

Abstract
Canonical-type transient receptor potential cation channel type 3 (TRPC3) allows the entry of extracellular Ca2+ and Na+ into various cells. In mouse skeletal myotubes, functional interaction between TRPC3 and RyR1 (ryanodine receptor type 1/Ca2+-release channel on sarcoplasmic reticulum membrane) regulates the gain of excitation–contraction coupling. Junctophilin-2 (JP2) is a TRPC3-interacting protein in mouse skeletal myotubes. Based on these knowledge from bona-fide TRPC3-expressing cells, to identify critical binding region(s) of JP2 that participate in binding to TRPC3, various JP2 portions were subjected to co-immunoprecipitation assay with intact TRPC3 from rabbit skeletal muscle. A region covering 143 to 234 amino acids of JP2 (F1-2) was the most efficient portion binding to TRPC3. Through mutational studies, we found that the binding ability of JP2 to TRPC3 was mainly due to glutamate in the F1-2 region (E227). This substantial binding between JP2 and TRPC3 suggests that JP2 can be a regulatory protein of TRPC3 and/or TRPC3-mediated Ca2+ homeostasis in skeletal muscle.
Keywords: RyR, Excitation–contraction coupling, Junctional complex
Introduction
Communications between intracellular and extracellular compartments of cells are significant events for cellular functions and the maintenance of intracellular Ca2+ homeostasis. In the case of skeletal muscle, a tight junction (called the triad junction) composed of t-tubule (invaginated plasma membrane) and sarcoplasmic reticulum (SR) membranes constitutes the structural element for efficient operation of excitation–contraction (EC) coupling, where dihydropyridine receptors on the t-tubule membrane sense membrane depolarization to control SR Ca2+ release via opening of the ryanodine receptor type 1 (RyR1/Ca2+-release channel) [1, 2]. We recently identified a role for TRPC3 (canonical-type transient receptor potential cation channel type 3) in EC coupling in mouse primary skeletal myotubes through a TRPC3 knock-down study; TRPC3 is required for full-gain skeletal EC coupling [3].
TRPC3 acts as a non-selective cation channel in the plasma membrane (PM), allowing extracellular Ca2+ and Na+ to enter into cells [4, 5]. Like the other six TRPC subtypes, TRPC3 has six transmembrane segments with a putative pore region between the fifth and sixth transmembrane segments, plus four ankyrin repeats in the N-terminus and a TRP box (EWKFAR) in the C-terminus. TRPC3 contributes to extracellular Ca2+ entry via two distinct pathways. First, it is involved in receptor-operated Ca2+ entry (ROCE) that is coupled to the activation of phospholipase C (PLC). Ligands that stimulate PLC lead to the generation of diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP2), and DAG can directly bind to TRPC3 to trigger Ca2+ entry. Secondly, TRPC3 participates in store-operated Ca2+ entry (SOCE), which is activated secondarily to Ca2+ depletion from the endoplasmic reticulum (ER)/SR by the activation of internal Ca2+-release channels, such as the inositol 1,4,5-trisphosphate receptor (IP3R) and RyR. SOCE is essential for the replenishment of internal Ca2+ stores and the maintenance of sustained Ca2+ signaling in response to repetitive cellular activities. Recently, researchers have tried to reveal how ER/SR Ca2+ depletion activates TRPC3 to induce SOCE in heterologously TRPC3-expressing cells such as HEK293 and COS7 cells: clustering of stromal interaction molecule 1 (STIM1) to the PM and ER junction in response to ER Ca2+ depletion and an indirect interaction between STIM1 and TRPC3 via Orai1 (a Ca2+-conducting channel located at PM) represent the elemental process for the activation of SOCE [6–8]; several investigators have shown that direct interaction between IP3Rand TRPC3 (competing with Ca2+/calmodulin binding to TRPC3 or homer1b/c-mediated gating of TRPC3 by IP3R) also contributes to SOCE [9–11]. In bona-fide TRPC3-expressing non-excitable cells, phospholipases (PLCγ2 and PLA2) have been known to be involved in the formation of the PM and ER junction and SOCE [12, 13], and identification of the molecular motifs that mediate the assembly of the PM and ER junction or triad junction has become one of central focuses in SOCE researches.
