidation of thiol groups in creatine kinase by iodine proposed an additional pathway to generate protein-SOH (20). Based on observations of reaction stoichiometry, the absence of iodine in the oxidized protein and small thiol-mediated reversibility, the authors concluded that SOH is a likely product. They proposed that the active site cysteine residue of creatine kinase is modified to SOH by iodine via the hydrolysis of an initial sulfenyl iodide intermediate.
Another crucial piece of the puzzle concerning the biological significance of protein-SOH formation would come two decades later. The previously mentioned studies all involved the oxidation of isolated target proteins by the addition of reagent oxidant compounds rather than enzymatically-generated oxidants. In 1991, it was demonstrated that the lone thiol group of albumin was oxidized upon incubation with xanthine oxidase and hypoxanthine (21). Experimental evidence revealed that disulfides were not formed, the modification was arsenite-reversible and co-incubation with catalase inhibited albumin thiol oxidation. These results clearly showed that H2O2 generated indirectly form the xanthine oxidase/hypoxanthine system oxidized the albumin thiol to an SOH group. As in previous studies, the cysteine modification progressively became irreversible with time, indicative of the further oxidation of the SOH intermediate to a sulfinic acid (SO2H) or sulfonic acid (SO3H) group. Based on the preference of these enzyme-generated oxidants for the albumin thiol compared to lipid targets and the ability of albumin to inhibit xanthine oxidase-mediated lipid peroxidation, the authors proposed that plasma protein thiols, mainly consisting of that of albumin, may act as sacrificial antioxidants in order to prevent lipid oxidation in the vasculature.
Hydrogen Peroxide
Roles have been described for H2O2 both as a second messenger in signal transduction and as a toxicant capable of damaging cellular components. H2O2 is generated endogenously and its’ levels are tightly regulated by peroxide-reducing enzymatic systems. This highly diffusible molecule is a secondary product of the superoxide generated by enzymes such as NADPH oxidase or during metabolic processes such as mitochondrial electron transport. Superoxide dismutates spontaneously to form H2O2 and this process can be accelerated by superoxide dismutase enzymes. Meanwhile, peroxide-metabolizing enzymes that utilize either heme iron or active site cysteines along with oxidant-induced transcriptional responses are able to balance fluxes in production. A paradigm has been established in which signaling pathways may be regulated by basal H2O2 levels (submicromolar concentrations) and stress responses can be initiated by higher concentrations. Much of the biological activity linked to this molecule is dependent on its ability to oxidize the thiol group of critical cysteine residues in proteins and as a two electron oxidant, the initial product formed by the reaction of H2O2 with thiols is an SOH (figure 1a). Since H2O2 does not react appreciably with protonated thiols in the absence of metals, cysteine pKa values will likely be a major determinant of H2O2 reactivity and may dictate specificity in signaling mediated by this molecule (22–23). It is also clear that factors beyond decreased pKa exert influence on the reactivity of particular protein cysteine residues with H2O2. For example, the peroxiredoxin (Prx) enzymes, in which the low pKa of reactive cysteine is not sufficient to explain their exceptional reactivity with H2O2, highlight how other features of these proteins are needed to account for their role as a biological scavenger of H2O2 (24).
Figure 1.
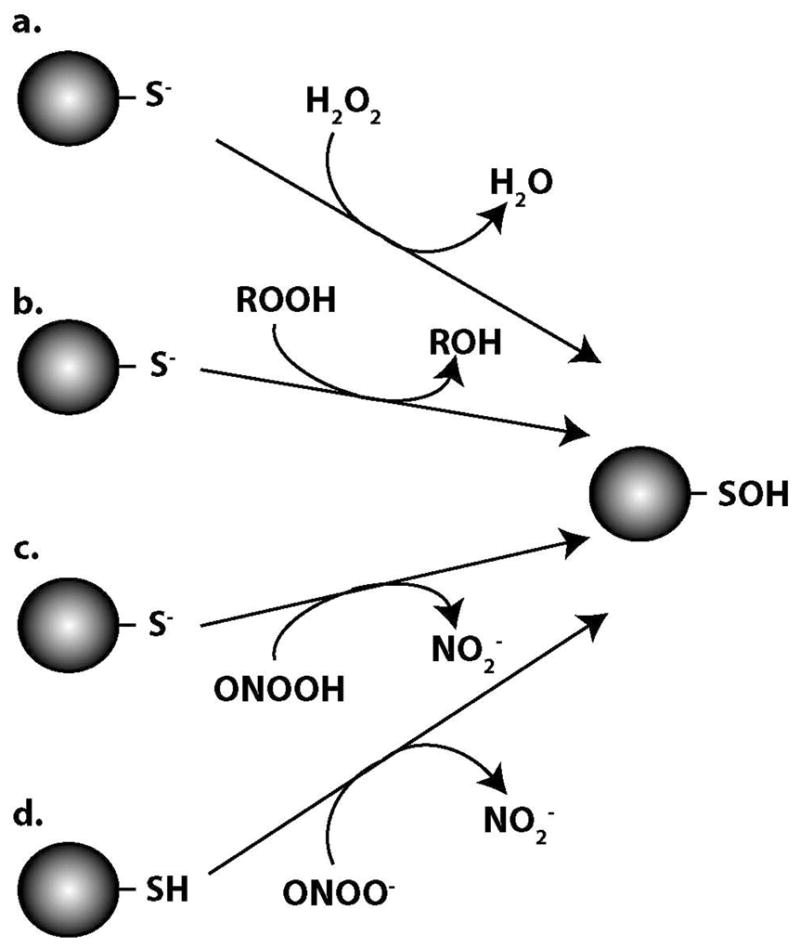
Proposed direct two-electron oxidation mechanisms of protein-SOH formation involving a) hydrogen peroxide, b) organic hydroperoxides, c) peroxynitrous acid and d) peroxynitrite anion.
H2O2 was shown to oxidize small thiol compounds such as glutathione to their corresponding disulfides and this oxidation was proposed to involve the intermediacy of an SOH derivative (10, 25). It later became apparent that H2O2 treatments led to the inhibition of thiol-dependent enzymes like GAPDH and papain through the oxidation of active site cysteine residues to disulfides and other oxidation products (15, 26). In the case of H2O2-mediated inactivation of papain, further analysis revealed that disulfides were not formed which led to the suggestion that inhibition occurred via the conversion of the thiol to an SOH group (16). Experimental evidence for H2O2-generated protein-SOH derivatives was provided shortly thereafter based on both the thiol oxidation stoichiometry and arsenite-reversibility of GAPDH inhibition by H2O2 (19). These investigators also observed a marked pH dependence of the rate of GAPDH inactivation that led them to propose that the mechanism involved the reaction of H2O2 with the deprotonated thiolate anion of the essential active site cysteine residue. This proposal was substantiated by a kinetic study on the H2O2-mediated oxidation of small thiol compounds to disulfides in which the rate limiting step was determined to be the nucleophilic attack of the thiolate on an oxygen atom of the H2O2 molecule resulting in the generation of the SOH and H2O (27). This initial step would be followed by the rapid reaction of the SOH intermediate with a second thiol molecule to generate the disulfide.
In more complex biological systems, investigators have detected H2O2 released from human neutrophils during phagocytosis (28–29). The H2O2 released from these cells was shown to be directly responsible for tissue damage associated with active inflammation as indicated by the destruction of cultured endothelial cells (30). Interestingly, H2O2 generated from activated neutrophils has also been demonstrated to oxidize serum protein thiol groups and deplete the levels of reduced glutathione in red blood cells (28, 31). Additionally, GAPDH in a native cellular environment underwent reversible thiol oxidation upon exposure of lung carcinoma cells to reagent H2O2 and xanthine oxidase-derived H2O2 readily oxidized plasma protein thiol groups (21, 32).
The decades since these initial discoveries have seen a multitude of studies focused on defining the impacts of H2O2-thiol interactions on cellular function and have resulted in a greater appreciation of the possible roles for protein-SOH intermediates. For example, H2O2-mediated protein-SOH formation has been shown to impact signal transduction cascades through both the inhibition of protein tyrosine phosphatases and activation of kinases leading to a general enhancement of phosphoprotein signaling cascades by H2O2 (33–34). Protein-SOH formation has also been described as an H2O2-sensing mechanism that can trigger altered gene expression in response to oxidative stress by either directly or indirectly activating transcription factors (3, 5, 35–36). Finally, protein-SOH derivatives have clearly been established as key intermediates in the cellular detoxification of H2O2 by a variety of active site thiol-containing peroxidase enzymes in which the two electron reduction of the peroxide is balanced by the two electron oxidation of the cysteine residue (37–39).
Organic Hydroperoxides
In addition to H2O2, organic-based hydroperoxides (ROOH) are also known to directly generate protein-SOH through a similar two electron oxidation of cysteine thiolates (figure 1b). Again, the mechanism involves the nucleophilic attack of the thiolate on an oxygen atom of the peroxide molecule leading to the oxidation of the cysteine to an SOH and the reduction of the ROOH to its corresponding alcohol. Early observations suggested that active site thiol-containing enzymes were especially sensitive to inhibition upon incubation with emulsions of fatty acid hydroperoxides (40). It was noted shortly thereafter that this inhibition of thiol-containing enzymes was due to fatty acid hydroperoxide-mediated oxidation of the protein thiol groups (41). As with the early studies of H2O2, these investigators collected data that indicated the oxidation products in many cases were not disulfides which again opened the possibility of the formation of protein-SOH. Studies on linoleic acid hydroperoxide (LAHP) mediated oxidation of thiols revealed that this compound generated monomeric protein oxidation products of the single cysteine-containing cytochrome c from S. oviformis and demonstrated that low pKa thiols were oxidized more rapidly upon incubation with LAHP (13, 42). These same investigators observed that LAHP oxidation of both protein and low molecular mass thiols was more rapid than that mediated by H2O2 and the 1:1 stoichiometry of thiols oxidized to peroxide added was consistent with the formation of SOH products (43). Several thiol-based cellular peroxidase enzymes, which catalyze the reduction of peroxides via formation of cysteine-SOH intermediates, are selective for the breakdown of organic hydroperoxides (39, 44–46). In addition to LAHP, other organic hydroperoxides such as ethyl hydroperoxide, benzoyl hydroperoxide, cumene hydroperoxide (CHP), and tert-butyl hydroperoxide (t-BHP) have been indicated as Prx substrates and generators of protein-SOH. For example, a sensing mechanism exists in which the reversible oxidation of a single conserved cysteine residue of the OhrR repressor in B. subtilis to cysteine-SOH upon exposure to CHP or t-BHP leads to derepression of the transcription of the organic peroxide resistance gene ohrA (47). Furthermore, widespread protein-SOH formation in response to both CHP and t-BHP treatments was demonstrated by the incorporation of an SOH-specific probe in E. coli (48).
Hypochlorous Acid and Chloramines
The production of chlorine-derived oxidants such as hypochlorous acid (HOCl) and N-chloramines (RNHCl) provide another pathway to protein-SOH formation. HOCl can be formed upon the peroxidase-catalyzed oxidation of Cl− by H2O2. The oxidizing equivalents of this species can be transferred via the secondary formation of RNHCl produced by the reaction of HOCl with deprotonated amines. Both of these compounds are thought to indirectly generate SOH following the hydrolysis of the initial sulfenyl chloride (RSCl) products formed upon their reaction with thiols (49–50) (figure 2b). The bactericidal action of chlorine was initially determined to be linked to the oxidation and subsequent inhibition of thiol-dependent enzymes (51). It was later confirmed using the myeloperoxidase/H2O2/chloride system that levels of thiol oxidation closely mirrored bactericidal action and that both HOCl and RNHCl were capable of oxidizing thiols (52).
