

Abstract
We previously demonstrated that mesenchymal stem/stromal cells (MSC) are recruited to tumors and that IFN-β produced by MSC inhibited tumor growth in xenograft models. Because of a deficient immune system, murine xenograft models cannot fully recapitulate tumor and immune cell interactions during progression. Therefore we investigated the capacity of MSC to migrate to and engraft into primary breast tumor sites and subsequently explore mechanisms of tumor inhibition by MSC-delivered IFN-β in a syngeneic, immunocompetent murine model. Herein we report that 1) systemically administrated MSC migrate to established 4 T1 breast cancer sites and localize among the tumor-stroma border and throughout the tumor mass; 2) high levels of IFN-β secreted by MSC are detectable in the tumor microenvironment but not in circulation; 3) intratumorally produced IFN-β inactivates constitutive phosphorylation of signal transducer activator transcription factor 3 (Stat3), Src, and Akt and down-regulates cMyc and MMP2 expression in 4 T1 cells, and 4) in mice with established breast cancer IFN-β expressing MSC administered systemically resulted in inhibition of primary cancer growth and in dramatic reduction of pulmonary and hepatic metastases. 5) MSC-IFN-β treated, but not control mice, maintained normal levels of splenic mature dendritic (DC), CD8+ T cells and CD4+/Foxp3+ regulatory T-cells (Treg). Our findings suggest that MSC are capable of migrating to tumor sites in an immunocompetent environment, that IFN-β produced by MSC suppresses breast cancer growth through inhibition of Stat3 signaling, and dramatically reduces pulmonary and hepatic metastases.
Electronic supplementary material
The online version of this article (doi:10.1007/s12307-010-0041-8) contains supplementary material, which is available to authorized users.
Keywords: IFN-β, MSC, Stat3, Breast cancer, Dendritic cell, T regulatory cell
Introduction
Mesenchymal stem/stromal cells (MSC) MSC are adult tissue resident progentior cells identified in many tissues/organs throughout the body. Initially isolated and described from bone marrow , MSC can differentiate into connective tissue lineages, such as bone, cartilage, and adipose as well as other tissue types [1, 2]. MSC were first characterized by our group as capable of selectively migrating to tumor sites through chemoattraction by tumor cell produced factors [3–5]. and local inflammatory chemokines and cytokines induced by tumor invasion [6, 7]. Solid tumor growth and invasion create a microenvironment that induces the secretion of factors that attract MSC to specifically migrate to the tumor sites [8]. This selective tropism for tumor microenvironments gives MSC the ability to selectively deliver growth inhibitory proteins such as interferon-β (IFN-β) and thus render the microenvironment inhospitable to tumor growth. In our previous studies using xenograft animal models [4, 5], we showed that IFN-β secreted by MSC inhibited melanoma xenografts in a SCID mouse. However, xenograft models may not be fully appropriate for evaluating the biologically complex interactions of this cell based delivery, given that the host lacks an immune response to the implanted cancer cells, the injected MSC, and to IFN-β locally delivered by MSC (MSC/IFN-β). Therefore, the interactions between cancer cells and normal cells cannot be fully evaluated, and the complexity of the observed effects remains poorly understood.
High concentrations of IFN-β induce apoptosis of cancer cell lines in vitro [9]. Although the exact mechanism remains unclear, it may involve the Jak1 and signal transduction activation transcription factor 1 (Stat1) intracellular signaling pathways [10]. It has also been reported that IFN-β may have anti-apoptotic effects, such as increasing Bcl2/Bcl-xL levels in B cells [11]. IFN-β also affects angiogenesis by down-regulating the expression of tumor-induced pro-angiogenic factors [12] such as basic fibroblast growth factor. However, because most IFN-β studies have been conducted using murine xenograft models [11–13], the biological effects of IFN-β have been difficult to evaluate. One report [14] suggested that IFN-α/β exerts little effect on apoptosis of T cells but instead predominantly functions through the inhibition of Stat3 and, to a lesser extent, Stat5 pathways. These findings suggest that IFN-β may exert anti-tumor effects via inhibition of signaling pathways in breast cancer cells and that the full biological effect should be assessed in a syngeneic, fully immunocompetent model.
Constitutively activated Stat3 has been reported to play an important role in tumorigenesis [15–17]. An early study with human breast cancer lines showed that five of nine cell lines tested had constitutively activated Stat3 [18]. A clinical investigation of human breast cancer specimens indicated that activated Stat3 was consistently found in tumor tissue but not in surrounding normal tissue cells [16]. Because knockout of the Stat3 gene is lethal for embryonic development in mice, it has not been possible to study the oncogenecity of Stat3 in a gene-deficient environment. In our previous studies, in a breast cancer transplant model in immunocompetent mice, both knockdown of Stat3 and pharmacological inactivation of Stat3 resulted in abrogation of breast tumor development [19, 20]. However, a more feasible approach to the inactivation of Stat3 is needed to determine the potential clinical applicability of this finding.
As the major clinical problem in breast cancer, mechanisms of metastasis have been investigated intensively. We previously demonstrated [19, 20] that primary breast tumors and metastases can be completely abrogated in immunocompetent mice after Stat3 knockdown or inactivation, prompting us to hypothesize that Stat3 plays a crucial role in breast cancer growth and metastasis. It is now well established that metastases are the result of interactions between cancer cells and their microenvironment. The process by which cancer cells establish tumors or migrate and settle in a distant tissue or organ is complicated, but it is believed that host cellular immunity is part of this process. However, malignant tumors usually escape host immune surveillance because of immune suppression induced by cancer cells [21–25]. Although mechanisms of this immune suppression have not been well characterized, strong evidence suggests that cancer cells circumvent host antigen presentation by preventing the maturation of DC [21, 26]. It has been reported that the number of mature DC is reduced during cancer development [21] and that cancer cells induce immunosuppression by stimulating regulatory T cells (Treg) [27–30]. Suppressing Treg has been shown to be effective in enhancing the immune response of host cells to cancer cells [31, 32]. Importantly, studies have shown that constitutively activated Stat3 is involved in these events [33]. Taken together, this evidence suggests that constitutively activated Stat3 not only functions as an initiator for oncogenesis but is also involved in interactions between cancer cells and their microenvironment.