Junctophilins (JPs) have been known to contribute to the formation of triad junctions in excitable cells both by interacting with PM/t-tubule via their N-terminal membrane occupation and recognition nexus (MORN) motifs (Tyr-Gln/Glu-Gly-Glu/Gln-Trp-x-Asn-Gly-Lys-x-His-Gly-Tyr-Gly) and by spanning ER/SR membranes via their C-terminus [14–16]. JP knock-out mice died shortly after birth (JP1 knock-out) or showed embryonic lethality (JP2 knock-out) due to deformed triad junctions and subsequent disturbance in Ca2+ homeostasis [14, 16]. According to our recent report using two bona-fide TRPC3-expressing systems, mouse skeletal myotubes, and rabbit skeletal muscle, JPs interact with both RyR1 and TRPC3 through subtype-specific interactions (JP1-RyR1 and JP2-TRPC3) [17].
In this study, we examined TRPC3-interacting site(s) in JP2 and found that the region from 143 to 234 amino acids of JP2 contained critical TRPC3-binding sites; the binding ability of JP2 to TRPC3 was mainly due to glutamate in the F1-2 region (E227).
Materials and methods
Materials
Anti-TRPC3 (used at 1:800 for immunoblot and 1:250 for co-immunoprecipitation assays), anti-JP2 (used at 1:2000), and anti-GST antibodies (used at 1:2000) were obtained from Santa Cruz Biotechnology. Protein G-Sepharose 4 Fast Flow affinity beads were obtained from Amersham Biosciences. Isopropyl-β-D-thiogalacto-pyranoside (IPTG) was obtained from Sigma-Aldrich.
Preparation of the triad vesicle sample from rabbit skeletal muscle
Triad vesicles were prepared from rabbit fast-twitch muscle (back and leg) using a procedure described previously [18]. The triad vesicles were solubilized with a lysis buffer (1% Triton X-100, 10 mM Tris–HCl, pH 7.4, 1 mM Na3VO4, 10% glycerol, 150 mM NaCl, 5 mM EDTA, and protease inhibitors (1 μM pepstatin, 1 μM leupeptin, 1 mM PMSF, and 1 μM trypsin inhibitor)) for 4h at 4°C. All surgical interventions and presurgical and postsurgical animal care were provided in accordance with the Laboratory Animals Welfare Act, the Guide for the Care and Use of Laboratory Animals and the Guidelines and Policies for Rodent Survival Surgery provided by the IACUC (Institutional Animal Care and Use Committee) in the College of Medicine, The Catholic University of Korea, Korea.
Preparation of various JP2 portions or mutated JP2s
Based on the mouse JP2 sequence (GenBank accession number AB024447), synthetic oligonucleotides (forward primers containing the nucleotide sequence corresponding to the 5′ end of a given fragment with the EcoR I site at the 5′ end; reverse primers containing the complementary nucleotide sequence corresponding to the 3′ end of a given fragment with Sal I site at 5′ end) were used for the amplification of the JP2 portions under the following PCR conditions: 30 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 90 s. The PCR fragments were sub-cloned into the pGEX-4T-1 vector at EcoR I and Sal I sites to make GST-fused proteins. Mutations were carried out using a site-directed mutagenesis system. In brief, by using F1-2 or wild type full-length JP2 in the pGEX-4T-1 vector as DNA templates, mutants were synthesized by PCR in the presence of a pair of complementary synthetic oligonucleotide primers containing the desired mutation using Pfu DNA polymerase. Following temperature cycling of PCR, the parental DNA template was digested by DpnI endonuclease (specific for cleaving methylated DNA), and the nicked vector DNA incorporating the desired mutations was then transformed into DH5α supercompetent cells. The DNA sequences of all mutants were confirmed by sequencing. Production of various GST-JP2 proteins was induced in log phase DH5α containing the desired plasmid by the addition of 0.1 mM IPTG for 6 h at 37°C, and bacterial cell lysates were prepared by sonication in a lysis buffer (1% Triton X-100, 10 mM Tris–HCl, pH 7.4, 1 mM Na3VO4, 10% glycerol, 150 mM NaCl, 5 mM EDTA, and protease inhibitors). After centrifugation of the lysates at 1400 g, the supernatants were used for immunoblot or co-immunoprecipitation assay.
