

Abstract
Left–right asymmetrical brain function underlies much of human cognition, behavior and emotion. Abnormalities of cerebral asymmetry are associated with schizophrenia and other neuropsychiatric disorders. The molecular, developmental and evolutionary origins of human brain asymmetry are unknown. We found significant association of a haplotype upstream of the gene LRRTM1 (Leucine-rich repeat transmembrane neuronal 1) with a quantitative measure of human handedness in a set of dyslexic siblings, when the haplotype was inherited paternally (P = 0.00002). While we were unable to find this effect in an epidemiological set of twin-based sibships, we did find that the same haplotype is overtransmitted paternally to individuals with schizophrenia/schizoaffective disorder in a study of 1002 affected families (P = 0.0014). We then found direct confirmatory evidence that LRRTM1 is an imprinted gene in humans that shows a variable pattern of maternal downregulation. We also showed that LRRTM1 is expressed during the development of specific forebrain structures, and thus could influence neuronal differentiation and connectivity. This is the first potential genetic influence on human handedness to be identified, and the first putative genetic effect on variability in human brain asymmetry. LRRTM1 is a candidate gene for involvement in several common neurodevelopmental disorders, and may have played a role in human cognitive and behavioral evolution.
Keywords: handedness, schizophrenia, association, imprinted gene, brain asymmetry
Introduction
Left–right asymmetrical function is a conserved feature of vertebrate central nervous systems.1 Asymmetrical brain function is pronounced in humans, and underlies much of our behavior, cognition and emotion.2 Approximately 90% of humans are right-handed.2 This is the strongest population-level bias in handedness for any primate.1,2 Left-handedness in humans is associated with reductions or reversals of normal brain asymmetries,3 particularly of cerebral cortical areas related to language perception and production. This association is likely to be partly genetic in etiology.4 Handedness and complex cognition in humans may, therefore, be related developmentally and evolutionarily, although the nature and extent of these relationships remain uncertain.
We performed previously the first genome-wide linkage scan for a quantitative measure of human handedness5 (relative hand skill; assessed with timed peg moving5), and found linkage on 2p12–q11.5 This analysis was performed in 222 reading-disabled siblings,6 although there was no relationship detectable between reading disability (RD) and relative hand skill in this sample.5,7 We then replicated the 2p12–q11 linkage,8 with the same measure of relative hand skill, in 105 pairs of left-handed brothers (P = 0.0009).8 We found that the linkage to relative hand skill, in the RD sibs, was derived entirely from paternal inheritance of 2p12–q11 (P = 0.0000037),9 whereas the maternally inherited locus was not linked.9 This suggested that the underlying gene was imprinted, and inactivated or downregulated on the maternally inherited chromosome.9 We did not have parental genotypes to assign parent-of-origin for analysis of the left-handed brother sample.9 However, under standard linkage analysis, the peak location of linkage to relative hand skill in the left-handed brothers was the same as the peak paternal-specific location in the dyslexic siblings,9 and was within the gene CTNNA2 (α-N-catenin) on 2p12.
Schizophrenia is a heterogeneous neuropsychiatric disorder that affects roughly 1% of the adult human population. Schizophrenia is associated with reductions or reversals of normal cerebral asymmetries, including the medial temporal lobe, superior temporal gyrus, planum temporale and the overall brain anterior–posterior torque.10,11 Schizophrenia is also associated with a slightly elevated rate of mixed or left-handedness.12,13 Crow drew attention to the association of abnormal brain asymmetry with schizophrenia-like psychoses,14 and was drawn to a human-specific rearrangement of the sex chromosomes and a candidate gene there,15 Protocadherin X-Y. However, this gene has not so far been shown to be linked to asymmetry, language or psychosis.
In a recent meta-analysis of 20 linkage screens for schizophrenia, 2p12–q22 was the only location to reach significance when adjusted for genome-wide testing.16 This linkage was derived primarily, but not exclusively, from a sample of affected sibling pairs of European descent that were recruited in New York and Oxford.17 We showed previously that the 2p12–q11 linkage in this sample was derived predominantly from paternal sharing (log of the odds, LOD = 4.72), not maternal sharing (LOD = 0.6).9 As parent-of-origin effects are unusual in the genome, we proposed in 2003 that a single paternally expressed gene was responsible for the linkages of 2p12–q11 to handedness and schizophrenia.9
In the present study we have used genetic association mapping and gene-functional analysis to identify a novel imprinted gene on 2p12, LRRTM1, that we propose is responsible for causing the linkages of this chromosomal region to human handedness and schizophrenia.