4 T1 is a well-characterized murine spontaneous breast cancer cell line originally derived from a BALB/c mouse that contains constitutively activated Stat3 [34]. The 4 T1/BALB/C syngeneic immunocompetent murine breast cancer model is useful for understanding breast cancer biology. In the present study, in order to examine our hypothesis, we used MSC isolated from BALB/c mice to deliver murine IFN-β to determine whether the concept of using MSC to deliver IFN-β can be further developed for the treatment for breast cancer.
Materials and Methods
Animals, female BALB/c mice (6 to 8 weeks old) were purchased from Charles River Laboratories (Wilmington, MA) and maintained in the M. D. Anderson conventional animal facility. Experiments were conducted under an appropriate protocol.
Cell line, antibodies, and reagents Firefly luciferase-tagged 4 T1 cells were a gift from Dr. Mien-Chie Hung (M. D. Anderson Cancer Center) and were maintained in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% fetal bovine serum. Mouse MSC (mMSC) were obtained from Dr. Darwin Prockop (Tulane University, New Orleans, LA). These MSC are fully characterized and tested for their ability to differentiate into three lineages before their release. The cells were cultured in alpha minimal essential medium supplemented with 10% horse serum, 10% fetal calf serum, 10% fetal bovine serum, L-glutamine, and a penicillin-streptomycin mixture. Passages 4–7 were used throughout these experiments. The MSC were negative for FLK1 (VEGF-R2), CD31 (PECAM), CD90 (Thy1), CD117 (c-kit), CD11b, Ter-119, CD45R/B220, Ly6G and Ly-6C, and CD3e, and were positive for CD106 (VCAM-1). cDNA of murine IFN-β was purchased from Invivogen. Lentiviral packaging, envelope, and gene transfer plasmids were gifts from Dr. Didio Trone (Geneva University, Geneva, Swaziland).Anti-p-Tyr-STAT3 (pTyr-705), STAT3, p-Src (pTyr 527),Src, p-Akt (pSer 473), Akt, cMyc, and matrix metalloproteinase 2 (MMP2) were purchased from Cell Signaling (Beverly, MA); anti-β-actin was from Sigma Life Science (St. Louis, MO). Cell Invasion Kit was from Chemicon (Temecula, CA). D-Luciferin for firefly luciferase was from Caliper LifeScience (Hopkinton, MA). Rhodamine 123 conjugated donkey anti rabbit secondary was from Calbiochem Int.
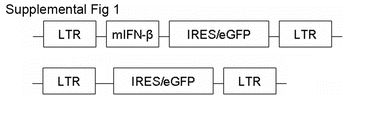
IFN-β lentiviral gene transfer plasmid construction, virus preparation and IFN-β gene transduction The cDNA of murine IFN-β was cleaved from pORF-mIFNβ plasmid from Invivogen (Cat# porf-mifnb). The fragment of IFN-β cDNA was sub-cloned into lentiviral gene transfer vector, pLOXiE [35]. The constructed lentiviral transfer plasmid was then verified by sequencing. The scheme of the plasmid is shown in supplemental Fig. 1. Lentivirus was prepared as described in our previously (Ling et al., 2005). MSC transductions by IFN-β/GFP or GFP alone as the control were done by lentiviral infections. The detailed procedure was described elsewhere [35]. Because all infected cells express GFP, as indicated in the gene transfer vector diagram in supplemental Fig. 1, MSC/IFN-β/GFP or MSC/GFP cells were selected by FACS using GFP as the marker. GFP-positive cells were subsequently expanded.
ELISA for IFN-β concentration After MSC were transduced with MSC/IFN-β/GFP or GFP and sorted, IFN-β levels in sorted MSC culture supernatant were measured by ELISA. ELISA was performed according to the manufacture’s instruction (PBL Biomedical Laboratories, New Brunswick, NJ).
Western blotting Western blotting was performed as previously described [19].
Cell proliferation assay 4 T1 cells were seeded onto six-well plates at a concentration of 5 × 106 cells per well in triplicate and were co-cultured with MSC/IFN-β/GFP or MSC/GFP (5 × 106 cells/well). The total number of viable 4 T1 cells in each well was determined using an automated analyzer (Vi-Cell, Beckman Coulter, Miami, FL).
Cell cycle analysis After 4 T1 cells were co-cultured with MSC/IFN-β/GFP, 4 T1 cell cycle analysis was performed as previously described [20]. Briefly, cells were fixed with 70% ice-cold ethanol and stained with propidium iodide (PI) solution (25 μg/mL PI, 180 U/mL RNase, 0.1% Triton X-100, and 30 mg/mL polyethylene glycol in 4 mM citrate buffer, pH 7.8; Sigma Chemical). DNA content was determined using a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and cell cycle distribution was analyzed using ModFit LT software (Verity Software House, Topsham, ME).
Matrix gel invasion assay The matrix gel invasion assay was conducted in matrix chambers according to the kit’s instructions (CHEMICON International, Billerica, MA). Briefly, 4 T1 cells were starving of fetal bovine serum overnight, then seeded onto upper culture wells (2 × 105cells/well) that contained no serum; the lower well was seeded with same amount of MSC/IFN-β/GFP and was with normal medium with 10% FBS. After 48 hours, the penetrated 4 T1 cells were collected and quantitated according to the manufacture instructions.
Mouse tumor formation assay For the in vivo studies, 7 × 103 4 T1 cells were injected into the mammary fat pads of 8-week-old female BALB/c mice. Tumor formation with or without treatment at the inoculation site was monitored using an in vivo imaging system (Xenogen-200; Xenogen Corp., Alameda, CA), and the tumor size was defined as the expression of luciferase as 4 T1 cell was tagged with luciferase prior to the injection.
MSC injection MSC/IFN-β/GFP or MSC/GFP were typsinized and washed with PBS three times, and then filtered using a 70-μM cell strainer. Mice were injected intravenously via the tail vein.
Bioluminescent imaging (BLI) Mice were injected intraperitoneally with 4 mg of D-luciferin and then imaged using the Xenogen-200 in vivo imaging system as described previously [20].