Figure 2.
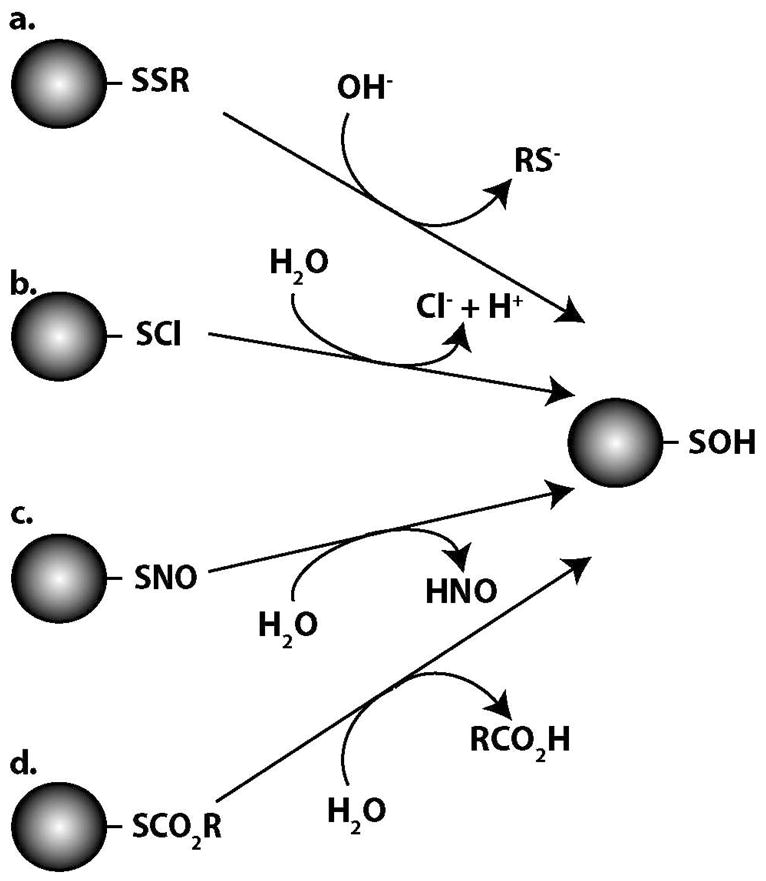
Proposed hydrolytic mechanisms of protein-SOH formation involving a) disulfide bonds, b) sulfenyl chloride intermediates, c) S-nitrosothiols and d) sulfenic esters.
It was also demonstrated that chloroperoxidase catalyzed the oxidation of a small thiol compound to its corresponding sulfenyl chloride in a chloride and H2O2-dependent manner and these results could be duplicated by substituting reagent HOCl for the enzyme system (53). Interestingly, the authors also found that this RSCl intermediate decomposed to form a stable SO3H which could also be generated by excess HOCl. Observations of the stoichiometry of oxidizing equivalent consumption (3 moles HOCl:1 mole thiol) also suggested the formation of SO3H products in bacterial proteins (52). The detection of the stable SO3H end products in each case supports the possible intermediacy of the SOH oxidation state. In fact, a more firm declaration of this mechanism appeared in a kinetic study of the oxidation of glutathione by HOCl in which the initial S-chloro derivative was proposed to undergo hydrolysis to yield the corresponding SOH (49). Others have invoked the hydrolysis of HOCl-generated RSCl to yield SOH in the context of myeloperoxidase-mediated oxidative cross-linking of low density lipoprotein and HOCl-mediated formation of protein-SOH has been demonstrated by the detection of protein-dimedone adducts by mass spectrometry (MS) (54–55).
In addition to the myeloperoxidase system, isolated neutrophils were shown to generate chloramines that were capable of oxidizing thiol compounds to their corresponding disulfides (56). More recently, observations of a 1:1 reaction stoichiometry of chloramines added to thiols oxidized for both isolated proteins and plasma were indicative of SOH formation (57–58). More conclusively, chloramine-mediated albumin-SOH formation was demonstrated by the detection of the distinctive absorbance peak at 347 nm following 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl)-adduct formation (57).
Nitric Oxide
Endogenous production of the free radical gas nitric oxide (NO) by the NO synthase enzymes is involved in a number of homeostatic regulatory pathways and stress-induced responses. While a significant portion of the signaling properties of NO are due to the cGMP-dependent cascades triggered by its interaction with the heme iron of soluble guanylyl cyclase, the interactions of NO and its reactive metabolites with thiols are also biologically significant. The formation of protein S-nitrosothiols (RSNO) has received particular attention in the context of NO-mediated signal transduction although several redox-based thiol post-translational modifications, including SOH formation may result from NO production.
Early evidence demonstrated that NO can oxidize a variety of thiol compounds to their corresponding disulfides (59). Based on their observations, these investigators proposed a mechanism in which the nucleophilic addition of a thiolate to NO is followed by protonation giving rise to a thionitroxide radical intermediate (RSNOH•) that upon radical coupling and product decomposition can generate the disulfide. A later study on the anaerobic reaction of NO with the thiol group of albumin established a possible route to protein-SOH formation in response to NO (60). The reported reaction stoichiometry, product reactivity with glutathione and protein-dimedone adduct formation indicated the conversion of the lone albumin thiol to a stable SOH product. The authors proposed the initial formation of a thionitroxide radical intermediate as well as the subsequent reaction of this intermediate with a second NO molecule to eventually generate albumin-SOH and N2O. The intermediacy of SOH was also invoked in the mechanism of glutathione disulfide formation in response to the anaerobic treatment of glutathione with NO (61). Crystal structure data for glutathione reductase and MS-based detection of dimedone adducts of cathepsin K following treatments with NO-donor compounds provide further evidence that, in addition to S-nitrosation, oxidation of protein thiols to SOH must be considered as a possible mechanism of action for NO and its various reactive metabolites (62–63). On a related note, an additional mechanism was proposed in which NO-mediated protein-SOH formation may occur through the hydrolysis of an initially formed RSNO (64) (figure 2c). While multiple chemical pathways are likely involved in the generation of RSNO from NO in aerobic systems, the contribution of the direct reaction of NO with thiolates as described in the initial step in NO-mediated SOH formation was negligible (65). Furthermore, the NO oxidation product nitrite has recently been demonstrated to generate SOH via oxygen atom transfer mediated by ferriheme compounds (66).
Peroxynitrite
Peroxynitrite, the term commonly used to refer to both the peroxynitrite anion (ONOO−) and the corresponding protonated peroxynitrous acid (ONOOH), is a potent oxidant formed by the rapid radical-radical reaction of NO with superoxide (O2.•−). The parallel generation of NO and O2•− by numerous cell types, especially during inflammatory responses, leads to peroxynitrite production in vivo. The oxidation of a wide range of biological targets is thought to potentiate the cytotoxic effects associated with peroxynitrite.
It was initially demonstrated that peroxynitrite efficiently oxidized both low molecular mass (cysteine) and protein (albumin) thiol groups (67). The observed end products were the disulfide for cysteine and irreversible higher oxides (likely SO2H or SO3H) for albumin. The pH dependence of the thiol oxidation suggested that ONOO− was the primary oxidizing species and that the reaction was with the protonated thiol group (figure 1d). While this initial observation of thiol oxidation supported a two-electron oxidation mechanism, other reports demonstrated that strong one-electron oxidants such as the hydroxyl radical and nitrogen dioxide radical could be produced during the decomposition of peroxynitrous acid therefore introducing additional indirect pathways for peroxynitrite-mediated thiol oxidation (68–69). On a related note, the peroxynitrite-mediated oxidation of both vitamin E and methionine were found to proceed through competing one and two-electron pathways although it appeared that the direct two-electron pathway was the dominant route (70–71). Interestingly, the one-electron oxidation product (thiyl radical (S•)) of albumin and glutathione was detected in EPR spin trapping studies of thiol oxidation by peroxynitrite (72–73). Again, it was emphasized that the thiyl radical was only a minor product (1–2%) of peroxynitrite-mediated thiol oxidation (73). The two possible reaction pathways were discussed at length by Quijano et al. in that 1) the ONOO− reacts with the protonated thiol involving a two-electron oxidation that yields an SOH intermediate and nitrite and 2) ONOOH or a secondary species derived from it participate in a one-electron oxidation generating the thiyl radical and nitrogen dioxide with both pathways ultimately leading to disulfide formation (74). A later proposal included two possible two-electron oxidation pathways in which the SOH can be generated; either the reaction of ONOO− with the thiol as described above or the reaction of ONOOH with the thiolate anion (75) (figure 1c).
Evidence for the formation of protein-SOH in response to peroxynitrite was initially provided in a description of the peroxynitrite reductase activity of the bacterial Prx AhpC (76). These investigators detected protein-NBD adducts by MS that were consistent with the formation of a Cys46-SOH intermediate following exposure of AhpC to peroxynitrite and, in doing so, demonstrated that the Prx family of proteins was able to catalytically detoxify peroxynitrite in addition to peroxides. Intermediate SOH formation was also detected as the primary reaction product at the active site cysteine of bovine 1-Cys Prx following peroxynitrite treatment based on analysis of 2-nitro-5-thiobenzoate (TNB)-reactivity (77). Subsequent observations of both reaction stoichiometry and end products following peroxynitrite-mediated oxidation of parasite, yeast and human Prxs further established a likely role for the direct two-electron oxidation of their active site cysteine residues to the SOH oxidation state during the catalytic decomposition of peroxynitrite (78–80). This idea is also supported by a study in which the expression of a mitochondrial Prx (Prx-3) was found to be neuroprotective against peroxynitrite-induced excitotoxic injury in vivo (81). Additionally, albumin-SOH formation has been confirmed by UV-visible spectral analysis of NBD adducts following peroxynitrite treatment (82). Since albumin-SOH formation was observed in both the presence and absence of carbon dioxide, which directs peroxynitrite reactivity towards one-electron oxidation mechanisms, additional radical-mediated pathways of SOH formation were proposed in which sulfinyl radical (RSO•) intermediates may be reduced by antioxidants or initially generated nitrothiols (RSNO2) may condense with water.
Hydrolysis of Disulfides
Another biochemical event that results in the formation of SOH intermediates is the hydrolysis of disulfide bonds. The existence of products of the SOH oxidation state has been discussed in the context of several hydrolytic mechanisms. Observations made in early studies on the decomposition of small disulfide-linked compounds led to suggestions of SOH generation in the initial step of hydrolysis (83–84).
An alkaline hydrolysis mechanism exists that involves the hydroxide anion (OH−) initiated cleavage of disulfides and is suggested to occur spontaneously at alkaline pH or be facilitated by enzymes at neutral pH (figure 2a). Structural analysis of products of alkali-mediated decomposition of small organic disulfides supported a mechanism involving the direct nucleophilic attack of OH− on a sulfur atom initiating disulfide decomposition and thereby generating a thiolate and an SOH-derivative (85). Additional studies on the alkaline hydrolysis of ovomucoid and albumin protein disulfides were also consistent with this reaction scheme (86–88). More recently, this mechanism has been proposed as the initial step in a scheme for microplasmin formation in alkaline solution that involves disulfide rearrangements in the parent plasmin protein (89). Similarly, the production of angiostatin from plasmin involves disulfide bond reduction and it was demonstrated that an enzyme is able to facilitate the same disulfide cleavage at neutral pH as OH− achieves at alkaline pH (90). It was subsequently shown that phosphoglycerate kinase (PGK) was responsible for this plasmin reductase activity and a scheme was proposed in which PGK facilitates cleavage of a specific plasmin disulfide bonds by OH− and results in the formation of an SOH intermediate and the opportunity for new disulfide arrangements (91–92).