Materials and methods
All work involving human samples and materials was approved by the appropriate institutional review boards, and appropriate informed consent was obtained from all human subjects.
Details of the Materials and methods are given in Supplementary information online.
Results
We genotyped 87 single-nucleotide polymorphisms (SNPs) in 222 RD siblings and their parents, within a region of paternal-specific linkage to relative hand skill that we had detected previously in this sample (Figure 1). The SNPs were targeted within four positional and functional candidate genes on 2p12–p11 (LRRTM4, CTNNA2, LRRTM1 and DNAH6) (Figure 1 and Table S1). Following our parent-of-origin linkage data (Figure 1), we tested for quantitative association of paternally inherited SNP alleles with relative hand skill (using QTDT18). Four SNPs in distinct locations (rs1517771, rs290015, rs2063436 and rs723524; Table S1), which were not in significant linkage disequilibrium (LD) with one another, showed nominally significant paternal-specific association with relative hand skill (0.05 > P > 0.01). This was no more than the expected false-positive rate.
Figure 1.
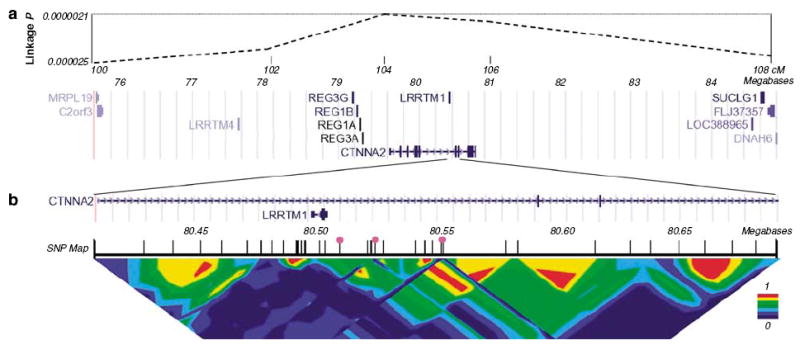
Mapping of an imprinted quantitative-trait-locus (QTL) for human handedness. (a) Top: pointwise significance of linkage derived from paternal sharing across 8 cM of chromosome 2p12-p11.9 This corresponds roughly to a 1-LOD unit support interval for the QTL. Significance of linkage is shown on a logarithmic scale. Bottom: known genes within the region (adapted from UCSC Genome Browser). Note LRRTM1 (transcribed proximal to distal (right to left)) sited within an intron of CTNNA2 (transcribed distal to proximal). (b) Close up of 286 kb around LRRTM1. Two exons of CTNNA2 are also visible. The final SNP map that we used in this region is shown (see also Table S1), and the three SNPs that showed the initial paternal-specific haplotype associations are highlighted by pink circles (rs1446109-rs1007371-rs723524). Pairwise intermarker LD (Cramer’s V) is shown at the bottom.
We then aimed to interrogate the underlying haplotype structure by testing haplotypes for association, that were constructed in ‘sliding windows’ representing all sets of three consecutive SNPs (disregarding any instances where SNPs were not in significant LD, that is, P > 0.05 based on χ2-test of pairwise LD). We used the total, paternal and multi-allelic options of QTDT together for the association tests. Significance levels for paternal-specific association were all P > 0.05, except for haplotypes derived from the SNPs rs1446109-rs1007371-rs723524 (P = 0.00002), and the overlapping window rs1007371-rs723524-rs1025947 (P = 0.0007). These SNPs are on 2p12 within a region of strong intermarker LD that spans at least the first exon and 137 kb upstream of the gene LRRTM1, which is located within an intron of CTNNA2 (Figure 1). There was no significant maternal-specific haplotype association at any location (all P > 0.05).