Co-immunoprecipitation assay
For co-immunoprecipitation assay [3, 18], the supernatants of the bacterial cell lysate containing each GST-JP2 protein (50 μg total protein) were incubated with solubilized SR samples containing intact TRPC3s (400 μg total protein) for 4 h at 4°C. The mixture was incubated with anti-TRPC3 antibody for 4 h at 4°C, followed by incubation with protein G-Sepharose affinity beads for 4 h at 4°C. The protein-bead complexes were washed three times with PBS containing protease inhibitors to remove nonspecifically bound proteins. The bound proteins to beads were eluted by boiling in the sodium dodecylsulfate (SDS) sample buffer and subjected to 10% polyacrylamide gel electrophoresis (SDS-PAGE) for immunoblot assay with anti-TRPC3, anti-GST, or anti-JP2 antibody. For immunoblot assay, the proteins on the gel were transferred onto PVDF (polyvinylidene fluoride) membrane at 100 V for 2 h. In order to obtain more precise band intensities, lower concentration of non-fat milk (2.5%) was used for blocking membranes. The membranes were incubated with one of primary antibodies, washed three times with PBS containing 0.1% Tween 20 (PBST), and then incubated with horseradish peroxidase-conjugated anti-rabbit (1:20,000) antibody for 45 min at room temperature. The membranes were washed three times with PBST and developed by using SuperSignal ultrachemiluminescent substrate (Pierce). The ratios of GST-mutant-band/TRPC3-band intensities were normalized by the ratio of GST-wild type-band/TRPC3 band intensities (which was set as 1) because there was a reasonable variation in the amount of co-immunoprecipitated TRPC3 s in samples (TRPC3 is a big and six-transmembrane protein).
Modeling of protein secondary structure
Secondary structures were predicted using a computer software program, Jpred3 Incorporating Jnet (that is the most wildly used program for secondary structure prediction), from Dundee University [19].
Statistical analysis
Results are given as mean ± S.E. of at least three independent experiments. Significant differences were analyzed using a paired t-test (GraphPad InStat, v2.04). Differences were considered to be significant at P<0.05. Graphs were prepared using Origin v7.
Results and discussion
JP is the protein that physically links the triad junction in striated muscle cells. It contains a transmembrane sequence at the C-terminus, which integrates into the SR/ER membrane. The N-terminus of JP contains a series of MORN motifs that adhere to the t-tubule membrane. Between these domains, JP contains a high degree of α-helical structures (in F2 region, Fig. 1a) that presumably account for an elastic coupling between t-tubule and SR membranes. In this study, detailed TRPC3-binding region(s) in JP2 were searched out by various GST-fused JP2 portions or mutants. Before testing the binding ability of each JP2 portion or mutant to TRPC3, full-length wild type JP2 as a GST-fused form was subjected to co-immunoprecipitation assay with intact TRPC3 from rabbit skeletal muscle (Fig. 1b). Successful co-immunoprecipitation of intact TRPC3 with GST-JP2 was shown by the immunoblot assay of the co-immunoprecipitants using both anti-GST and anti-JP2 antibodies, which suggests that GST-tag does not interfere with the interaction between TRPC3 and JP2, and again confirms the physical interaction between TRPC3 and JP2 as we observed in vivo previously [17].
Fig. 1.

Schematic diagram of mouse JP2 primary sequence, and co-immunoprecipitation of TRPC3 and GST-JP2. a Eight MORN motifs and a transmembrane (TM) domain exist. Amino acids are numbered. ‘+’ or ‘−’ indicates positively or negatively charged residue. b Solubilized rabbit triad sample containing intact TRPC3 was incubated with E. coli lysate expressing GST-JP2 and then subjected to co-immunoprecipitation assay with anti-TRPC3 antibody followed by immunoblot assay with anti-TRPC3, anti-GST, or anti-JP2 antibody. GST-JP2 was successfully co-immunoprecipitated with TRPC3. To show the specificity of anti-TRPC3 antibody for the recognition of TRPC3, “Input” without antibody served as a negative control for co-immunoprecipitation assay in Figs. 1–5. IP, immunoprecipitation; IB, immunoblot
Considering the location of TRPC3 (in t-tubule membrane) and the length of JP2 (696 amino acids), it is likely that the half of JP2 toward the N-terminus containing t-tubule membrane-attached MORN motifs is involved in binding to TRPC3. Accordingly, two fragments of JP2, F1 and F2, were constructed as GST-fused proteins (Fig. 1a). F1 (a.a. 143–284) corresponds to the joining region between two groups of MORN motifs. F2 (a.a. 322–423) corresponds to the elastic α-helical region starting from just after the last MORN motif. Regions for MORN motifs were not considered because it has been known that they have selective binding affinity to plasma membrane [20] and their function is target JPs to the plasma membrane [16]. In addition, two-thirds of residues in a MORN motif is composed of nonpolar/aliphatic or aromatic amino acids, suggesting that MORN motifs are not suitable to be exposed to hydrophilic conditions or interacting with hydrophilic surfaces of certain molecules. Unlike F2, F1 showed strong binding ability to intact TRPC3 (Fig. 2). The F1 region is one of the two divergent regions between JP1 and JP2 isoforms (with only 29.6% homology in amino acid sequences of F1 regions from mouse JP1 and JP2). For example, only the F1 region from JP2 has a putative binding motif (a.a. 176-APDSPAA-182) to Src homology domain 3 that is a prevalent protein binding module to recognize proline-rich sequences.