Histological analysis For histological analysis, breast tumors from mice treated with and without MSC/IFNb/GFP were excised and fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, and stained with H&E. The pStat3 immunostaining was performed on paraffin-embedded tumor sections, and the pAkt immunostaining was performed on tumor cryosections. The slides were analyzed under an Olympus BX 41 microscope with a digital capture camera (Olympus DP70).
Results
MSC/IFN-β cells selectively engraft in breast tumor sites Immunohistochemical staining for MSC/IFN-β/GFP cells with anti-murine IFN-β antibody indicated that MSC exhibit tropism for 4 T1 breast tumor sites (Fig. 1a I–III) after tail vein injection in mice. High levels of IFN-β (dark brown color) were demonstrated along the border of breast tumor and stromal tissue (Fig. 1a II–III). This phenomenon is consistent with our previous findings in a both breast cancer and melanoma xenograft models [7]. No specific staining was seen in control mice injected, i.v. with MSC/GFP (Fig. 1a IV), indicating IFN-β secretion by gene-modified MSC at the tumor sites. To demonstrate that MSC were producing and secreting the IFN-β, we reacted MSC/IFN-β/GFP cells attached on slides with anti-mouse IFN-β antibody (the secondary antibody labled with Rhodamine, showing in red fluorescent color) Fig. 1 VI, As MSC/IFN-β/GFP cells co-expressing GFP, Fig. 1V, MSC were seen in green color as well; and a merged picture showed in Fig. 1 VII, indicating that the MSC we injected into mice were expressing GFP and murine IFN-β. As 4 T1 cells were tagged with GFP as marker; therefore, we were unable to distinguish 4 T1 cells from MSC/IFN/GFP in tumor tissues. Additionally, ELISA demonstrated the secretion of high levels of IFN-β after transduction with murine IFN-β cDNA (Fig. 1b left panel) in vitro. Very low levels of IFN-β (0.58 IU/ml) were found in the serum of mice after i.v. MSC/IFN-β/GFP cell injection (Fig. 1b right panel). This finding is consistent with our previous data that also showed low levels of IFN-β levels in the serum of MSC-IFN-β injected mice [7].
Fig. 1.

MSC/IFNβ/GFP cells home to 4 T1 breast tumors and express high levels of IFNβ. 1 × 106 MSC/IFN-/GFP and MSC/GFP cells were injected into 4 T1 tumor established mice through tail vein. Tissues were collected in 3 days after MSC/IFN/GFP and MSC/GFP cell administration and slides sections were prepared. Immunohistochemical staining with anti-mouse IFN-β antibody in Fig. 1 I-IV indicates that high levels of IFNβ were expressed by MSC especially around the border of the tumor and stromal tissue. Magnifications of the pictures are as indicated. Mice injected with MSC/GFP served as controls, Fig. 1 IV, absence of IFN-β positivity after injection of MSC/GFP cells. Staining attached MSC/IFN-β/GFP cells on slides were probed with anti mouse IFN-β antibody, the secondary antibody is conjugated Rhodamine (Abcam, Cambridge, MA), Fig. 1 VI; or without anti IFN-β antibody, Fig. 1 V. Fig. 1 VII is the merging result for Fig. 1 V and VI. b left, high levels (45 IU/ml) of IFNβ in MSC/IFN-β/GFP culture medium determined by ELISA b right, low levels of IFN-β (0.5-1 IU/ml) in mouse serum collected 3 days after MSC/IFN-β/GFP injection (n = 3)
Co-culture of 4 T1 with MSC/IFN-β/GFP reduces cell invasion 4 T1 cells are strongly invasive in vitro and in vivo [19, 20]. In order to assess the effect of MSC/IFN-β/GFP on the invasion of 4 T1 cells, we performed in vitro cell migration assays, and found that the migratory capacity of 4 T1 cells was significantly inhibited after cells were co-cultured with MSC/IFN-β/GFP compared with 4 T1 cells co-cultured with MSC/GFP (P < 0.01), (Fig. 2). In addition, cellular migration was inhibited to a similar extent in Stat3 knockdown 4 T1 cells (Ling et al., 2005) and in 4 T1 cells treated with the Stat3 inhibitor CDDO-Me, a small molecule that, as we demonstrated, inactivates Stat3 [20]. Recombinant murine IFN-β was used as a control for MSC/IFN-β/GFP.
Fig. 2.

MSC/IFN-β/GFP inhibits migration of 4 T1 cells in co-culture. 4 T1 cell migration was significantly inhibited (p < 0.01) when cells were co-cultured with MSC/IFNβ/GFP. In contrast, co-cultures with MSC/GFP, or treatment with scrambled shStat3 had no effect on migration of 4 T1 cells. Recombinant murine IFNβ (50 IU) was used as control for MSC/IFNβ/GFP
IFN-β inactivates Stat3 in 4 T1 cells in vitro and in vivo Constitutively activated Stat3 has been reported to be a key regulator in breast cancer. In order to examine how IFN-β affects Stat3 activation, we investigated Stat3 signaling following co-culture of 4 T1 cells with MSC/IFN-β/GFP. As shown in Fig. 3a, Stat3 was inactivated when 4 T1 cells were co-cultured with MSC/IFN-β/GFP for 48 h, and Src, the upstream regulator of Stat3, was found to be inactivated. Interestingly, the downstream target of Src, Akt, was inactivated under the same conditions. Furthermore, the expression of c-Myc and MMP2 was completely abrogated after co-culture with MSC/IFN-β as well. In order to determine whether IFN-β expressed by MSC can effectively inactivate 4 T1 breast cancer in vivo, immunohistochemical staining was performed using anti-phospho-Stat3 (pStat3 705) antibody. In addition, we observed similar results from 4 T1 cells after treatment with murine recombinant IFN-β, Fig. 3b. We also examined effect of co-culturing 4 T1 MSC/IFN-β/GFP and murine recombinant IFN-β on total Stat3 and pStat3, the results showed in Fig. 3c, indicating MSC/IFN-β/GFP and murine recombinant IFN-βconditions had a similar effect on Stat3. As shown in Fig. 3dright panel, pStat3 in 4 T1 breast tumors was found to be inactivated 3 days after MSC/IFN-β/GFP cells were administrated systemically via tail vein injection (1 × 106cells/mouse).
Fig. 3.