Other disulfide hydrolytic mechanisms have been proposed that also involve the generation of protein-SOH intermediates. An acid-base assisted hydrolysis mechanism was discussed in the context of ligand activation of the delta opioid receptor. This mechanism involves a disulfide bridge between extracellular loops of the receptor as well as a neighboring aspartic acid residue (93). Interaction of both the carboxylic acid of the aspartic acid and a primary amine of the opioid ligand with the protein disulfide was proposed to polarize and cleave the bond thereby resulting in a conformational change and receptor activation. The initial sulfenic ester product generated upon disulfide cleavage was proposed to hydrolyze to generate an SOH-derivative (figure 2d). Finally, the formation of SOH derivatives as a result of hydroxyl radical (OH•) mediated cleavage of protein disulfides was recently described in the context of hinge cleavage in recombinant human IgG1 antibodies (94). This radical-induced cleavage of the disulfide bond between two hinge cysteines was proposed to lead to the formation of a thiyl radical on one cysteine and an SOH on the other.
Additional Mechanisms
Several other biochemical pathways to SOH formation have been proposed in studies on the reactivity of thiols or thiol-based intermediates with a number of compounds. The reaction of small thiosulfinate compounds (RS(O)SR) with protein cysteine residues has been shown to form protein mixed disulfides with the additional generation of SOH-derivative by-products. For example, mixed disulfide formation was detected following the reaction of the bioactive garlic component allicin with papain and the reaction of glutathione disulfide-S-oxide with rat brain proteins (95–96). In both cases sulfur exchange reactions were proposed in which a protein mixed disulfide is generated and an SOH derivative of the small thiol compound is a leaving group however the possibility of similar reactions producing protein-SOH were not addressed. Complex free radical chemistry can also create additional routes to SOH formation as discussed in the context of thiol-based radical repair mechanisms (97). The initial generation of a thiyl radical alone offers multiple biochemical pathways to the formation of SOH products. The thiyl radical may directly combine with a hydroxyl radical to generate an SOH. Additionally, the reaction of the thiyl radical with O2 produces a thiylperoxyl radical (RSOO.) which upon further reaction with another thiol generates an SOH and an RSO•. Likewise, the reaction of the RSO• with a thiol will form an SOH and an S•. An RSO• species may also be reduced by antioxidants thereby generating an SOH and a secondary radical (82). While these additional mechanisms of SOH formation have been proposed, the likelihood and extent to which they are operative as biological pathways of protein-SOH formation has yet to be established.
3. Reactivity of Protein-SOH Intermediates
The protein-SOH oxidation state is generally thought to be a transient intermediate in pathways leading to the formation of more stable thiol oxidation products. The reactivity of this functional group as both a nucleophile and an electrophile leads to a range of possible reactions that will be discussed below. Previous reviews of protein-SOH by Claiborne and colleagues outlined the major factors that are likely to influence the stability of this modification based on the structural properties of individual proteins that were found to form more stable SOH adducts (98–99). These factors include; 1) limited solvent access to restrict reactions with cellular reductants, 2) the absence of proximal cysteine thiols that would react with the SOH to generate a disulfide, and 3) the presence of hydrogen bonding with neighboring amino acid residues favoring ionization to a sulfenate (SO−) derivative that is considered slightly more stable. However, the majority of protein-SOH that forms in biological systems is likely to participate in any one of a number of possible reactions that result in either secondary oxidation products or a regeneration of the parent thiol/thiolate.
Although indirect evidence had supported a possible role for SOH intermediacy in various pathways of thiol oxidation, NMR data collected in a study on the oxidation of a small thiol compound provided biophysical evidence that the SOH oxidation state was in fact an intermediate in the formation of both disulfides and higher sulfur oxides (100). A further study demonstrated that the most likely reaction in a homogenous mixture of small SOH compounds was that of a self-condensation leading to the formation of RS(O)SR (101). However, the relevance of this type of reaction for protein-SOH, especially in complex biological systems, is not clear due to both the low abundance of SOH present at any given time as well as steric hindrances at the sites of modification in many proteins. Several early studies of protein-SOH formation demonstrated the ability of mild reducing agents to reverse this modification and, in turn, regenerate the cysteine thiol at the site of modification (figure 3a). Trivalent arsenicals such as sodium arsenite and phenyl arsene oxide reduced GAPDH-SOH and the specificity of these reductants for SOH versus disulfides was a strong early indication for the existence of protein-SOH (17, 19, 102). Other mild reducing agents like sodium borohydride, ascorbate and azide were also shown to reverse the SOH modification (19, 103). A handful of small thiol compounds including dithiothreitol, glutathione and cysteine, as well as thiosulfate and thiourea were demonstrated to regenerate the active site thiol of GAPDH following SOH formation (17, 19, 102). While SOH repair by dithiols like dithiothreitol proceeds by a rapid concerted reaction, repair by monothiols is a two step process that involves initial mixed disulfide formation followed by a thiol-disulfide exchange reaction (104). Reactions of the SOH functional group with thiols to generate disulfides are arguably the most biologically significant reactions in which protein-SOH participate.
Figure 3.
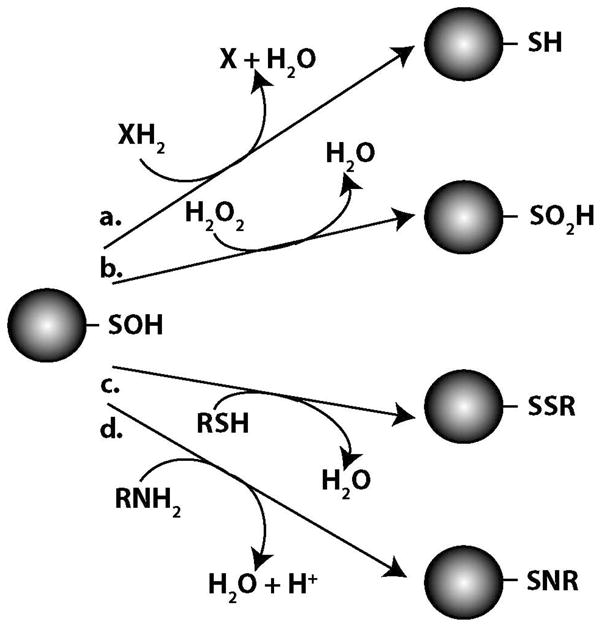
Potential reactions of protein-SOH intermediates. a) two-electron reduction to regenerate protein thiol. b) further oxidation to form sulfinic acid. c) reaction with second thiol to generate disulfide. d) reaction with amine to form sulfenamide.
The initial oxidation of cysteine and glutathione to the SOH oxidation state was proposed to be immediately followed by the reaction of these intermediates with another thiol compound to form disulfide-linked oxidation products (10). Similarly, observations of the 2:1 (SH:oxidant) reaction stoichiometry of H2O2-mediated oxidation of p-mercaptobenzoate to its corresponding disulfide suggested the intermediacy of an SOH derivative (105). Upon the formation of the SOH intermediate, a rapid reaction involving a secondary thiol group leads to the generation of a disulfide bond (figure 3c). This reactivity of the SOH functional group with thiols has since been exploited in attempts to detect protein-SOH. For example, the incorporation of the aromatic thiol compound TNB as a disulfide, along with the spectrophotometric changes that accompany this reaction, were used as evidence of SOH formation in NADH peroxidase (106). A genetically-encoded probe for tracking protein-SOH formation in vivo is also based on disulfide-mediated incorporation of a His-tagged domain of the Yap1 protein containing an SOH-reactive cysteine residue (48). Protein-SOH formation in biological systems likely leads to the generation of any one or all of the following disulfide linkages; 1) intraprotein disulfides upon reaction of the SOH with another cysteine within the same protein, 2) interprotein disulfides upon reaction of the SOH with a cysteine residue in a second protein or 3) mixed disulfides upon reaction of the SOH with a low molecular mass thiol-containing compound.
Reversible intraprotein disulfide formation through the intermediacy of cysteine-SOH has been demonstrated as the mechanism of activation of the OxyR transcription factor and the subsequent induction of antioxidant defenses in response to hydrogen peroxide (5). Additionally, the mechanism of methionine sulfoxide reduction catalyzed by the methionine sulfoxide reductase enzymes was described to involve the formation of a cysteine-SOH intermediate followed by the resolution of this derivative by a second cysteine thiol within the enzyme thereby generating an intramolecular disulfide (107). As opposed to these intramolecular disulfides generated upon reaction of an SOH derivative with a resolving cysteine thiol located within the same protein, interprotein disulfides can be generated when a resolving cysteine residue from a second protein is involved. For example, initial protein-SOH formation upon hydroperoxide reduction by Prx enzymes from S. cerevisiae and S. typhimurium lead to intersubunit dimerization through the formation of a disulfide bond (37, 108). Similar intersubunit disulfide formation through the intermediacy of an SOH has recently been described as a mechanism of H2O2-mediated activation of cGMP-dependent protein kinase isoform 1-alpha (34). In addition to descriptions of homodimer formation, interprotein disulfide formation between different proteins following the initial formation of an SOH derivative has been demonstrated to trigger a disulfide relay system in the activation of genes in response to oxidative stress (3). The generation of an SOH at the reactive active site cysteine residue of the Orp1 protein upon peroxide reduction transduces a signal through interprotein disulfide formation with the Yap1 transcription factor that subsequently leads to Yap1 intraprotein disulfide formation and activation of antioxidant gene expression (36).
The generation of protein mixed disulfides, broadly referred to as S-thiolation, describes the formation of disulfide linkages with various small thiol compounds. This includes the highly abundant cellular small thiol glutathione whose incorporation with proteins via mixed disulfide formation is referred to as S-glutathionylation. Roles for protein mixed disulfide formation have been strongly implicated in redox-regulated signal transduction (109). As with intra- and interprotein disulfide formation, mixed disulfide formation can certainly proceed through intermediacy of protein-SOH derivatives. Small thiol compounds bound to proteins via disulfides have been detected in cells under basal conditions (110). Additionally, oxidant challenges presented in both whole animal and cell culture studies were associated with extensive protein mixed disulfide formation (111–112). Interestingly, a report on oxidant-mediated protein mixed disulfide formation presented evidence in support of a formation mechanism that involves the reaction of an initially oxidized protein cysteine residue with a reduced small thiol compound rather than a thiol-disulfide exchange mechanism preceded by the formation of small disulfide compounds (113). The respiratory burst associated with phagocytosis in human neutrophils coincided with rapid protein mixed disulfide formation and was correlated to observations of microtubule disassembly (114). These results could be duplicated by peroxide treatments and presented a possible regulatory mechanism for protein oxidation in the control of cellular function. This protein mixed disulfide formation associated with the respiratory burst was later found to be chiefly mediated by H2O2 and again was likely to be independent of thiol-disulfide exchange (115–116). These data were suggestive of a route to mixed disulfide formation in which specific reactive cysteine residues in proteins are susceptible to an initial oxidative insult, thereby generating a cysteine-SOH intermediate, which can participate in a secondary reaction with any one of a number of compounds in the cellular pool of small thiols to produce the mixed disulfide. Along those lines, a number of studies have demonstrated that the generation of peroxide-mediated S-glutathionylated proteins and other mixed disulfides in key redox regulatory pathways involve the intermediacy of protein-SOH (117–120). Importantly, the conversion of protein-SOH to either intra-, inter-, or mixed disulfides retains the reversible aspects of the thiol modification through which a multitude of cellular reducing systems (both enzyme and small molecule based) can recycle the proteins to their native state.