Fig. 2.
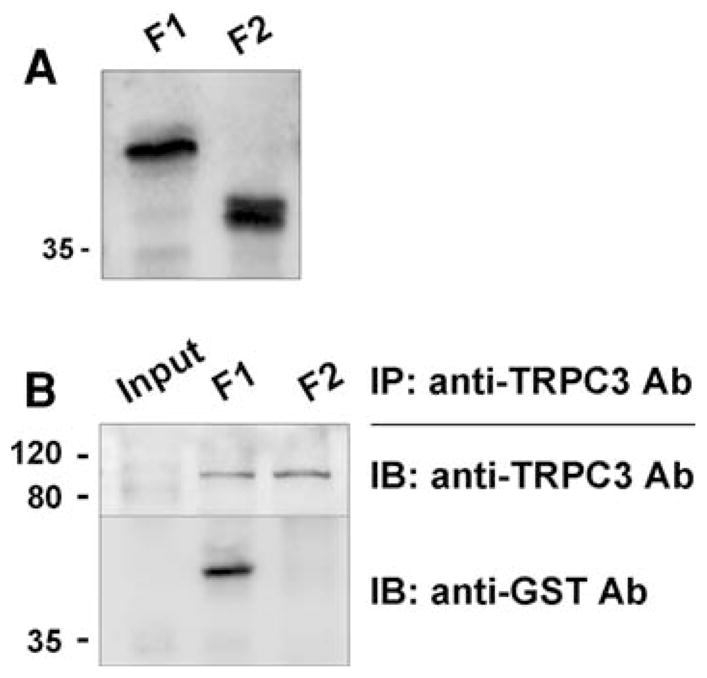
Co-immunoprecipitation of TRPC3 and GST-JP2 fragments. a Successful expression of GST-JP2 fragments (F1 and F2) was confirmed by immunoblot assay with anti-GST antibody. b Solubilized rabbit triad sample containing intact TRPC3 was incubated with E. coli lysate expressing F1 or F2, and then subjected to co-immunoprecipitation assay with anti-TRPC3 antibody followed by immunoblot assay with anti-TRPC3 or anti-GST antibody. F1 was strongly co-immunoprecipitated with TRPC3 but not F2. IP, immunoprecipitation; IB, immunoblot
In order to find critical residue(s) that causes the strong binding ability of F1 to TRPC3, the F1 region was narrowed to shorter forms: F1-1 (a.a. 143–244), F1-2 (a.a. 143–234), and F1-3 (a.a. 143–215) (Fig. 1a). These three shorter forms of F1 were subjected to co-immunoprecipitation assay with intact TRPC3 from rabbit skeletal muscle (Fig. 3). F1-1 and F1-2 still sustained strong binding ability to TRPC3. However, the shortest form, F1-3, showed greatly decreased binding ability to TRPC3 (0.25 ± 0.22 compared with that of F1-1). Therefore, F1-2 is the minimum-sized fragment to sustain effective binding to TRPC3. Non-specific bands near the size marker for 35 kDa in Figs. 3 and 4 disappeared under blocking membranes with 5% non-fat milk instead of 2.5% during immunoblot assay.
Fig. 3.