Changes in Intracellular signal transduction in 4 T1 cells in co-culture with MSC/IFNβ/GFP. 4 T1 breast cancer cells were co-cultured with MSC/IFNβ/GFP for 48 hours. Western blot were performed to examine changes in expression levels of several oncoproteins and proteins involved in metastasis., pStat3, pSrc and pAKT were inactivated; and expression of c-Myc and mmp2 was completely abolished (a). (Quantitative analysis is shown in lower panel of a). Similar results were observed when 4 T1 cells were treated with recombinant murine IFNβ, b (quantitative analysis in b, lower panel). c shows inhibition of Stat3 activation after 4 T1 cells were co-cultured with MSC/IFN-β/GFP or murine recombinant IFN-β. d: immunohistochemical staining of pStat3 in 4 T1 tumor in vivo after systemic administration of MSC/IFN-β/GFP cells in mice, d, right panel; mice treated with MSC/GFP, as the control, showed a strong pStat3 activity in 4 T1 tumors, (left panel)
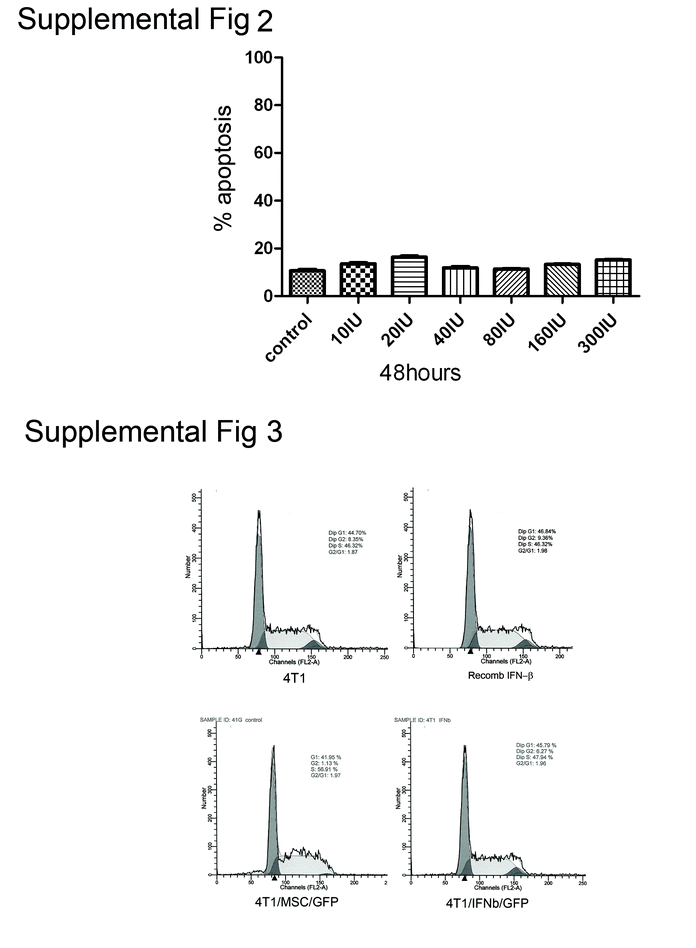
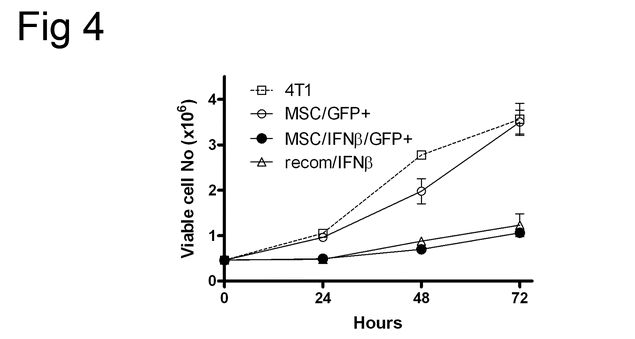
MSC/IFN-β inhibits 4 T1 cell proliferation in vitro It has been reported that Stat3 inhibition can induce apoptosis [36, 37]. However, in our previous studies [19, 20], we did not detect apoptosis after either knockdown or inhibition of Stat3. We therefore examined 4 T1 cell apoptosis by measuring Annexin V and propidium iodide (PI) positivity, using flow cytometry, after cells had been co-cultured with MSC/IFN-β/GFP: no significant apoptosis was observed (supplemental data, Fig. 2), while 4 T1 cell growth was inhibited 3-fold (supplemental data Fig. 3). However, we did not observe significant changes in cell cycle (supplemental data, Fig. 4).
Systemic injection of MSC/IFN-β inhibits 4 T1 breast cancer growth and metastases in vivo As mentioned earlier, 4 T1 is a spontaneous breast cancer cell line derived from the BALB/c mouse. It is a very aggressive breast cancer and often exhibits pulmonary and hepatic metastases after transplantation into the BALB/c mammary gland fat pad. In order to examine the effect on breast cancer of locally produced high levels of IFN-β, 7 × 103 firefly luciferase-tagged 4 T1 cells were injected into the mouse mammary gland fat pad, and MSC/IFN-β/GFP (1 × 106/mouse) or control MSC/GFP cells (1 × 106/mouse) were injected into mice through the tail vein 1 day after 4 T1 inoculation. Mice were monitored for 30 days, and mice with tumors larger than 2.5 cm were euthanized and a survival curve was plotted as shown in Fig. 4a. The difference between the two group is significant, P < 0.001 (Log-rank test). All MSC/IFN-β/GFP—treated mice remained breast cancer free for 103 days after the tumor cells were transplanted (Fig. 4b right panel), and were then terminated for histological analysis, whereas all MSC/GFP-treated mice developed aggressive breast cancer and had to be euthanized because of the size of breast tumors (over 2.5 cm) (Fig. 4b left panel, and 5c left panel). The tumor size in live mice was determined by expression of luciferase in 4 T1 cells (Fig. 4b, c left panel) and by direct examination of metastases (Fig. 4c middle panel, lung and right panel, liver), whereas mice treated with MSC/IFN-β/GFP had neither tumors nor metastases. In order to confirm that the control MSC/GFP did not enhance 4 T1 breast tumor growth, we also compared tumor sizes in mice that were administered control MSC/GFP, Fig. 4d, no effect of MSC/GFP on 4 T1 tumor growth was observed. As shown in supplemental data, Fig. 5, no significant difference between the two groups (P = 0.2844) in tumor growth was seen when either no cells or MSC/GFP were injected in animals bearing 4 T1 tumors.We also explored the effects of MSC/ IFN-β/GFP on 4 T1 breast tumor growth when MSC/ IFN-β/GFP treatment was delayed and MSC were injected i.v. on day 5 after tumor implantation. As shown in Fig. 5a and b, significant inhibition of tumor growth was observed on day 13 after tumor cell implantation (P = 0.02). In addition, tumors grew faster in the MSC/GFP group than in the MSC/ IFN-β/GFP group (Fig. 5c). Histological results show that lungs (Fig. 5d upper panel) livers (Fig. 5d lower panel) from MSC/ IFN-β/GFP treated mice had dramatically reduced metastatic nodules than those from the control group.