Under conditions of either prolonged or more severe oxidative stresses, protein-SOH can be further oxidized to SO2H (figure 3b), which in turn can be oxidized to SO3H. These higher oxides are generally considered to be more stable derivatives and, in all but a few cases, biologically irreversible. Therefore, formation of these species in biological systems is more likely indicative of oxidative damage rather than regulatory processes since recovery of the native protein would require degradation of the modified species and new protein translation. Exposure of NADH peroxidase to non-physiological concentrations of H2O2 generated irreversible cysteine oxidation products corresponding to the SO2H and/or SO3H oxidation states based on observations of an acidic shift in isoelectric focusing and subsequent MS analysis as well as 13C NMR data (106, 121). Crystal structure data demonstrated the formation of higher cysteine oxides at the active sites of both glutathione reductase and protein tyrosine phosphatase 1B (62, 122). MS analysis of protein tyrosine phosphatase 1B following peroxide treatment detected the formation of SO2H and SO3H derivatives while the lack of arsenite or dithiothreitol reversibility of GAPDH inactivation by peroxynitrite was also used as evidence for higher oxide formation (123–124). An investigation of alpha-1 antitrypsin modification by H2O2 utilizing both NBD-Cl reactivity and isoelectric focusing at several time points established that SO2H and SO3H were generated via the intermediacy of the SOH oxidation state (125). The so-called overoxidation of Prx enzymes to SO2H and SO3H observed during oxidative stress is likely due to increased demand for peroxide decomposition and has garnered the most attention of modifications of this type from investigators. Methods involving either MS analysis of acidic variant spots on two-dimensional electrophoretic gels or oxidation state-specific antibodies have been developed to follow these modifications in cellular systems (126–127). While the generation of protein SO2H or SO3H derivatives on the majority of proteins is irreversible as indicated above, Prx-SO2H was found to be reversed to the parent thiol in cells following the removal of the oxidant (128). This reduction of Prx-SO2H derivatives was later demonstrated to be catalyzed by an enzyme subsequently named sulfiredoxin (129).
Interactions with amine groups are the final secondary reactions involving protein-SOH to be discussed here. The SOH derivative of GAPDH was shown to react with benzylamine to form a sulfenamide (RSNR’) product (130) (figure 3d). While this reaction with a small amine-containing compound was useful for identifying the SOH oxidation state of this protein in vitro, reactions of protein-SOH with amines present in neighboring amino acid residues within the protein itself may prove more likely in biological systems. For example, parallel reports of the crystal structure of protein tyrosine phosphatase 1B revealed that the initial SOH intermediate formed at the active site cysteine residue upon enzyme oxidation is rapidly converted to a sulfenyl-amide product (122, 131). This conversion was proposed to proceed via the nucleophilic attack of a main chain nitrogen atom of a neighboring serine residue on the electrophilic sulfur of the SOH group. The formation of the sulfenyl-amide derivative was found to be accompanied by a large conformational change in the protein and was also found to be reversible by small thiol-based reduction. A similar sulfenyl-amide derivative was described following the initial formation of a protein-SOH intermediate in the organic peroxide sensor OhrR (120). Stable sulfinamide (RS(O)NR’) cross-links have also been detected following further reactions of protein-SOH. MS analysis of the S100A8 protein following HOCl-mediated oxidation suggested the formation of sulfinamide bonds upon nucleophilic substitution of a cysteine-SOH intermediate by the amine of a neighboring lysine residue (55). A further MS-based analysis of peptides exposed to HOCl revealed a variety of possible sulfur-nitrogen cross-links including both sulfenamides and sulfinamides following reaction of oxidized cysteine residues with lysine, arginine or N-terminal amines (54). Sulfinamide products have also been detected following initial SOH formation in peptides corresponding to the active site of both matrix metalloproteinase-7 and protein tyrosine phosphatase 1B (132–133).
4. Strategies for the Detection of Protein-SOH
Given the relative instability of protein-SOH based on their propensity to undergo the range of reactions discussed above, detection of these species has proven to be a difficult task and a majority of efforts to discover new targets utilize indirect methods of detection. There are no characteristic UV-visible spectral properties that can be used to identify protein-SOH and both the lack of stability and small size of this post-translational modification would appear to prevent the development of an antibody to specifically detect these proteins. The majority of evidence collected in support of the existence of protein-SOH has been based on their reactivity with chemical probes, selective reduction/reversal of the modification, or the detection of more stable thiol oxidation products that imply the intermediacy of SOH. However, a limited number of direct biophysical methods for protein-SOH detection are available (figure 4a). Both the electron density map collected via x-ray crystallography and 13C NMR chemical shift data support the existence of a SOH at Cys42 in the enzyme NADH peroxidase (121, 134). Several other protein crystal structure reports have indicated the presence of SOH-modified cysteine residues (62, 135–136). Additionally, protein-SOH formation has been directly detected by mass spectrometry as an m/z shift equivalent to the addition of one oxygen atom (+16 Da) in several studies of cysteine oxidation (47, 107, 137). In general, these direct observations of protein-SOH formation are likely to be limited to in vitro analysis of purified proteins. Perhaps not surprisingly, a correlation exists between the stability of a particular protein-SOH and its experimental detection using either direct methods or those involving chemical probes. Recent analyses of SOH reactivity have showcased the plethora of possible reactions for SOH in biological systems that will impact stability and be in competition with any chemical probes (138–140). Therefore solvent exposed and highly reactive protein-SOH will be especially challenging to trap and detect using probes which have to compete with these alternative reactions for the SOH group. Supplemental table 1 presents a comprehensive list of 63 unique proteins that have thus far been found to form cysteine-SOH using any of 9 indicated biochemical methods. These include the direct means mentioned above as well as strategies and probes based on the SOH biochemistry detailed below.
Figure 4.

Common methods for protein-SOH detection. a) direct detection using biophysical techniques. b) sodium arsenite-mediated selective reduction followed by assays indicative of reappearance of cysteine thiol group or incorporation of thiol-reactive probes to facilitate isolation or visualization. c) specific reactivity with dimedone can be observed by mass addition or blockade of SOH-dependent secondary reactions and dimedone derivatives combine specific reactivity with various detection or isolation capabilities. d) products generated upon reaction of protein-SOH or protein-SH with NBD-chloride can be differentiated based on either spectral properties or mass.
Some of the initial evidence, in addition to observations of thiol to oxidant reaction stoichiometry, collected in support of protein-SOH formation was based on the reversibility of the thiol modification by mild reducing agents like sodium arsenite (figure 4b). Arsenite is suggested to be a selective reductant for SOH as compared to disulfides and can therefore be used to differentiate these modifications experimentally. GAPDH that was inactivated by o-iodosobenzoate or iodine monochloride could be reactivated by arsenite treatments and this evidence was used as an indication of protein-SOH formation (17). Arsenite-mediated reversibility of thiol modifications has since been used by others to detect SOH formation on specific proteins and has also been coupled to biotin switch labeling for proteome-wide analysis of SOH formation (19, 21, 141–142). Reversibility of protein thiol oxidation by other mild reducing agents such as ascorbate and small thiol-containing compounds have also been used previously as evidence to suggest cysteine modification to the SOH oxidation state (102–103).
Perhaps the most extensively-used method for the discovery of SOH-containing proteins stems from the initial description of SOH reactivity with nucleophilic compounds (143). Since this reactivity is unique to SOH as compared to their parent thiols or disulfides, it can be used to distinguish between these cysteine redox states. Dimedone (5,5-dimethyl-1,3-cyclohexanedione) and its various derivatives have become the centerpieces in the arsenal of tools for SOH detection (figure 4c). It was first demonstrated that dimedone covalently reacts with SOH to form a thioether-linked derivative (144). These investigators were able to follow incorporation of 14C-labeled dimedone at the active site cysteine of oxidized GAPDH that was absent in the reduced enzyme. Since this key finding, others have been able to detect dimedone-protein adducts by mass spectrometry as indicated by an m/z shift associated with the addition of 140 Da per molecule of dimedone incorporated (i.e. per site of cysteine-SOH formation) (63, 137, 145–146). More recently, a number of fluorescent and affinity-based conjugates of dimedone have been developed to improve protein-SOH detection capabilities by facilitating the identification of target proteins from complex biological samples (147–149). These dimedone derivatives render dimedone-based studies applicable to gel electrophoresis-based analysis rather than the radioisotope or biophysical techniques previously required to detect adduct formation. Fluorescent probes facilitate both microscopy-based studies and direct in-gel visualization of labeled proteins while biotinylated compounds provide an avenue for affinity isolation of proteins. Finally, antibodies have now been developed to detect protein-dimedone adducts by at least two independent research groups. One recent study examined protein-SOH formation in tumor cells and demonstrated the utility of this antibody for immunoblots and immunofluorescence microscopy, as well as protein microarray analysis (150). A second report tracked both GAPDH-SOH levels and the general extent of protein-SOH formation in myocytes under basal conditions and upon H2O2 exposure (151).
Dimedone reactivity has been used in other ways to tease out roles for protein-SOH in various experimental systems. For example, the inhibition of mixed disulfide (S-glutathionylation) formation in the presence of dimedone has been used as evidence indicative of SOH intermediacy in this process (60, 119, 152–153). Similarly, dimedone-mediated inhibition of probe incorporation at sites of SOH formation has been used to confirm specificity of the methodologies for this modification in proteomic screens (48, 148). The disruption of T-cell function by dimedone exposure has also recently been reported as an indication that SOH formation is a necessary regulatory mechanism in cell signaling and proliferative pathways (154).
Although dimedone has been the most frequently used nucleophilic compound for the detection of protein-SOH formation, other nucleophiles have also been used as effective SOH probes. These strategies make use of some of the common biological SOH reactivities discussed above. The ability of SOH to react with amines to form sulfenamides facilitated the detection of GAPDH-SOH by incorporation of 14C following adduct formation between radiolabeled benzylamine and the oxidized enzyme (130). The well-characterized reaction of SOH with thiols to produce disulfides has also been utilized to detect SOH formation in proteins. The aromatic thiol compound TNB has a characteristic absorbance maximum at 412 nm. The decrease in A412 upon anaerobic incubation of oxidized NADH peroxidase, as well as its subsequent increase after DTT addition, was indicative of mixed disulfide formation by reaction of TNB with an active site cysteine-SOH (106). Therefore an assay for protein-SOH using simple spectrophotometry could be devised for the discovery of target proteins. This same protocol was employed to demonstrate the initial formation of a cysteine-SOH derivative in the catalysis of peroxide reduction by bacterial Prx enzyme AhpC (38).