The rs1446109-rs1007371-rs723524 haplotype 2-2-2 (where 2 is the minor allele of each SNP) was responsible primarily for the paternal-specific association observed with haplotypes derived from these three SNPs. The 2-2-2 haplotype had 9% frequency, and was associated with a mean shift of 1.1 s.d. toward left-handedness in the relative hand skill distribution5,7 when inherited paternally, compared to all other haplotypes. Genotyping of 32 more SNPs, including 12 within 137 kb upstream of LRRTM1, confirmed that rs1446109-rs1007371-rs723524 haplotype 2-2-2 represents a distinct haplotype clade for at least 76 kb upstream of, and including, the predicted promoter of LRRTM1 (Figure S1 and Table S1).
We screened the first two exons and predicted promoter of LRRTM1 for polymorphisms in 26 left-handers from the RD sample by denaturing high-performance liquid chromatography and sequencing, but we did not detect any polymorphisms that tagged rs1446109-rs1007371-rs723524 haplotype 2-2-2, or that had overt disruptive effects on the predicted LRRTM1 protein (entirely coded within exon 2). The only novel SNP that we identified was upstream of the first LRRTM1 exon (Table S1).
We analyzed the SNPs rs1007371 and rs723524, which together can be used to construct a close proxy to the ‘risk’ haplotype described above (see Figure S1), in a sample of normal twin-based sibships from Brisbane, Australia, that was derived from 215 independent families. We found no significant evidence for paternal association of the risk haplotype with our quantitative measure of human handedness (P > 0.1).
We genotyped the SNPs rs1446109 and either rs723524 or rs718466 (the latter are equivalent tagging SNPs according to international HapMap data) in four family samples of white European descent that included individuals with schizophrenia or poor-outcome schizoaffective disorder. These comprised a subset of the New York/Oxford sample17 (226 families), an Irish ‘High Density’ sample19 (236 families), a sample collected in Montreal20 (124 families) and an Afrikaner sample21 (416 families) (Table 1). The 2-2 haplotype defined by the rare alleles of these SNPs is equivalent to the risk haplotype for left-handedness defined above (Figure S1). The 2-2 haplotype varied in frequency between 7.6 and 12.1% in the four sample sets. We found that haplotype 2-2 was overtransmitted paternally to affected individuals in a combined analysis of the four samples, P = 0.0014 (one-tailed test, 38 transmissions to 16 nontransmissions; Table 1). This was a specific hypothesis test that requires no statistical adjustment. There was no significant paternal overtransmission of any other haplotype, nor was there maternal overtransmission of any haplotype. The paternal 2-2 result was derived roughly equally from three of the four samples (Table 1).
Table 1.
Transmission disequilibrium statistics for two-marker haplotypes (see main text), for families affected with schizophrenia/schizoaffective disorder
|
Paternal |
Maternal |
Both parents |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | TR | NT | χ2 | TR | NT | χ2 | TR | NT | χ2 | |
| New York/Oxford | |||||||||||
| Haplo.1 | 504 | 81.8 | 18 | 27 | 1.80 | 21 | 19 | 0.10 | 40 | 47 | 0.56 |
| Haplo.2 | 50 | 8.1 | 15 | 6 | 3.86 | 8 | 9 | 0.06 | 23 | 15 | 1.68 |
| Haplo.3 | 61 | 9.9 | 13 | 13 | 0.00 | 15 | 14 | 0.03 | 29 | 28 | 0.02 |
| Haplo.4 | 1 | 0.2 | 0 | 0 | 0.00 | 0 | 2 | 2.00 | 0 | 2 | 2.0 |
| Irish high density | |||||||||||
| Haplo.1 | 336 | 79.6 | 6 | 14 | 3.20 | 15 | 17 | 0.12 | 23 | 33 | 1.79 |