Fig. 4.

MSC/IFN-β/GFP inhibits breast tumor progression in 4 T1 tumor-bearing mice. Mice were I.V. injected MSC/IFN-β/GFP or control MSC/GFP, 1 × 106 cells/mouse through tail vein on day one after 4 T1 cell injection. 4 T1 tumors in the control group grew rapidly and all mice were euthanized on day 35 after the 4 T1 cell injection, as shown in bleft panels. Survival analysis was performed on 4 T1 tumor bearing mice treated with MSC/IFN-β/GFP compared to other two groups, P < 0.01 demonstrating a statistically significant increase in survial. Mice treated with MSC/IFN-βGFP had no 4 T1 breast tumor detectable up to 103 days after tumor cell injection, (bright panel). A representative 4 T1 breast tumor is photographed in situ (cleft panel), with metastatic tumor nodules in lungs (c, middle panel) and liver (cright panel). The survival curves of the two groups are shown in a displaying a significant difference (P < 0.001, log rank test). d shows mice injected either MSC/GFP (left panel) or not (right panel) did not affect 4 T1 tumor growth
Fig. 5.

Effect of delayed MSC/IFN-β/GFP treatment on 4 T1 breast tumor progression. Mice were injected with MSC/GFP or MSC/IFN-β/GFP (1 × 106 cells/mouse) 5 days after 4 T1 implantation. Significant inhibition of tumor growth was observed on day 13 after tumor cell injection in the MSC/IFN-β/GFP as compared with the control group, p = 0.025, (a).Tumors were observed to grow rapidly in the control group (bleft panel) while tumor size was reduced in MSC/IFN-β/GFP treated mice (bright panel). As determined by BLI, tumor size was size significantly larger in MSC/GFP treated mice (cleft panel), while that in MSC/IFN-β/GFP treated mice were significantly reduced (cright panel). Extensive Metastatic tumors (arrows) were observed in lungs (dleft upper panel) and liver (dleft lower panel) of control mice. In contrast, few metastatic nodules were seen in the lungs from MSC/IFN-β/GFP treated mice (dright upper panel) and no metastatic nodules were observed in the livers from MSC/IFN-β/GFP treated mice (dright lower panel)
Treatment with MSC/IFN-β/GFP stimulates cell-mediated immunity Mature splenic DCs were analyzed after mice were treated with MSC/IFN-β/GFP. Mature murine DCs were identified as major histocompatibility complex II and 33D1 double positive cells (Fig. 6aI–II). 33D1 is a marker of mature DC [38]. MSC/IFN-β/GFP—treated mice had significantly more mature splenic DCs than MSC/GFP-treated mice (P < 0.01). This set of experiments was performed using mice that started MSC/IFN-β/GFP treatment (a single injection of MSC/IFN-β/GFP, 1 × 106 cells/mouse via the tail vein 1 day after 4 T1 injection) 1 day after tumor cell implantation. We also evaluated splenic CD8+ T cells after treatment with MSC/IFN-β/GFP. As indicated in Fig. 6a III, the CD8+ T cell population after treatment with MSC/IFN-β/GFP was significantly greater than that in control (MSC/GFP) mice (P < 0.01). In addition, Treg cells were examined after MSC/IFN-β/GFP treatment by monitoring CD4 and Foxp3 double-positive splenic cells. As shown in Fig. 6a IV, the percentage of Treg cells in the control group was significantly higher than that in the MSC/IFN-β/GFP—treated group (P < 0.01). Immunohistochemical staining showed fewer Foxp3+ cells in MSC/IFN-β/GFP—treated (Fig. 6b) than in MSC/GFP-treated mice. Immunohistochemical staining also indicated more CD8+ T cells in spleens of MSC/IFN-β/GFP than in MSC/GFP-treated mice (Fig. 6c). Finally, more mature DCs were observed in spleen sections from mice treated with MSC/IFN-β/GFP Fig. 6d right panel) than in spleen sections from mice treated with MSC/GFP (Fig. 6d left panel).
Fig. 6.


Systemic treatment with MSC/IFNβ/GFP improves the cellular immunity in breast tumor-bearing mice. Mature DCs in spleen were analyzed by flow cytomery after MSC/IFNβ/GFP injection. Mature DC (a I–II ) and CD8+ T cell (Fig. 6a III ) frequencies were significantly increased compared with mice treated with MSC/GFP, p < 0.01. Importantly, levels of Tregs in MSC/IFNβ/GFP mice remained the same as in normal mice but were elevated in MSC/GFP treated mice, p < 0.01, a IV. Immunohistochemical analysis of splenic Foxp3+ T cells (b), CD 8+ T cells (c) and mature DCs (d) support these results. Fewer mature DC are noted in MSC/GFP treated mouse spleens (d, left panel) than in MSC/IFN-β/GFP treated spleens (d, right panel)
Discussion
In this study we have demonstrated that in an fully syngeneic immunocompetent model BALB/c mice, BALB/c mMSC home specifically to 4 T1 (from BALB/c) breast cancer sites and deliver murine IFN-β to the tumors sufficient to inhibit breast cancer growth through inactivation of the Stat3 signaling pathway. In addition, MSC/IFN-β/GFP treatment also showed signs of an improvement in cell-mediated immunity as indicated by increased numbers of splenic mature DC and decreased numbers of Treg cells. Because a fully immunocompetent and syngeneic system was used this study, problems associated with xenograft models, such as lack of interaction between cancer and host immune cells were avoided, and more biologically relevant information was generated. To the best of our knowledge, this is the first report showing the biological effects of IFN-β delivered by MSC specifically to breast cancer sites in an immunocompetent model. In addition, our in vivo and in vitro data indicate that the mechanism of this effect is through inactivation of constitutively activated Stat3 in breast cancer cells, which in turn improves the breast cancer host’s cell-mediated immunity. Our present findings are not only consistent with our previous work [5, 7] in xenograft models showing MSC homing to the tumor microenvironment and evidence that IFN-β delivered by MSC inhibits human cancer xenografts but also illuminates some of the molecular mechanisms that convey the observed anti-tumor effects of IFN-β on breast cancer in vivo and extend our previous work on the efficiency of systemically delivered MSC/IFN-β. These results further support the translation of this novel therapeutic concept into clinical trials, which is under development [3].