Limitations associated with the lack of an easily-distinguishable dimedone-SOH reaction product initially necessitated either radiolabeling or mass spectrometry capabilities to probe for adduct formation. While the recently-developed fluorescent and affinity conjugates of dimedone have since addressed some of these applicability issues, another reagent has been utilized for protein-SOH detection. While the electrophilic compound NBD-Cl reacts with both thiols and SOH, the adducts can clearly be distinguished by both characteristic spectroscopic/fluorescent properties and masses (155) (figure 4d). The product formed upon reaction of NBD-Cl with SOH (R-S(O)-NBD) is either a sulfoxide or sulfenate ester and has a unique absorbance maximum at 347 nm. This is in contrast to the thioether product (R-S-NBD) formed upon reaction of NBD-Cl with thiols that maximally absorbs at 420 nm and possesses fluorescent properties (156). Therefore, the absorbance peak shift, or lack thereof, observed upon reaction of NBD-Cl with oxidant-treated proteins has been used extensively to confirm the presence or absence of the SOH post translational modification in a number of isolated proteins (33, 36, 47, 82, 125, 157). In addition to its utility as a spectral probe, retention of the SOH oxygen atom upon reaction with NBD-Cl results in a product that can differentiated form the thiol-NBD-Cl reaction product by mass spectrometry analysis (155).
5. Proteomic Studies of Protein-SOH
While several assays now exist for the detection of protein-SOH formation, most have been limited in their application to pre-selected isolated protein systems. These methods have been able to establish roles for protein-SOH as key intermediates in enzyme catalysis and cellular responses to oxidative stress. However, an understanding of the properties that dictate the susceptibility of cysteine residues to SOH formation, or oxidative modifications in general, and an appreciation of the extent to which these occur in biological systems remains unclear. With that being said, the degree of nucleophilicity and consequently the reactivity of a particular cysteine, is known to be dependent upon the protonation state of its thiol group. This is due to the increased nucleophilicity and reactivity of the deprotonated thiolate anion compared to that of the parent thiol. The extent to which a given protein cysteine residue exists in the thiol or thiolate form is determined by its pKa. The enhanced reactivity of thiolate anions with oxidants such as hydrogen peroxide has been used as a rationale to suggest that depressed cysteine pKa value may provide a basis for specific targeting in redox signal transduction, but a complete characterization of the protein structural features dictating cysteine pKa is still missing. Therefore, studies of SOH formation on a proteome-wide scale and the implementation of other bioinformatics-based approaches seem well-poised to address some of the outstanding issues regarding protein thiol oxidation. Interestingly, a recent computational analysis in which functional site profiling and electrostatic features of a large set of protein cysteine residues known to be modifiable to SOH was performed in order to determine common properties that may underlie specific targeting of this post-translational modification in particular (158). This general analysis of known modification sites revealed that 1) solvent accessibility is not a key determining feature, 2) neighboring cysteine residues is not important for reactivity, 3) nearby titratable polar residues may enhance the likelihood of modification, 4) proximal histidines seem to be a common feature and 5) interactions with threonine residues may be important for decreasing pKa values.
Proteomic analysis of general oxidative thiol modifications have rapidly evolved over the past decade and typically involve differential (control vs. oxidative treatment) thiol-trapping strategies aimed at detecting proteins with cysteines labeled following an ex vivo reduction step (i.e. DTT-reversible modifications). These studies have utilized either radiolabeled, fluorescent, or biotinylated thiol-reactive probes to facilitate detection and/or isolation of labeled proteins (159–161). Similar labeling protocols have recently been applied to more advanced difference gel electrophoresis and isotope coded affinity tag techniques (162–163). The so-called thiol redox proteome uncovered by these methods represents a broader population of modifications that encompasses SOH as well as a multitude of disulfides that may or may not have formed via the intermediacy of SOH. In contrast, studies designed specifically to detect protein-SOH are in their relative infancy.
Eaton and co-workers published the first proteomic analysis of SOH formation (142). In this study, tissue protein-SOH formation was detected following perfusion of isolated rat hearts with 0.1 mM H2O2 for 5 minutes. These investigators employed a strategy based on the biotin switch protocol originally developed to label S-nitrosated proteins in which a biotin tag is selectively exchanged at the sites of thiol modification (164). By altering the original method via the replacement of ascorbate with the SOH-specific reducing agent sodium arsenite, the assay was adapted for SOH detection. Briefly, their protocol involved 1) blocking/capping of free thiols with maleimide to prevent both reactions of SOH with thiols and possible non-specific labeling, 2) arsenite-mediated reduction of protein-SOH and 3) labeling of nascent thiols with a thiol-reactive biotin-maleimide probe. Biotin incorporation at the sites of SOH formation facilitated streptavidin-mediated affinity isolation of target proteins. Following SDS-PAGE separation and in-gel digestion, 17 proteins were identified by MALDI-TOF MS analysis. Although extensive sample processing under denaturing conditions may have limited the number of SOH-containing proteins identified and biased the selection towards the most stable protein-SOH, the lack of biotin labeling in untreated control samples and the blockade of biotin incorporation by dimedone pre-treatment greatly increased confidence in the specificity of the methodology for detection of SOH formation induced by increases in cellular H2O2 levels.
A second proteomic study of SOH formation in isolated rat hearts from the same research group followed a different labeling strategy (149). In this report, the authors utilized a biotinylated dimedone analog that was synthesized to probe for protein-SOH. Therefore, the selective-reactivity of dimedone towards SOH was directly combined with the biotin moiety as a handle for subsequent protein isolation. Loading of the heart tissue with this cell-permeable probe was accomplished by perfusing the heart with 0.05 mM biotinylated dimedone for 5 minutes immediately prior to oxidant challenge with 0.05 mM H2O2 for an additional 5 minutes. Proteins forming SOH in response to H2O2 treatment would then have incorporated the probe via reaction with the dimedone functional group and labeled proteins present in the tissue homogenate could then be affinity purified through the interaction of the biotin portion of the tag with streptavidin-agarose. SDS-PAGE separation of the isolated proteins, trypsin digestion of resultant protein bands and LC-MS/MS analysis led to the identification of 24 unique proteins that appear to form SOH upon H2O2 treatment. As a cautionary note related to this and the other proteomic studies discussed here, two of the identified target candidates did not actually contain any cysteine residues, demonstrating the inherent risk of false positive identifications arising from issues with either probe specificity or affinity isolation procedures. Therefore, these proteome-wide screens serve to uncover a tentative list of potential targets while MS detection of the labeled cysteine-containing peptide is required for definitive identification.
Our research group presented a proteomic study using a novel genetically-encoded probe to follow SOH formation induced by H2O2 exposure in E. coli (48). In this case, the strategy was to utilize the well-established reactivity of cysteine 598 of the S. cerevisiae transcription factor Yap1 toward the SOH formed at cysteine 36 of the redox sensing protein Orp1 as a general trap for proteins that form SOH. The probe consists of 85 amino acids of the C-terminal cysteine rich domain of Yap1 along with a His-tag as a handle for purifications. Two of the three native cysteine residues in this domain were mutated (C620A and C629T) leaving a single cysteine (C598) that can incorporate as a mixed disulfide at sites of SOH formation and facilitate Ni+ affinity column purification of the target proteins via the His-tag portion of the probe. E. coli cultures expressing the His-tagged Yap1 domain were treated with 0.5 mM H2O2 for 2.5 minutes and those proteins co-purified on a Ni+ affinity column (i.e. those that formed SOH and subsequently a disulfide with the probe) were eluted via DTT-mediated disulfide reduction and separated by SDS-PAGE. Tryptic digests of protein bands were analyzed by LC-MS/MS and 32 candidate targets were identified. Further alterations of this probe using molecular genetics techniques to incorporate subcellular targeting sequences are currently underway to expand its utility as an organelle-specific trap for SOH formation. This will also provide a means to delve deeper into this sub-proteome by limiting sample complexity in order to uncover less abundant and/or less extensively-modified proteins that may be missed in broader proteomic analyses. This Yap1-based probe remains the only biological-based system designed to capture SOH-containing proteins without the necessity to add exogenous chemical trapping agents. It is also possible to adapt this probe to examine protein-SOH formation in any biological system for which standard molecular genetics manipulations are possible.
The most recent proteomic analysis of SOH formation was performed on cultured HeLa cells using a novel dimedone-based probe (165). This compound, 4-(3-azidopropyl)cyclohexane-1,3-dione (DAz-2), as well as a similar probe examined previously possesses superior cell permeability compared with more bulky biotinylated dimedone analogs (148). DAz-2 consists of a dimedone-like portion that provides the selective SOH reactivity coupled with an azide functional group that facilitates chemical manipulations (i.e. addition of affinity tags) following the preparation of cell extracts. In this study, HeLa cells in suspension were loaded with the probe via 120 minute incubation with 5 mM DAz-2. After washing away excess probe, cells were incubated an additional 60 minutes in culture media. No exogenous oxidant was added suggesting that all proteins that incorporated the label form SOH under basal conditions. Lysate fractions were prepared and azide-reactive phosphine-biotin was used to biotinylate (via amide linkage) proteins that were DAz-2-labeled in order to facilitate avidin-based purification of the modified proteins. SDS-PAGE separation, tryptic digestion and LC-MS/MS analysis yielded a list of 193 potential SOH-containing proteins in this tumor cell line.
Several tools and strategies now exist that can be used in combination to aid investigators in uncovering the set of proteins that form SOH intermediates in both ROS-mediated cysteine oxidation and cysteine-dependent enzyme catalysis. The high reactivity and subsequent short-lived nature of this post-translational modification still present challenges to overcome in cataloging this sub-proteome. It also underscores that these studies would best be performed in parallel with and compared to proteomic analysis of more stable thiol oxidation products like mixed disulfides that may follow initial SOH formation in order to gain a more complete understanding of the physiological and pathological roles of protein thiol oxidation. Since the probability that these various probes and experimental strategies selectively capture different subsets of SOH-containing proteins is quite high, it seems clear that they each warrant further exploration. In future studies, these methods may be applied to less complex biological systems (i.e. organelle-specific analysis) as a means to delve deeper into this proteome and uncover less abundant or less extensively-modified targets. It will also be of interest to compare the SOH proteome following exposure to different oxidative stimuli (i.e. classes of oxidants) so that any specificity in cysteine targeting can be defined and mechanistic details underlying the triggering of divergent cellular responses can be clarified. Another issue to consider while compiling the SOH proteome is the relative extent to which a specific protein is modified under the given experimental conditions. Possible quantitative aspects along with difficulties associated with these types of analyses were recently addressed using both TNB and dimedone-based probes to examine plasma protein-SOH (166). Finally, the SOH proteome generated in response to the addition of exogenous oxidants will need to be compared to that generated by endogenously-produced oxidants as well as that present under basal conditions in order to differentiate pathways that may utilize SOH intermediates in normal functional processes and those that incorporate SOH formation to sense and transmit stress responses.
Supplementary Material
Acknowledgments
This work was funded by a grant from the American Heart Association (0635328N). NJK was funded by Award Number T32ES007059 from the National Institute of Environmental Health Sciences. We would like to thank Christina L. Takanishi and Heather M. Bolstad for careful reading of the manuscript.