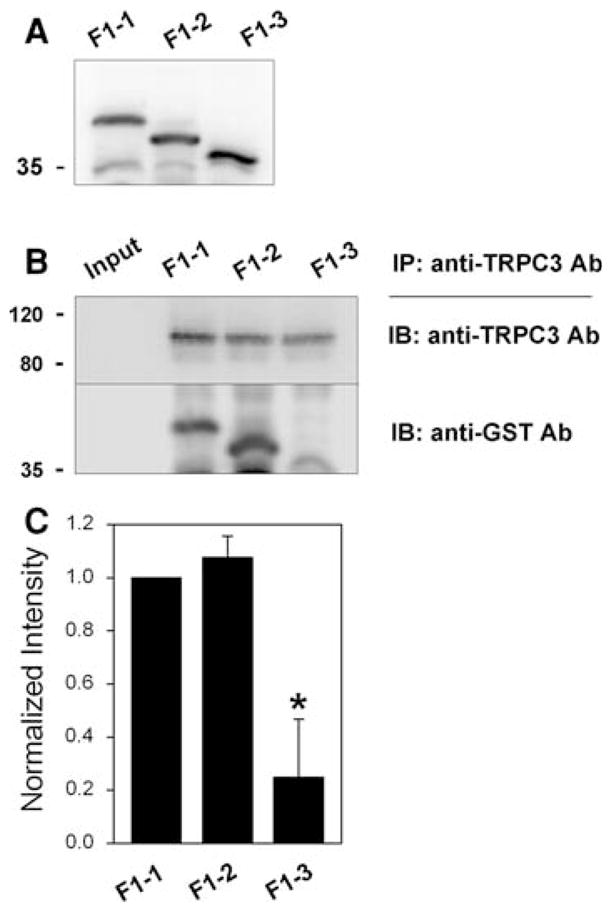
Co-immunoprecipitation of TRPC3 and shorter F1 forms. a Successful expression of GST-F1 shorter forms (F1-1, F1-2 and F1-3) was confirmed by immunoblot assay with anti-GST antibody. b Solubilized rabbit triad sample containing intact TRPC3 was incubated with E. coli lysate expressing one of the shorter forms, and then subjected to co-immunoprecipitation assay with anti-TRPC3 antibody followed by immunoblot assay with anti-TRPC3 or anti-GST antibody. F1-3 was much more weakly co-immunoprecipitated with TRPC3, compared with F1-1 and F1-2. IP, immunoprecipitation; IB, immunoblot. c The results of co-immunoprecipitation assays in (b) are summarized as bar graphs. The ratios of GST-F1 shorter form-band/TRPC3-band intensities were normalized by the ratio of GST-F1-1-band/TRPC3 band intensities (which was set as 1). Statistical significance compared to F1-1 is indicated by an asterisk
Fig. 4.
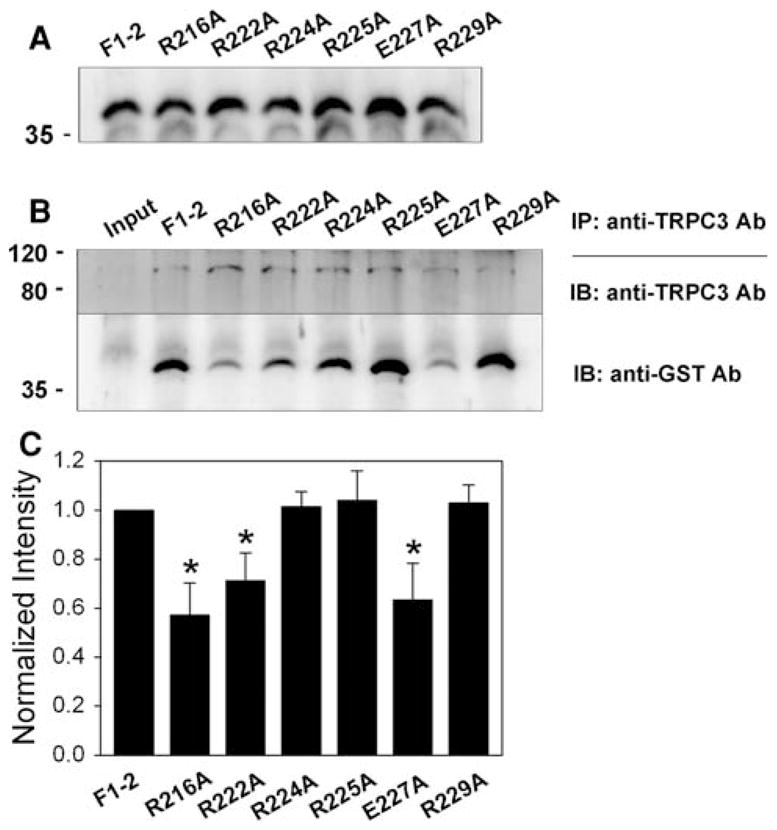
Co-immunoprecipitation of TRPC3 and single mutants of F1-2. a Successful expression of GST-F1-2 mutants (R216A, R222A, R224A, R225A, E227A, and R229A) was confirmed by immunoblot assay with anti-GST antibody. b Solubilized rabbit triad sample was incubated with E. coli lysate expressing one of six F1-2 mutants, and then subjected to co-immunoprecipitation assay with anti-TRPC3 antibody followed by immunoblot assay with anti-TRPC3 or anti-GST antibody. R216A, R222A, and E227A show significantly decreased binding ability to TRPC3. IP, immunoprecipitation; IB, immunoblot. c The results of co-immunoprecipitation assays in (b) are summarized as bar graphs. The ratios of GST-mutant-band/TRPC3-band intensities were normalized by the ratio of GST-wild type (F1-2)-band/TRPC3 band intensities (which was set as 1). Statistically significant decrease compared to F1-2 is indicated by asterisks
The region from 216 to 234 amino acids, which is missing in F1-3 but included in F1-2, contains six positively or negatively charged residues (R216, R222, R224, R225, E227, and R229 in Fig. 1a) that cover almost one-third of the region (31.6%, which is much higher than the random occurrence rate of charged amino acids in a given peptide). Based on this unusual fact, each of six charged residues was mutated to alanine and subjected to co-immunoprecipitation assay with intact TRPC3 from rabbit skeletal muscle (Fig. 4). Compared to wild type (F1-2), there was no change in the binding ability of R224A, R225A, or R229A to TRPC3. Unlike these three mutants, R216A, R222A, and E227A showed significantly decreased binding ability to TRPC3, suggesting that these three residues are critical and enable the F1-2 region to sustain strong binding ability to TRPC3. However, peptide fragments in solution have the potential to present folding issues and adopt conformations that may differ from that adopted in the full-length context. Therefore, we also expressed full-length JP2 mutants (R216A, R222A, E227A, and the triple mutant (R216A/R222A/E227A)) and examined their binding ability to TRPC3 by co-immunoprecipitation