MSC contribute to the maintenance and regeneration of connective tissues [1]. After systematic infusion, MSC appear to engraft depending on the production of incompletely understood paracrine signals in the tissue microenvironment [39]. Solid tumor growth and invasion create a microenvironment similar to that described in wound healing, which also induces MSC to specifically migrate to the wound (or site of inflammation) [40, 41]. This feature gives MSC the ability to deliver molecules such as IFN-β and thus render the microenvironment inhospitable to tumor growth. Recently, it is also reported that MSC may involve in transition to tumor-associated fibroblasts and contributes to fibrovascular network expansion and tumor progression [52].
The injection of recombinant IFN-β into humans has shown no or limited effects on the growth of several different cancer types [42]. Because of IFN-β’s well described short half life [43], it is not expected that sufficient levels of injected recombinant IFN-β can be maintained at the tumor site to affect tumor growth before the protein is degraded. However, using MSC to secrete IFN-β in situ would overcome this issue. As shown in Fig. 1 by immunohistochemical staining, mMSC were localized along the tumor/stroma border, expressing high levels of IFN-β. In contrast we were not able to detect MSC or MSC produced IFN-β in other tissues, suggesting the exquisite selectivity of MSC for tumors and the impact of the MSC-produced IFN-β on tumor growth. Importantly, constitutively activated Stat3 in 4 T1 was inactivated after MSC/IFN-β/GFP were administered systemically (Fig. 3b), even while plasma levels of IFN-β were extremely low.
It has been reported that IFN-β has an inhibitory effect on cancer cell growth in vitro and in xenograft models [7]. However, other evidence has also shown that IFN-β is a strong immune modulator in vivo. Therefore, the entire spectrum of anti-tumor mechanisms of IFN-β has not been fully characterized under physiological conditions [44]. Using our immunocompetent syngeneic model, we were able to show that constitutively activated Stat3 in 4 T1 cells was inactivated when those cells were co-cultured with MSC/IFN-β/GFP (Fig. 3a), the same effect was achieved by co-cuture with recombinant IFN-β protein. Moreover, downstream targets of Stat3, such as c-Myc and MMP2, were down-regulated. It was interesting that Akt, a downstream target of Src, was inactivated as well. Knockdown of Stat3 in 4 T1 cells led to Src inactivation by a feedback loop in the absence of pStat3 (19); thus, it is reasonable to assume that the inactivation of Akt occurred through Src dephosphorylation by Stat3 (19, 20). It has been documented that constitutively activated Stat3 in cancer cells drives a number of factors, including cytokines and chemokines, that induce host immune tolerance to cancer cells (45). Therefore, it is possible that IFN-β inactivates epidermal growth factor receptor (EGFR) signaling, acting as an antagonist for EGFR or VEFGR, which are the upstream regulators of Src. The inactivation of Src induces inactivation of Stat3 and therefore Akt. Furthermore, the inactivation of Stat3 leads to decreased expression of c-Myc and MMP2. Because c-Myc has been characterized as a potent factor for cell proliferation (46) and MMP2 is closely associated with cancer invasion and metastasis (47), it is possible that these combined effects contribute to the observed growth and metastasis inhibition of breast cancer in mice.
Studies have shown that both cancer-bearing animals and human cancer patients retain relatively intact cell-mediated immunity during the early stages of disease (48). However, this immunity is gradually lost as the cancer develops. During the early stages of cancer, the host’s immune tolerance of cancer cells is considered one of the major problems of the disease (48). Intensive studies of cancer immunosuppression have indicated that cancer usually induces immunosuppression in the host. A larger than normal population of Treg has been observed in patients with various types of cancer and in animal cancer models (48). One therapeutic strategy for overcoming this immunosuppression induced by cancer cells has been to stimulate cancer antigen presentation by DC, professional antigen-presenting cells. However, it has been reported that DCs are impaired during cancer development (21). A logical goal is to help patients regain normal function in DCs, allowing CD8+ T cells to be primed by specific cancer antigen-presenting DCs thus leading to a strong immune response and cytotoxic clearance of tumor cells.
IFN-β functions as an immunomodulator in stimulating innate immunity to viral infections by activating natural killer cells and macrophages (49). Reports have also indicated that IFN-β can stimulate CD8+ T cells (50). Therefore, it is possible that high local levels of IFN-β in the tumor environment favor the function of both DCs and CD8+ T cells. Hence, the biological effect of IFN-β in this contest would inhibit tumor growth, engraftment, and metastasis. Our results further suggest an effective alternative to the currently used systemic administration of recombinant IFN-β protein, the clinical effects of which are reduced because of its rapid degradation in vivo.
Recent evidence has suggested that Stat3 plays a critical role in the development of both primary and metastatic breast cancer (16, 51). Inactivation of Stat3 also led to a virtual abrogation of both primary and metastatic breast tumor development in a mouse transplant model (20). Studies have shown that Stat3 plays a central role in regulating the expression or activation of potent oncoproteins such as Src, Akt, c-Myc, and Twist (19), it is very likely that a combined effect after the inactivation of Stat3 leads to blunted tumor growth. Recent evidence has also indicated that cancer cells inhibit the maturation of host DCs (21) and that immature DCs further stimulate the Treg population; as a result, immune tolerance of the host is induced and cancer development is facilitated. In previous studies (20), similar results were observed when CDDO-Me was employed to inactivate Stat3 in the same 4 T1/BALB/c breast cancer model used here. Taken together, our data and those of others suggest that inactivation of Stat3 would be an efficient way to suppress and perhaps to abrogate breast cancer growth and metastases.