Abbreviations
- TNB
2-nitro-5-thiobenzoate
- DAz-2
4-(3-azidopropyl)cyclohexane-1,3-dione
- NBD-Cl
7-chloro-4-nitrobenzo-2-oxa-1,3-diazole
- CHP
cumene hydroperoxide
- LAHP
linoleic acid hydroperoxide
- OH−
hydroxide anion
- HOCl
hypochlorous acid
- RNHCl
N-chloramines
- NO
nitric oxide
- ROOH
organic-based hydroperoxides
- ONOO−
peroxynitrite anion
- ONOOH
peroxynitrous acid
- Prx
peroxiredoxin
- PGK
phosphoglycerate kinase
- RSNO
S-nitrosothiols
- RSNR’
sulfenamide
- SOH
sulfenic acid
- RSCl
sulfenyl chloride
- RS(O)NR’
sulfinamide
- SO2H
sulfinic acid
- RSO
sulfinyl radical
- SO3H
sulfonic acid
- O2•−
superoxide
- t-BHP
tert-butyl hydroperoxide
- RSNOH•
thionitroxide radical intermediate
- RS(O)SR
thiosulfinate compounds
- RSOO•
thiylperoxyl radical
- S•
thiyl radical
References
- 1.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 2.Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol. 2004;14:679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 4.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 6.Cooper CE, Patel RP, Brookes PS, Darley-Usmar VM. Nanotransducers in cellular redox signaling: modification of thiols by reactive oxygen and nitrogen species. Trends Biochem Sci. 2002;27:489–492. doi: 10.1016/s0968-0004(02)02191-6. [DOI] [PubMed] [Google Scholar]
- 7.Wood MJ, Storz G, Tjandra N. Structural basis for redox regulation of Yap1 transcription factor localization. Nature. 2004;430:917–921. doi: 10.1038/nature02790. [DOI] [PubMed] [Google Scholar]
- 8.Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE. 2000;2000:PE1. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- 9.Matsuyama Y, Terawaki H, Terada T, Era S. Albumin thiol oxidation and serum protein carbonyl formation are progressively enhanced with advancing stages of chronic kidney disease. Clin Exp Nephrol. 2009;13:308–315. doi: 10.1007/s10157-009-0161-y. [DOI] [PubMed] [Google Scholar]
- 10.Pirie NW. The oxidation of sulphydryl compounds by hydrogen peroxide: Catalysis of oxidation of cysteine by thiocarbamides and thiolglyoxalines. Biochem J. 1933;27:1181–1188. doi: 10.1042/bj0271181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toennies G. Oxidation of cysteine in non-aqueous media: The “sulfenic acid” as the primary oxidation product. J Biol Chem. 1937;122:27–47. [Google Scholar]
- 12.Basford RE, Huennekens FM. Studies on Thiols 1. Oxidation of Thiol Groups by 2,6–Dichlorophenol Indophenol. J Am Chem Soc. 1955;77:3873–3877. [Google Scholar]
- 13.Little C, O'Brien PJ. Oxidation of Small Thiols by Lipid Peroxides. Biochem J. 1967;102:10p. [Google Scholar]
- 14.Fraenkel-Conrat H. The reaction of tobacco mosaic virus with iodine. J Biol Chem. 1955;217:373–381. [PubMed] [Google Scholar]
- 15.Pihl A, Lange R. The interaction of oxidized glutathione, cystamine monosulfoxide, and tetrathionate with the-SH groups of rabbit muscle D-glyceraldehyde 3-phosphate dehydrogenase. J Biol Chem. 1962;237:1356–1362. [PubMed] [Google Scholar]
- 16.Glazer AN, Smith EL. The Sulfur Distribution of Papain. J Biol Chem. 1965;240:201–208. [PubMed] [Google Scholar]
- 17.Parker DJ, Allison WS. The mechanism of inactivation of glyceraldehyde 3-phosphate dehydrogenase by tetrathionate, o-iodosobenzoate, and iodine monochloride. J Biol Chem. 1969;244:180–189. [PubMed] [Google Scholar]
- 18.Ehring R, Colowick SP. The two-step formation and inactivation of acylphosphatase by agents acting on glyceraldehyde phosphate dehydrogenase. J Biol Chem. 1969;244:4589–4599. [PubMed] [Google Scholar]
- 19.Little C, O'Brien PJ. Mechanism of peroxide-inactivation of the sulphydryl enzyme glyceraldehyde-3-phosphate dehydrogenase. Eur J Biochem. 1969;10:533–538. doi: 10.1111/j.1432-1033.1969.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 20.Trundle D, Cunningham LW. Iodine oxidation of the sulfhydryl groups of creatine kinase. Biochemistry. 1969;8:1919–1925. doi: 10.1021/bi00833a023. [DOI] [PubMed] [Google Scholar]
- 21.Radi R, Bush KM, Cosgrove TP, Freeman BA. Reaction of xanthine oxidase-derived oxidants with lipid and protein of human plasma. Arch Biochem Biophys. 1991;286:117–125. doi: 10.1016/0003-9861(91)90016-c. [DOI] [PubMed] [Google Scholar]
- 22.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 23.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 25.Pirie NW. The oxidation of sulphydryl compounds by hydrogen peroxide: Catalysis of oxidation of cysteine and glutathione by iron and copper. Biochem J. 1931;25:1565–1579. doi: 10.1042/bj0251565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanner T, Pihl A. Studies on the active--SH group of papain and on the mechanism of papain activation by thiols. J Biol Chem. 1963;238:165–171. [PubMed] [Google Scholar]
- 27.Barton JP, PJ, Sims RJ. Kinetics of the Reaction of Hydrogen Peroxide with Cysteine and Cysteamine. J Chem Soc Perkin II. 1973;1973:1547–1549. [Google Scholar]
- 28.Baehner RL, Nathan DG, Castle WB. Oxidant injury of caucasian glucose-6-phosphate dehydrogenase-deficient red blood cells by phagocytosing leukocytes during infection. J Clin Invest. 1971;50:2466–2473. doi: 10.1172/JCI106747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Root RK, Metcalf J, Oshino N, Chance B. H2O2 release from human granulocytes during phagocytosis. I. Documentation, quantitation, and some regulating factors. J Clin Invest. 1975;55:945–955. doi: 10.1172/JCI108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss SJ, Young J, LoBuglio AF, Slivka A, Nimeh NF. Role of hydrogen peroxide in neutrophil-mediated destruction of cultured endothelial cells. J Clin Invest. 1981;68:714–721. doi: 10.1172/JCI110307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall ND, Maslen CL, Blake DR. The oxidation of serum sulph-hydryl groups by hydrogen peroxide secreted by stimulated phagocytic cells in rheumatoid arthritis. Rheumatol Int. 1984;4:35–38. doi: 10.1007/BF00683883. [DOI] [PubMed] [Google Scholar]
- 32.Brodie AE, Reed DJ. Reversible oxidation of glyceraldehyde 3-phosphate dehydrogenase thiols in human lung carcinoma cells by hydrogen peroxide. Biochem Biophys Res Commun. 1987;148:120–125. doi: 10.1016/0006-291x(87)91084-9. [DOI] [PubMed] [Google Scholar]
- 33.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 34.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 35.Aslund F, Zheng M, Beckwith J, Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma LH, Takanishi CL, Wood MJ. Molecular mechanism of oxidative stress perception by the Orp1 protein. J Biol Chem. 2007;282:31429–31436. doi: 10.1074/jbc.M705953200. [DOI] [PubMed] [Google Scholar]
- 37.Chae HZ, Uhm TB, Rhee SG. Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc Natl Acad Sci U S A. 1994;91:7022–7026. doi: 10.1073/pnas.91.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 39.Baker LM, Poole LB. Catalytic mechanism of thiol peroxidase from Escherichia coli. Sulfenic acid formation and overoxidation of essential CYS61. J Biol Chem. 2003;278:9203–9211. doi: 10.1074/jbc.M209888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wills ED. Effect of unsaturated fatty acids and their peroxides on enzymes. Biochem Pharmacol. 1961;7:7–16. doi: 10.1016/0006-2952(61)90119-8. [DOI] [PubMed] [Google Scholar]
- 41.Lewis SE, Wills ED. The Destruction of SH Groups of Proteins and Amino Acids by Peroxides of Unsaturated Fatty Acids. Biochem Pharmacol. 1962;11:901–912. [Google Scholar]
- 42.Little C, O'Brien PJ. Products of oxidation of a protein thiol group after reaction with various oxidizing agents. Arch Biochem Biophys. 1967;122:406–410. doi: 10.1016/0003-9861(67)90212-3. [DOI] [PubMed] [Google Scholar]
- 43.Little C, O'Brien PJ. The effectiveness of a lipid peroxide in oxidizing protein and non-protein thiols. Biochem J. 1968;106:419–423. doi: 10.1042/bj1060419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobson FS, Morgan RW, Christman MF, Ames BN. An alkyl hydroperoxide reductase from Salmonella typhimurium involved in the defense of DNA against oxidative damage. Purification and properties. J Biol Chem. 1989;264:1488–1496. [PubMed] [Google Scholar]
- 45.Jeong W, Cha MK, Kim IH. Thioredoxin-dependent hydroperoxide peroxidase activity of bacterioferritin comigratory protein (BCP) as a new member of the thiol-specific antioxidant protein (TSA)/Alkyl hydroperoxide peroxidase C (AhpC) family. J Biol Chem. 2000;275:2924–2930. doi: 10.1074/jbc.275.4.2924. [DOI] [PubMed] [Google Scholar]
- 46.Lee J, Spector D, Godon C, Labarre J, Toledano MB. A new antioxidant with alkyl hydroperoxide defense properties in yeast. J Biol Chem. 1999;274:4537–4544. doi: 10.1074/jbc.274.8.4537. [DOI] [PubMed] [Google Scholar]
- 47.Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci U S A. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takanishi CL, Ma LH, Wood MJ. A genetically encoded probe for cysteine sulfenic acid protein modification in vivo. Biochemistry. 2007;46:14725–14732. doi: 10.1021/bi701625s. [DOI] [PubMed] [Google Scholar]
- 49.Folkes LK, Candeias LP, Wardman P. Kinetics and mechanisms of hypochlorous acid reactions. Arch Biochem Biophys. 1995;323:120–126. doi: 10.1006/abbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 50.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 51.Knox WE, Stumpf PK, Green DE, Auerbach VH. The Inhibition of Sulfhydryl Enzymes as the Basis of the Bactericidal Action of Chlorine. J Bacteriol. 1948;55:451–458. [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas EL. Myeloperoxidase-hydrogen peroxide-chloride antimicrobial system: effect of exogenous amines on antibacterial action against Escherichia coli. Infect Immun. 1979;25:110–116. doi: 10.1128/iai.25.1.110-116.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silverstein RM, Hager LP. The chloroperoxidase–catalyzed oxidation of thiols and disulfides to sulfenyl chlorides. Biochemistry. 1974;13:5069–5073. doi: 10.1021/bi00722a001. [DOI] [PubMed] [Google Scholar]
- 54.Fu X, Mueller DM, Heinecke JW. Generation of intramolecular and intermolecular sulfenamides, sulfinamides, and sulfonamides by hypochlorous acid: a potential pathway for oxidative cross-linking of low-density lipoprotein by myeloperoxidase. Biochemistry. 2002;41:1293–1301. doi: 10.1021/bi015777z. [DOI] [PubMed] [Google Scholar]
- 55.Raftery MJ, Yang Z, Valenzuela SM, Geczy CL. Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem. 2001;276:33393–33401. doi: 10.1074/jbc.M101566200. [DOI] [PubMed] [Google Scholar]