assay with anti-TRPC3 antibody (Fig. 5). E227A and the triple mutant in full-length JP2 showed significantly decreased binding ability to TRPC3 compared with wild type full-length JP2, suggesting that the binding ability of JP2 to TRPC3 was mainly due to glutamate at position 227 (E227). These results are the first to address a TRPC3-binding protein at the level of pinpointing a critical region and residues that participate in TRPC3-binding. There has been no previous report of a TRPC3-binding site in TRPC3-interacting proteins, although >10 proteins have been suggested as TRPC3-interacting/regulating proteins in endogenously or heterologously TRPC3-expressing systems. In addition, unlike other proteins found in heterologously TRPC3-expressing cells such as HEK293 or CHO cells, JP2 is one of TRPC3-interacting proteins that have been identified in “bona-fide TRPC3-expressing mouse and rabbit skeletal muscle cells” [17].
Fig. 5.

Co-immunoprecipitation of TRPC3 and full-length JP2 mutants. a Successful expression of full-length JP2s (wild type, R216A, R222A, E227A, and the triple mutant) was confirmed by immunoblot assay with anti-GST antibody. b Solubilized rabbit triad sample was incubated with E. coli lysate expressing one of full-length JP2s, and then subjected to co-immunoprecipitation assay with anti-TRPC3 antibody followed by immunoblot assay with anti-TRPC3 or anti-GST antibody. E227A and the triple mutant showed significantly decreased binding ability to TRPC3. IP, immunoprecipitation; IB, immunoblot. c The results of co-immunoprecipitation assays in (b) are summarized as bar graphs. The ratios of GST-mutant-band/TRPC3-band intensities were normalized by the ratio of GST-wild type JP2-band/TRPC3 band intensities (which was set as 1). Statistically significant decrease compared to wild type JP2 is indicated by asterisks
It is interesting that the three charged residues of JP2 (R216, R222, and E227) are highly conserved in JP1. According to our previous studies, there is no physical interaction between JP1 and TRPC3 [17]. Thus, it is likely that JP1 and JP2 have different three-dimensional conformations that may discriminate JP2 from JP1 in terms of TRPC3-binding. Indeed, in-silico modeling studies reveal that the F1 region of mouse JP2 is highly structured, with a charged region flanked by two amphipathic α-helical structures, and there is no α-helix just after the three corresponding charged residues in the F1 region of mouse JP1 (Fig. 6). Modeling for the triple mutant of F1 region at the three charged residues (R216A/R222A/E227A) shows an additional α-helix on the sites for the triple mutation, suggesting that the additional secondary structure makes the F1 region of JP2 lose its binding ability to TRPC3. The corresponding triple mutant on the basis of rabbit JP2 sequences showed the same tendency to change secondary structures (Supplementary data 1). Thus, the coordinated interaction between the α-helical domains of the F1 region may determine the isoform-specific interaction between TRPC3 and JP2 (not JP1).
Fig. 6.