In conclusion, the findings presented herein indicate that MSC/IFN-β/GFP specifically engraft at the sites of breast cancer in syngeneic and fully immunocompetent mice. Those mMSC secrete levels of IFN-β sufficient to inhibit the development of both primary and metastatic tumors by inactivating Stat3. However, further investigations are needed to explore 1) the long-term biological effects of MSC for the breast cancer host and 2) the dose effect of IFN-β delivered by MSC. Results of this study suggest that expression of IFN-β in situ by MSC can inhibit or abrogate cancer growth by inactivation of constitutively activated Stat3, which drives multiple oncogenes and oncoproteins. Importantly, the inactivation of Stat3 also resulted in an improvement in breast cancer host immune tolerance by enhancing the mature DC population and reducing Treg cells, which, as the biological consequence of this interaction, helps the breast cancer host to eliminate breast cancer cells via multiple direct and indirect mechanisms.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
(GIF 5 kb)
{kind=link}
(GIF 48 kb)
{kind=link}
(GIF 11 kb)
{kind=link}
(GIF 10 kb)
Footnotes
Supported in part by grants from the National Cancer Institute (CA-55164, CA-16672, CA-49639,P30 and CA016672) and the Paul and Mary Haas Chair in Genetics (to MA). XL is supported in part by P50 CA116199. FM is supported in part by RC1CA146381, CA-109451 and CA-116199 and a grant from the Susan G Komen Breast Cancer Foundation (BCTR0504372).
References
- 1.Oreffo RO, Cooper C, Mason C, Clements M. Mesenchymal stem cells: lineage, plasticity, and skeletal therapeutic potential. Stem Cell Rev. 2005;1(2):169–178. doi: 10.1385/SCR:1:2:169. [DOI] [PubMed] [Google Scholar]
- 2.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 3.Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Handb Exp Pharmacol. 2007;180:263–283. doi: 10.1007/978-3-540-68976-8_12. [DOI] [PubMed] [Google Scholar]
- 4.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas3. Cancer Res. 2005;65(8):3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 5.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors1. Cancer Res. 2002;62(13):3603–3608. [PubMed] [Google Scholar]
- 6.Ponte AL, Marais E, Gallay N, Langonne A, Delorme B, Herault O, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25(7):1737–1745. doi: 10.1634/stemcells.2007-0054. [DOI] [PubMed] [Google Scholar]
- 7.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96(21):1593–1603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 8.Birnbaum T, Roider J, Schankin CJ, Padovan CS, Schichor C, Goldbrunner R, et al. Malignant gliomas actively recruit bone marrow stromal cells by secreting angiogenic cytokines. J Neurooncol. 2007;83(3):241–247. doi: 10.1007/s11060-007-9332-4. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida J, Mizuno M, Wakabayashi T. Interferon-beta gene therapy for cancer: basic research to clinical application. Cancer Sci. 2004;95(11):858–865. doi: 10.1111/j.1349-7006.2004.tb02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bianchini F, Mannini A, Mugnai G, Ruggieri S, Calorini L. Expression of a metastatic phenotype in IFNs-primed/TNFalpha-activated B16 murine melanoma cells: role of JAK1/PKCdelta signal transduction factors. Clin Exp Metastasis. 2006;23(3–4):203–208. doi: 10.1007/s10585-006-9030-1. [DOI] [PubMed] [Google Scholar]
- 11.Sharief MK, Semra YK. Down-regulation of survivin expression in T lymphocytes after interferon beta-1a treatment in patients with multiple sclerosis. Arch Neurol. 2002;59(7):1115–1121. doi: 10.1001/archneur.59.7.1115. [DOI] [PubMed] [Google Scholar]
- 12.Yoshiji H, Kuriyama S, Noguchi R, Yoshii J, Ikenaka Y, Yanase K, et al. Combination of interferon-beta and angiotensin-converting enzyme inhibitor, perindopril, attenuates the murine liver fibrosis development. Liver Int. 2005;25(1):153–161. doi: 10.1111/j.1478-3231.2005.01038.x. [DOI] [PubMed] [Google Scholar]
- 13.Genka S, Shitara N, Tsujita Y, Kosugi Y, Takakura K. Effect of interferon-beta on the cell cycle of human glioma cell line U-251 MG: flow cytometric two-dimensional (BrdU/DNA) analysis. J Neurooncol. 1988;6(4):299–307. doi: 10.1007/BF00177424. [DOI] [PubMed] [Google Scholar]
- 14.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94(8):3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19(21):2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 16.Clevenger CV. Roles and regulation of stat family transcription factors in human breast cancer. Am J Pathol. 2004;165(5):1449–1460. doi: 10.1016/S0002-9440(10)63403-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darnell JE., Jr STATs and gene regulation. Science. 1997;277(5332):1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 18.Garcia R, Yu CL, Hudnall A, Catlett R, Nelson KL, Smithgall T, et al. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997;8(12):1267–1276. [PubMed] [Google Scholar]
- 19.Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65(7):2532–2536. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- 20.Ling X, Konopleva M, Zeng Z, Ruvolo V, Stephens LC, Schober W, et al. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67(9):4210–4218. doi: 10.1158/0008-5472.CAN-06-3629. [DOI] [PubMed] [Google Scholar]
- 21.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4(12):941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 22.Gervais A, Leveque J, Bouet-Toussaint F, Burtin F, Lesimple T, Sulpice L, et al. Dendritic cells are defective in breast cancer patients: a potential role for polyamine in this immunodeficiency. Breast Cancer Res. 2005;7(3):R326–R335. doi: 10.1186/bcr1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nefedova Y, Gabrilovich DI. Targeting of Jak/STAT pathway in antigen presenting cells in cancer. Curr Cancer Drug Targets. 2007;7(1):71–77. doi: 10.2174/156800907780006887. [DOI] [PubMed] [Google Scholar]