- 56.Thomas EL, Grisham MB, Jefferson MM. Myeloperoxidase-dependent effect of amines on functions of isolated neutrophils. J Clin Invest. 1983;72:441–454. doi: 10.1172/JCI110992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peskin AV, Winterbourn CC. Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic Biol Med. 2006;40:45–53. doi: 10.1016/j.freeradbiomed.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 58.Summers FA, Morgan PE, Davies MJ, Hawkins CL. Identification of plasma proteins that are susceptible to thiol oxidation by hypochlorous acid and N-chloramines. Chem Res Toxicol. 2008;21:1832–1840. doi: 10.1021/tx8001719. [DOI] [PubMed] [Google Scholar]
- 59.Pryor WAC, DF, Govindan CK, Crank G. Oxidation of Thiols by Nitric Oxide and Nitrogen Dioxide: Synthetic Utility and Toxicological Implications. J Org Chem. 1982;47:156–159. [Google Scholar]
- 60.DeMaster EG, Quast BJ, Redfern B, Nagasawa HT. Reaction of nitric oxide with the free sulfhydryl group of human serum albumin yields a sulfenic acid and nitrous oxide. Biochemistry. 1995;34:11494–11499. doi: 10.1021/bi00036a023. [DOI] [PubMed] [Google Scholar]
- 61.Hogg N, Singh RJ, Kalyanaraman B. The role of glutathione in the transport and catabolism of nitric oxide. FEBS Lett. 1996;382:223–228. doi: 10.1016/0014-5793(96)00086-5. [DOI] [PubMed] [Google Scholar]
- 62.Becker K, Savvides SN, Keese M, Schirmer RH, Karplus PA. Enzyme inactivation through sulfhydryl oxidation by physiologic NO-carriers. Nat Struct Biol. 1998;5:267–271. doi: 10.1038/nsb0498-267. [DOI] [PubMed] [Google Scholar]
- 63.Percival MD, Ouellet M, Campagnolo C, Claveau D, Li C. Inhibition of cathepsin K by nitric oxide donors: evidence for the formation of mixed disulfides and a sulfenic acid. Biochemistry. 1999;38:13574–13583. doi: 10.1021/bi991028u. [DOI] [PubMed] [Google Scholar]
- 64.Stamler JS, Hausladen A. Oxidative modifications in nitrosative stress. Nat Struct Biol. 1998;5:247–249. doi: 10.1038/nsb0498-247. [DOI] [PubMed] [Google Scholar]
- 65.Keszler A, Zhang Y, Hogg N. Reaction between nitric oxide, glutathione, and oxygen in the presence and absence of protein: How are S-nitrosothiols formed? Free Radic Biol Med. 2010;48:55–64. doi: 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heinecke J, Ford PC. Formation of cysteine sulfenic acid by oxygen atom transfer from nitrite. J Am Chem Soc. 2010;132:9240–9243. doi: 10.1021/ja102221e. [DOI] [PubMed] [Google Scholar]
- 67.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 68.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hogg N, Darley-Usmar VM, Wilson MT, Moncada S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem J. 1992;281(Pt 2):419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hogg N, Joseph J, Kalyanaraman B. The oxidation of alpha-tocopherol and trolox by peroxynitrite. Arch Biochem Biophys. 1994;314:153–158. doi: 10.1006/abbi.1994.1423. [DOI] [PubMed] [Google Scholar]
- 71.Pryor WA, Jin X, Squadrito GL. One- and two-electron oxidations of methionine by peroxynitrite. Proc Natl Acad Sci U S A. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gatti RM, Radi R, Augusto O. Peroxynitrite-mediated oxidation of albumin to the protein-thiyl free radical. FEBS Lett. 1994;348:287–290. doi: 10.1016/0014-5793(94)00625-3. [DOI] [PubMed] [Google Scholar]
- 73.Karoui H, Hogg N, Frejaville C, Tordo P, Kalyanaraman B. Characterization of sulfur-centered radical intermediates formed during the oxidation of thiols and sulfite by peroxynitrite. ESR-spin trapping and oxygen uptake studies. J Biol Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 74.Quijano C, Alvarez B, Gatti RM, Augusto O, Radi R. Pathways of peroxynitrite oxidation of thiol groups. Biochem J. 1997;322(Pt 1):167–173. doi: 10.1042/bj3220167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trujillo M, Radi R. Peroxynitrite reaction with the reduced and the oxidized forms of lipoic acid: new insights into the reaction of peroxynitrite with thiols. Arch Biochem Biophys. 2002;397:91–98. doi: 10.1006/abbi.2001.2619. [DOI] [PubMed] [Google Scholar]
- 76.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 77.Peshenko IV, Shichi H. Oxidation of active center cysteine of bovine 1-Cys peroxiredoxin to the cysteine sulfenic acid form by peroxide and peroxynitrite. Free Radic Biol Med. 2001;31:292–303. doi: 10.1016/s0891-5849(01)00579-2. [DOI] [PubMed] [Google Scholar]
- 78.Trujillo M, Budde H, Pineyro MD, Stehr M, Robello C, Flohe L, Radi R. Trypanosoma brucei and Trypanosoma cruzi tryparedoxin peroxidases catalytically detoxify peroxynitrite via oxidation of fast reacting thiols. J Biol Chem. 2004;279:34175–34182. doi: 10.1074/jbc.M404317200. [DOI] [PubMed] [Google Scholar]
- 79.Ogusucu R, Rettori D, Munhoz DC, Netto LE, Augusto O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: rate constants by competitive kinetics. Free Radic Biol Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 80.Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, Kissner R, Koppenol WH, Rees JF, Knoops B. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 2004;571:161–165. doi: 10.1016/j.febslet.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 81.Hattori F, Murayama N, Noshita T, Oikawa S. Mitochondrial peroxiredoxin-3 protects hippocampal neurons from excitotoxic injury in vivo. J Neurochem. 2003;86:860–868. doi: 10.1046/j.1471-4159.2003.01918.x. [DOI] [PubMed] [Google Scholar]
- 82.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42:9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 83.Challenger F, Rawlings AA. The formation of organo-metalloidal and similar compounds by micro-organisms. Part V. Methylated alkyl sulphides. The fission of the disulphide link. J Chem Soc. 1937;1937:868–875. [Google Scholar]
- 84.Toennies G. Relations of thiourea, cysteine, and the corresponding disulfides. J Biol Chem. 1937:297–313. [Google Scholar]
- 85.Danehy JP, Hunter WE. The alkaline decomposition of organic disulfides. II. Alternative pathways as determined by structure. J Org Chem. 1967;32:2047–2053. [Google Scholar]
- 86.Donovan JW. Spectrophotometric observation of the alkaline hydrolysis of protein disulfide bonds. Biochem Biophys Res Commun. 1967;29:734–740. doi: 10.1016/0006-291x(67)90279-3. [DOI] [PubMed] [Google Scholar]
- 87.Donovan JW, White TM. Alkaline hydrolysis of the disulfide bonds of ovomucoid and of low molecular weight aliphatic and aromatic disulfides. Biochemistry. 1971;10:32–38. doi: 10.1021/bi00777a005. [DOI] [PubMed] [Google Scholar]
- 88.Andersson LO. Hydrolysis of disulfide bonds in weakly alkaline media. II. Bovine serum albumin dimer. Biochim Biophys Acta. 1970;200:363–369. doi: 10.1016/0005-2795(70)90178-9. [DOI] [PubMed] [Google Scholar]
- 89.Wu HL, Shi GY, Wohl RC, Bender ML. Structure and formation of microplasmin. Proc Natl Acad Sci U S A. 1987;84:8793–8795. doi: 10.1073/pnas.84.24.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stathakis P, Lay AJ, Fitzgerald M, Schlieker C, Matthias LJ, Hogg PJ. Angiostatin formation involves disulfide bond reduction and proteolysis in kringle 5 of plasmin. J Biol Chem. 1999;274:8910–8916. doi: 10.1074/jbc.274.13.8910. [DOI] [PubMed] [Google Scholar]
- 91.Lay AJ, Jiang XM, Kisker O, Flynn E, Underwood A, Condron R, Hogg PJ. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature. 2000;408:869–873. doi: 10.1038/35048596. [DOI] [PubMed] [Google Scholar]
- 92.Lay AJ, Jiang XM, Daly E, Sun L, Hogg PJ. Plasmin reduction by phosphoglycerate kinase is a thiol-independent process. J Biol Chem. 2002;277:9062–9068. doi: 10.1074/jbc.M111531200. [DOI] [PubMed] [Google Scholar]
- 93.Brandt W, Golbraikh A, Tager M, Lendeckel U. A molecular mechanism for the cleavage of a disulfide bond as the primary function of agonist binding to G-protein-coupled receptors based on theoretical calculations supported by experiments. Eur J Biochem. 1999;261:89–97. doi: 10.1046/j.1432-1327.1999.00224.x. [DOI] [PubMed] [Google Scholar]
- 94.Yan B, Yates Z, Balland A, Kleemann GR. Human IgG1 hinge fragmentation as the result of H2O2-mediated radical cleavage. J Biol Chem. 2009;284:35390–35402. doi: 10.1074/jbc.M109.064147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rabinkov A, Miron T, Konstantinovski L, Wilchek M, Mirelman D, Weiner L. The mode of action of allicin: trapping of radicals and interaction with thiol containing proteins. Biochim Biophys Acta. 1998;1379:233–244. doi: 10.1016/s0304-4165(97)00104-9. [DOI] [PubMed] [Google Scholar]
- 96.Li J, Huang FL, Huang KP. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J Biol Chem. 2001;276:3098–3105. doi: 10.1074/jbc.M008260200. [DOI] [PubMed] [Google Scholar]
- 97.Wardman P. Evaluation of the “radical sink” hypothesis from a chemical-kinetic viewpoint. J Radioanalytical and Nuclear Chemistry. 1998;232:23–27. [Google Scholar]
- 98.Claiborne A, Miller H, Parsonage D, Ross RP. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993;7:1483–1490. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 99.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 100.Davis FA, Billmers RL. Chemistry of sulfenic acids. 4. The first direct evidence for the involvement of sulfenic acids in the oxidation of thiols. J Am Chem Soc. 1981;103:7016–7018. [Google Scholar]
- 101.Davis FA, Jenkins LA, Billmers RL. Chemistry of sulfenic acids. 7. Reason for the high reactivity of sulfenic acids. Stabilization by intramolecular hydrogen bonding and electronegativity effects. J Org Chem. 1986;51:1033–1040. [Google Scholar]
- 102.Allison WS, Connors MJ. The activation and inactivation of the acyl phosphatase activity of glyceraldehyde-3-phosphate dehydrogenase. Arch Biochem Biophys. 1970;136:383–391. doi: 10.1016/0003-9861(70)90209-2. [DOI] [PubMed] [Google Scholar]
- 103.You KS, Benitez LV, McConachie WA, Allison WS. The conversion of glyceraldehyde-3-phosphate dehydrogenase to an acylphosphatase by trinitroglycerin and inactivation of this activity by azide and ascorbate. Biochim Biophys Acta. 1975;384:317–330. doi: 10.1016/0005-2744(75)90033-9. [DOI] [PubMed] [Google Scholar]
- 104.Armstrong DA, Buchanan JD. Reactions of O-.2, H2O2 and other oxidants with sulfhydryl enzymes. Photochem Photobiol. 1978;28:743–755. doi: 10.1111/j.1751-1097.1978.tb07011.x. [DOI] [PubMed] [Google Scholar]
- 105.Entsch B, Ballou DP, Husain M, Massey V. Catalytic mechanism of p-hydroxybenzoate hydroxylase with p-mercaptobenzoate as substrate. J Biol Chem. 1976;251:7367–7369. [PubMed] [Google Scholar]
- 106.Poole LB, Claiborne A. The non-flavin redox center of the streptococcal NADH peroxidase. II. Evidence for a stabilized cysteine-sulfenic acid. J Biol Chem. 1989;264:12330–12338. [PubMed] [Google Scholar]
- 107.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J Biol Chem. 2000;275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 108.Poole LB. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 2. Cystine disulfides involved in catalysis of peroxide reduction. Biochemistry. 1996;35:65–75. doi: 10.1021/bi951888k. [DOI] [PubMed] [Google Scholar]