Secondary structure modeling of F1 regions. Secondary structure modeling for F1 regions (a.a. 143–284 of mouse JP2, a.a. 143–280 of mouse JP1) was conducted using a secondary structure prediction program, Jpred3 Incorporating Jnet [19]. “H” and “E” indicate α-helix and β-strand, respectively. No indication means random coil. The charged region, including R216, R222, and E227 of JP2, at which mutations induced a significant decrease in the TRPC3-binding ability in Fig. 4 is flanked by two amphipathic α-helical structures. In the case of the F1 region of JP1, there is no α-helix just after the three corresponding charged residues (indicated by a gray box in JP2). Modeling for the triple mutant at the three charged residues on the basis of the F1 region of mouse JP2 (R216A/R222A/E227A) shows an additional α-helix on the sites of triple mutation (indicated by a black box), suggesting that the additional secondary structure makes the F1 region of JP2 lose its binding ability to TRPC3
It has been shown that, like the interaction between TRPC3 and JPs, FKBPs (FK-506 binding proteins) also have isoform-specific interactions with RyR isoforms (FKBP12-RyR1, FKBP12.6-RyR2) [18, 21, 22]. The interaction between RyR1 and FKBP12 is mediated mainly by glutamine (Q3) of FKBP12, but totally different residues of FKBP12.6 are involved in RyR2-binding (E31, D32, and W59), although FKBP12 and FKBP12.6 have >85% amino acid homology and extremely high similarity in their three-dimensional structures [23, 24].
Several mutations in JP2 have been found in human hypertrophic cardiomyopathy patients, and these JP2 mutants induce attenuated spontaneous Ca2+ waves when they are expressed in HL1 cardiac myocytes [25, 26]. One of these mutations occurred at S165, which is located in the F1-2 region that we suggest as the critical TRPC3-binding region in this study. Interestingly, S165 is only a putative phosphorylation site in JP2 by protein kinase C. Based on the fact that JP2 is also abundantly expressed in skeletal muscle, as in cardiac muscle [27], it is possible that, in addition to a simple binding between JP2 and TRPC3, JP2 can be a regulatory protein of TRPC3 and/or TRPC3-mediated Ca2+ homeostasis in skeletal muscle. This idea is supported by earlier reports; JPs have been known to contribute to the formation of triad junctions in excitable cells [14, 15], and the acute suppression of JP1 and JP2 in adult myotubes using RNA interference (around 75% reduction) causes deformed triad junctions and reduced SOCE [28]. However, whether the binding of JP2 to TRPC3 plays a more important role in muscle (directly mediating SOCE or other calcium signaling rather than contributing the formation of triad junction for normal SOCE) remains to be proven because our conclusion has been based on results from co-immunoprecipitation assay that is solution chemistry and not necessarily biology. Further functional studies on the relationship between TRPC3 and JPs will enhance the understanding of how SOCE is related to the assembly of PM/ER or triad junctions and complex molecular mechanism behind Ca2+ homeostasis in various cells including cardiac and skeletal muscle cells as well as how JPs play an important role in defining the architecture of the triadic junction in striated muscle.
Supplementary Material
Acknowledgments
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2007-331-E00014 to E.H.L.), the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MOST) (M10503010001-08N0301-00110 to D.H.K.), and NIH grants to J.M.
Abbreviations
- TRPC3
Canonical-type transient receptor potential cation channel type 3
- RyR1
Ryanodine receptor type 1/Ca2+-release channel
- JP
Junctophilin
- EC coupling
Excitation–contraction coupling
- PM
Plasma membrane
- PLC
Phospholipase C
- DAG
Diacylglycerol
- ER
Endoplasmic reticulum
- SR
Sarcoplasmic reticulum
- SOCE
Store-operated Ca2+ entry
- ROCE
Receptor-operated Ca2+ entry
- IP3R
Inositol 1,4,5-trisphosphate receptor
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11010-009-0070-0) contains supplementary material, which is available to authorized users.
Contributor Information
Jin Seok Woo, Department of Physiology, College of Medicine, The Catholic University of Korea, Seoul 137-701, Korea.
Ji-Hye Hwang, Department of Physiology, College of Medicine, The Catholic University of Korea, Seoul 137-701, Korea.
Jae-Kyun Ko, Department of Physiology and Biophysics, UMDNJ-Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA.
Do Han Kim, Department of Life Science, Gwangju Institute of Science and Technology, Gwangju 500-712, Korea.
Jianjie Ma, Department of Physiology and Biophysics, UMDNJ-Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA.
Eun Hui Lee, Email: EHUI@catholic.ac.kr, Department of Physiology, College of Medicine, The Catholic University of Korea, Seoul 137-701, Korea.