- 24.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Carbone DP. Tumor-host immune interactions and dendritic cell dysfunction. Adv Cancer Res. 2004;92:13–27. doi: 10.1016/S0065-230X(04)92002-7. [DOI] [PubMed] [Google Scholar]
- 27.Elpek KG, Lacelle C, Singh NP, Yolcu ES, Shirwan H. CD4 + CD25+ T regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B cell lymphoma model. J Immunol. 2007;178(11):6840–6848. doi: 10.4049/jimmunol.178.11.6840. [DOI] [PubMed] [Google Scholar]
- 28.Filaci G, Fenoglio D, Fravega M, Ansaldo G, Borgonovo G, Traverso P, et al. CD8+CD28-T Regulatory Lymphocytes Inhibiting T Cell Proliferative and Cytotoxic Functions Infiltrate Human Cancers. J Immunol. 2007;179(7):4323–4334. doi: 10.4049/jimmunol.179.7.4323. [DOI] [PubMed] [Google Scholar]
- 29.Schabowsky RH, Madireddi S, Sharma R, Yolcu ES, Shirwan H. Targeting CD4 + CD25 + FoxP3+ regulatory T-cells for the augmentation of cancer immunotherapy. Curr Opin Investig Drugs. 2007;8(12):1002–1008. [PubMed] [Google Scholar]
- 30.Wang HY, Wang RF. Regulatory T cells and cancer. Curr Opin Immunol. 2007;19(2):217–223. doi: 10.1016/j.coi.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Carpentier AF, Meng Y. Recent advances in immunotherapy for human glioma. Curr Opin Oncol. 2006;18(6):631–636. doi: 10.1097/01.cco.0000245321.34658.f4. [DOI] [PubMed] [Google Scholar]
- 32.Lizee G, Radvanyi LG, Overwijk WW, Hwu P. Improving antitumor immune responses by circumventing immunoregulatory cells and mechanisms. Clin Cancer Res. 2006;12(16):4794–4803. doi: 10.1158/1078-0432.CCR-06-0944. [DOI] [PubMed] [Google Scholar]
- 33.Pallandre JR, Brillard E, Crehange G, Radlovic A, Remy-Martin JP, Saas P, et al. Role of STAT3 in CD4 + CD25 + FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol. 2007;179(11):7593–7604. doi: 10.4049/jimmunol.179.11.7593. [DOI] [PubMed] [Google Scholar]
- 34.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52(6):1399–1405. [PubMed] [Google Scholar]
- 35.Ling X, Ma G, Sun T, Liu J, Arlinghaus RB. Bcr and Abl interaction: oncogenic activation of c-Abl by sequestering Bcr. Cancer Res. 2003;63(2):298–303. [PubMed] [Google Scholar]
- 36.Burke WM, Jin X, Lin HJ, Huang M, Liu R, Reynolds RK, et al. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells1. Oncogene. 2001;20(55):7925–7934. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 37.Nam S, Buettner R, Turkson J, Kim D, Cheng JQ, Muehlbeyer S, et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells1. Proc Natl Acad Sci U S A. 2005;102(17):5998–6003. doi: 10.1073/pnas.0409467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nussenzweig MC, Steinman RM, Witmer MD, Gutchinov B. A monoclonal antibody specific for mouse dendritic cells. Proc Natl Acad Sci U S A. 1982;79(1):161–165. doi: 10.1073/pnas.79.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parekkadan B. van PD, Megeed Z, Kobayashi N, Tilles AW, Berthiaume F, et al. Immunomodulation of activated hepatic stellate cells by mesenchymal stem cells. Biochem Biophys Res Commun. 2007;363(2):247–252. doi: 10.1016/j.bbrc.2007.05.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and woundhealing. N Engl J Med. 1986;315(26):1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 41.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 42.Nakanishi H, Mizutani Y, Kawauchi A, Ukimura O, Shiraishi T, Hatano M, et al. Significant antitumoral activity of cationic multilamellar liposomes containing human IFN-beta gene against human renal cell carcinoma. Clin Cancer Res. 2003;9(3):1129–1135. [PubMed] [Google Scholar]
- 43.Goldstein D, Sielaff KM, Storer BE, Brown RR, Datta SP, Witt PL, et al. Human biologic response modification by interferon in the absence of measurable serum concentrations: a comparative trial of subcutaneous and intravenous interferon-beta serine. J Natl Cancer Inst. 1989;81(14):1061–1068. doi: 10.1093/jnci/81.14.1061. [DOI] [PubMed] [Google Scholar]
- 44.Kroger A, Koster M, Schroeder K, Hauser H, Mueller PP. Activities of IRF-1. J Interferon Cytokine Res. 2002;22(1):5–14. doi: 10.1089/107999002753452610. [DOI] [PubMed] [Google Scholar]
- 45.Pfitzner E, Kliem S, Baus D, Litterst CM. The role of STATs in inflammation and inflammatory diseases. Curr Pharm Des. 2004;10(23):2839–2850. doi: 10.2174/1381612043383638. [DOI] [PubMed] [Google Scholar]
- 46.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 47.Mendes O, Kim HT, Lungu G, Stoica G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin Exp Metastasis. 2007;24(5):341–351. doi: 10.1007/s10585-007-9071-0. [DOI] [PubMed] [Google Scholar]
- 48.Herber DL, Nagaraj S, Djeu JY, Gabrilovich DI. Mechanism and therapeutic reversal of immune suppression in cancer. Cancer Res. 2007;67(11):5067–5069. doi: 10.1158/0008-5472.CAN-07-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gigli G, Caielli S, Cutuli D, Falcone M. Innate immunity modulates autoimmunity: type 1 interferon-beta treatment in multiple sclerosis promotes growth and function of regulatory invariant natural killer T cells through dendritic cell maturation. Immunology. 2007;122(3):409–417. doi: 10.1111/j.1365-2567.2007.02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robbins SH, Bessou G, Cornillon A, Zucchini N, Rupp B, Ruzsics Z, et al. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 2007;3(8):e123. doi: 10.1371/journal.ppat.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carraway KL, III, Sweeney C. Co-opted integrin signaling in ErbB2-induced mammary tumor progression. Cancer Cell. 2006;10(2):93–95. doi: 10.1016/j.ccr.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 52.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4(4):e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
(GIF 5 kb)
(GIF 48 kb)
(GIF 11 kb)
(GIF 10 kb)