- 109.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 110.Modig H. Cellular mixed disulphides between thiols and proteins, and their possible implication for radiation protection. Biochem Pharmacol. 1968;17:177–186. doi: 10.1016/0006-2952(68)90321-3. [DOI] [PubMed] [Google Scholar]
- 111.DeLucia AJ, Mustafa MG, Hussain MZ, Cross CE. Ozone interaction with rodent lung. III. Oxidation of reduced glutathione and formation of mixed disulfides between protein and nonprotein sulfhydryls. J Clin Invest. 1975;55:794–802. doi: 10.1172/JCI107990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Collison MW, Beidler D, Grimm LM, Thomas JA. A comparison of protein S-thiolation (protein mixed-disulfide formation) in heart cells treated with t-butyl hydroperoxide or diamide. Biochim Biophys Acta. 1986;885:58–67. doi: 10.1016/0167-4889(86)90038-8. [DOI] [PubMed] [Google Scholar]
- 113.Park EM, Thomas JA. S-thiolation of creatine kinase and glycogen phosphorylase b initiated by partially reduced oxygen species. Biochim Biophys Acta. 1988;964:151–160. doi: 10.1016/0304-4165(88)90161-4. [DOI] [PubMed] [Google Scholar]
- 114.Burchill BR, Oliver JM, Pearson CB, Leinbach ED, Berlin RD. Microtubule dynamics and glutathione metabolism in phagocytizing human polymorphonuclear leukocytes. J Cell Biol. 1978;76:439–447. doi: 10.1083/jcb.76.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ravichandran V, Seres T, Moriguchi T, Thomas JA, Johnston RB., Jr S-thiolation of glyceraldehyde-3-phosphate dehydrogenase induced by the phagocytosis-associated respiratory burst in blood monocytes. J Biol Chem. 1994;269:25010–25015. [PubMed] [Google Scholar]
- 116.Chai YC, Ashraf SS, Rokutan K, Johnston RB, Jr, Thomas JA. S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: evidence against a role for glutathione disulfide. Arch Biochem Biophys. 1994;310:273–281. doi: 10.1006/abbi.1994.1167. [DOI] [PubMed] [Google Scholar]
- 117.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 118.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 119.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee JW, Soonsanga S, Helmann JD. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc Natl Acad Sci U S A. 2007;104:8743–8748. doi: 10.1073/pnas.0702081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Crane EJ, 3rd, Vervoort J, Claiborne A. 13C NMR analysis of the cysteine-sulfenic acid redox center of enterococcal NADH peroxidase. Biochemistry. 1997;36:8611–8618. doi: 10.1021/bi9707990. [DOI] [PubMed] [Google Scholar]
- 122.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 123.DeGnore JP, Konig S, Barrett WC, Chock PB, Fales HM. Identification of the oxidation states of the active site cysteine in a recombinant protein tyrosine phosphatase by electrospray mass spectrometry using on-line desalting. Rapid Commun Mass Spectrom. 1998;12:1457–1462. doi: 10.1002/(SICI)1097-0231(19981030)12:20<1457::AID-RCM346>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 124.Souza JM, Radi R. Glyceraldehyde-3-phosphate dehydrogenase inactivation by peroxynitrite. Arch Biochem Biophys. 1998;360:187–194. doi: 10.1006/abbi.1998.0932. [DOI] [PubMed] [Google Scholar]
- 125.Griffiths SW, King J, Cooney CL. The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. J Biol Chem. 2002;277:25486–25492. doi: 10.1074/jbc.M203089200. [DOI] [PubMed] [Google Scholar]
- 126.Wagner E, Luche S, Penna L, Chevallet M, Van Dorsselaer A, Leize-Wagner E, Rabilloud T. A method for detection of overoxidation of cysteines: peroxiredoxins are oxidized in vivo at the active-site cysteine during oxidative stress. Biochem J. 2002;366:777–785. doi: 10.1042/BJ20020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Woo HA, Kang SW, Kim HK, Yang KS, Chae HZ, Rhee SG. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J Biol Chem. 2003;278:47361–47364. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]
- 128.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 129.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 130.Allison WS, Benitez LV, Johnson CL. The formation of a protein sulfenamide during the inactivation of the acyl phosphatase activity of oxidized glyceraldehyde-3-phosphate dehydrogenase by benzylamine. Biochem Biophys Res Commun. 1973;52:1403–1409. doi: 10.1016/0006-291x(73)90657-8. [DOI] [PubMed] [Google Scholar]
- 131.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 132.Shetty V, Spellman DS, Neubert TA. Characterization by tandem mass spectrometry of stable cysteine sulfenic acid in a cysteine switch peptide of matrix metalloproteinases. J Am Soc Mass Spectrom. 2007;18:1544–1551. doi: 10.1016/j.jasms.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shetty V, Neubert TA. Characterization of novel oxidation products of cysteine in an active site motif peptide of PTP1B. J Am Soc Mass Spectrom. 2009;20:1540–1548. doi: 10.1016/j.jasms.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yeh JI, Claiborne A, Hol WG. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 A resolution. Biochemistry. 1996;35:9951–9957. doi: 10.1021/bi961037s. [DOI] [PubMed] [Google Scholar]
- 135.Choi HJ, Kang SW, Yang CH, Rhee SG, Ryu SE. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nat Struct Biol. 1998;5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- 136.Nakamura T, Yamamoto T, Abe M, Matsumura H, Hagihara Y, Goto T, Yamaguchi T, Inoue T. Oxidation of archaeal peroxiredoxin involves a hypervalent sulfur intermediate. Proc Natl Acad Sci U S A. 2008;105:6238–6242. doi: 10.1073/pnas.0709822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. J Biol Chem. 2006;281:15110–15120. doi: 10.1074/jbc.M600837200. [DOI] [PubMed] [Google Scholar]
- 138.Turell L, Botti H, Carballal S, Ferrer-Sueta G, Souza JM, Duran R, Freeman BA, Radi R, Alvarez B. Reactivity of sulfenic acid in human serum albumin. Biochemistry. 2008;47:358–367. doi: 10.1021/bi701520y. [DOI] [PubMed] [Google Scholar]
- 139.Nagy P, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the oxidation of cysteine by hypohalous acid to give cysteine sulfenic acid. J Am Chem Soc. 2007;129:14082–14091. doi: 10.1021/ja0737218. [DOI] [PubMed] [Google Scholar]
- 140.Nagy P, Lemma K, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the reaction of cysteine thiosulfinate ester with cysteine to give cysteine sulfenic acid. J Org Chem. 2007;72:8838–8846. doi: 10.1021/jo701813f. [DOI] [PubMed] [Google Scholar]
- 141.Ishii T, Sunami O, Nakajima H, Nishio H, Takeuchi T, Hata F. Critical role of sulfenic acid formation of thiols in the inactivation of glyceraldehyde-3-phosphate dehydrogenase by nitric oxide. Biochem Pharmacol. 1999;58:133–143. doi: 10.1016/s0006-2952(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 142.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kice JL, Cleveland JP. Acid- and nucleophile-catalyzed oxygen-18 exchange of phenyl benzenethiolsulfinate. New insight into the chemistry of sulfenic acids and sulfenyl derivatives. J Am Chem Soc. 1970;92:4757–4758. [Google Scholar]
- 144.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 145.Willett WS, Copley SD. Identification and localization of a stable sulfenic acid in peroxide-treated tetrachlorohydroquinone dehalogenase using electrospray mass spectrometry. Chem Biol. 1996;3:851–857. doi: 10.1016/s1074-5521(96)90071-x. [DOI] [PubMed] [Google Scholar]
- 146.Conway ME, Poole LB, Hutson SM. Roles for cysteine residues in the regulatory CXXC motif of human mitochondrial branched chain aminotransferase enzyme. Biochemistry. 2004;43:7356–7364. doi: 10.1021/bi0498050. [DOI] [PubMed] [Google Scholar]
- 147.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reddie KG, Seo YH, Muse WB, Iii, Leonard SE, Carroll KS. A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol Biosyst. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Charles RL, Schroder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 150.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid–specific antibodies. Proc Natl Acad Sci U S A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maller NC, Schroder E, Eaton P. Gapdh Is Unlikely to Mediate Hydrogen Peroxide Signaling: Studies with a Novel Anti-Dimedone Sulfenic Acid Antibody. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3149. [DOI] [PubMed] [Google Scholar]
- 152.Sadidi M, Geddes TJ, Kuhn DM. S-thiolation of tyrosine hydroxylase by reactive nitrogen species in the presence of cysteine or glutathione. Antioxid Redox Signal. 2005;7:863–869. doi: 10.1089/ars.2005.7.863. [DOI] [PubMed] [Google Scholar]
- 153.Johansson M, Lundberg M. Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. BMC Biochem. 2007;8:26. doi: 10.1186/1471-2091-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 155.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 156.Birkett DJ, Price NC, Radda GK, Salmon AG. The reactivity of SH groups with a fluorogenic reagent. FEBS Lett. 1970;6:346–348. doi: 10.1016/0014-5793(70)80095-3. [DOI] [PubMed] [Google Scholar]
- 157.Silva GM, Netto LE, Discola KF, Piassa-Filho GM, Pimenta DC, Barcena JA, Demasi M. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J. 2008;275:2942–2955. doi: 10.1111/j.1742-4658.2008.06441.x. [DOI] [PubMed] [Google Scholar]
- 158.Salsbury FR, Jr, Knutson ST, Poole LB, Fetrow JS. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci. 2008;17:299–312. doi: 10.1110/ps.073096508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Baty JW, Hampton MB, Winterbourn CC. Proteomic detection of hydrogen peroxide-sensitive thiol proteins in Jurkat cells. Biochem J. 2005;389:785–795. doi: 10.1042/BJ20050337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Le Moan N, Clement G, Le Maout S, Tacnet F, Toledano MB. The Saccharomyces cerevisiae proteome of oxidized protein thiols: contrasted functions for the thioredoxin and glutathione pathways. J Biol Chem. 2006;281:10420–10430. doi: 10.1074/jbc.M513346200. [DOI] [PubMed] [Google Scholar]
- 162.Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J Biol Chem. 2007;282:22040–22051. doi: 10.1074/jbc.M703591200. [DOI] [PubMed] [Google Scholar]
- 163.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A. 2008;105:8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 165.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 166.Rehder DS, Borges CR. Possibilities and pitfalls in quantifying the extent of cysteine sulfenic acid modification of specific proteins within complex biofluids. BMC Biochem. 2010;11:25. doi: 10.1186/1471-2091-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.