References
- 1.Sandow A. Excitation–contraction coupling in skeletal muscle. Pharmacol Rev. 1965;17:265–320. [PubMed] [Google Scholar]
- 2.Lee EH, Lopez JR, Li J, Protasi F, Pessah IN, Kim DH, Allen PD. Conformational coupling of DHPR and RyR1 in skeletal myotubes is influenced by long-range allosterism: evidence for a negative regulatory module. Am J Physiol Cell Physiol. 2004;286:C179–C189. doi: 10.1152/ajpcell.00176.2003. [DOI] [PubMed] [Google Scholar]
- 3.Lee EH, Cherednichenko G, Pessah IN, Allen PD. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J Biol Chem. 2006;281:10042–10048. doi: 10.1074/jbc.M600981200. [DOI] [PubMed] [Google Scholar]
- 4.Owsianik G, D’Hoedt D, Voets T, Nilius B. Structure–function relationship of the TRP channel superfamily. Rev Physiol Biochem Pharmacol. 2006;156:61–90. doi: 10.1007/s10254-005-0006-0. [DOI] [PubMed] [Google Scholar]
- 5.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 6.Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci USA. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA. 2008;105:2895–2900. doi: 10.1073/pnas.07122 88105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 9.Kim JY, Zeng W, Kiselyov K, Yuan JP, Dehoff MH, Mikoshiba K, Worley PF, Muallem S. Homer 1 mediates store- and inositol 1,4,5-trisphosphate receptor-dependent translocation and retrieval of TRPC3 to the plasma membrane. J Biol Chem. 2006;281:32540–32549. doi: 10.1074/jbc.M602496200. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci USA. 2001;98:3168–3173. doi: 10.1073/pnas.051632698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem. 2001;276:21303–21310. doi: 10.1074/jbc. M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 13.Nishida M, Sugimoto K, Hara Y, Mori E, Morii T, Kurosaki T, Mori Y. Amplification of receptor signalling by Ca2+ entry-mediated translocation and activation of PLCgamma2 in B lymphocytes. EMBO J. 2003;22:4677–4688. doi: 10.1093/emboj/cdg457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 2001;154:1059–1067. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee EH, Kim DH, Allen PD. Interplay between intra- and extracellular calcium ions. Mol Cells. 2006;21:315–329. [PubMed] [Google Scholar]
- 16.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/S1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 17.Woo JS, Kim DH, Allen PD, Lee EH. TRPC3-interacting triadic proteins in skeletal muscle. Biochem J. 2008;411:399–405. doi: 10.1042/BJ20071504. [DOI] [PubMed] [Google Scholar]
- 18.Lee EH, Rho SH, Kwon SJ, Eom SH, Allen PD, Kim DH. N-terminal region of FKBP12 is essential for binding to the skeletal ryanodine receptor. J Biol Chem. 2004;279:26481–26488. doi: 10.1074/jbc.M309574200. [DOI] [PubMed] [Google Scholar]
- 19.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisleder N, Takeshima H, Ma J. Immuno-proteomic approach to excitation–contraction coupling in skeletal and cardiac muscle: molecular insights revealed by the mitsugumins. Cell Calcium. 2008;43:1–8. doi: 10.1016/j.ceca.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samso M, Shen X, Allen PD. Structural characterization of the RyR1-FKBP12 interaction. J Mol Biol. 2006;356:917–927. doi: 10.1016/j.jmb.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 22.Xin HB, Rogers K, Qi Y, Kanematsu T, Fleischer S. Three amino acid residues determine selective binding of FK506-binding protein 12.6 to the cardiac ryanodine receptor. J Biol Chem. 1999;274:15315–15319. doi: 10.1074/jbc.274.22.15315. [DOI] [PubMed] [Google Scholar]
- 23.Deivanayagam CC, Carson M, Thotakura A, Narayana SV, Chodavarapu RS. Structure of FKBP12.6 in complex with rapamycin. Acta Crystallogr D Biol Crystallogr. 2000;56:266–271. doi: 10.1107/S0907444999016571. [DOI] [PubMed] [Google Scholar]
- 24.Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 25.Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, Wehrens XH, Claycomb WC, Ko JK, Hwang M, Pan Z, Ma J, Ackerman MJ. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. doi: 10.1016/j.yjmcc. 2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, Ikeda A, Kamatani N, Takeshima H, Matsuoka R. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet. 2007;52:543–548. doi: 10.1007/s10038-007-0149-y. [DOI] [PubMed] [Google Scholar]
- 27.Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273:920–927. doi: 10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- 28.Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, Zhao X, Thornton A, Komazaki S, Takeshima H, Ma J, Pan Z. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.