| Haplo.2 | 51 | 12.1 | 10 | 1 | 7.36 | 11 | 6 | 1.47 | 22 | 8 | 6.53 |
| Haplo.3 | 30 | 7.1 | 5 | 5 | 0.00 | 5 | 8 | 0.69 | 11 | 14 | 0.36 |
| Haplo.4 | 5 | 1.2 | 0 | 1 | 1.00 | 1 | 1 | 0.00 | 1 | 2 | 0.33 |
| Montreal | |||||||||||
| Haplo.1 | 327 | 86.5 | 4 | 4 | 0.00 | 11 | 9 | 0.20 | 15 | 13 | 0.14 |
| Haplo.2 | 31 | 8.2 | 3 | 3 | 0.00 | 5 | 12 | 2.88 | 8 | 15 | 2.13 |
| Haplo.3 | 19 | 5.0 | 2 | 2 | 0.00 | 7 | 2 | 2.78 | 9 | 4 | 1.92 |
| Haplo.4 | 1 | 0.3 | 0 | 0 | 0.00 | 0 | 0 | 0.00 | 0 | 0 | 0.00 |
| Afrikaner | |||||||||||
| Haplo.1 | 1013 | 81.0 | 16 | 19 | 0.26 | 21 | 18 | 0.23 | 39 | 39 | 0.00 |
| Haplo.2 | 95 | 7.6 | 10 | 6 | 1.00 | 4 | 14 | 5.56 | 15 | 21 | 1.00 |
| Haplo.3 | 118 | 9.4 | 11 | 9 | 0.20 | 15 | 8 | 2.13 | 27 | 18 | 1.80 |
| Haplo.4 | 24 | 1.9 | 2 | 5 | 1.29 | 3 | 3 | 0.00 | 5 | 8 | 0.69 |
| Total sample | |||||||||||
| Haplo.1 | 2180 | 81.8 | 44 | 64 | 3.70 | 68 | 63 | 0.19 | 117 | 132 | 0.90 |
| Haplo.2 | 227 | 8.5 | 38 | 16 | 8.96 | 28 | 41 | 2.45 | 68 | 59 | 0.64 |
| Haplo.3 | 228 | 8.6 | 31 | 29 | 0.07 | 42 | 32 | 1.35 | 76 | 64 | 1.03 |
| Haplo.4 | 31 | 1.2 | 2 | 6 | 2.00 | 4 | 6 | 0.40 | 6 | 12 | 2.00 |
χ2, χ2 statistic; haplo.1, haplotype 1-1; haplo.2, haplotype 2-2; haplo.3, haplotype 1-2; haplo.4, haplotype 2-1; N, number of instances of the haplotype in the sample; NT, number of non-transmitted haplotypes from heterozygous parents; TR, number of transmitted haplotypes from heterozygous parents; %, haplotype frequency.
The left-handedness risk haplotype 2-2 is highlighted in bold.
We genotyped rs1446109 (almost tagging for the risk haplotype; Figure S1), in two case–control collections of European descent (461 cases and 459 controls from Munich, Germany, and 429 cases and 428 controls from Scotland (all cases had Diagnostic and Statistical Manual of Mental Disorders, fourth edition diagnoses of schizophrenia, and both sample sets were recruited according to the same protocol)).22 As the parental origin of the alleles could not be established and the paternal and maternal alleles were confounded, we expected this analysis to be low-powered to detect an imprinted effect. Nonetheless, this SNP showed a trend toward association (P = 0.09) when tested under a genotypic 2-df logistic regression model using covariates for gender and the collection site (Munich/Aberdeen). The additive component of this model was significant at P = 0.036, and the direction of allelic effect was the same as in the family samples. When we repeated the analysis using only those cases (151) who reported a positive family history of schizophrenia or bipolar disorder, with the aim of removing sporadic environmental cases, rs1446109 showed significant association with P = 0.013, and the additive component of the model showed P = 0.0038, again in the expected direction. We also tested for association in a sample of 270 Han Chinese families23 but we found no significant bias in paternal or maternal transmission of any haplotype to schizophrenic people (there were 65 paternal transmissions to 78 paternal nontransmissions of the 2-2 haplotype).
No imprinted genes were known previously on chromosome 2p. We found that human LRRTM1 is imprinted (paternal-only expression) in hybrid A9 cells24 (mouse cell lines containing single human chromosomes of known parental origin) (Figure 2). Despite certain limitations with hybrid cells in the context of imprinting studies,25 mouse A9 cells have been used for the reliable identification and verification of human imprinted genes.24 To validate the chromosome 2 hybrid cells, we analyzed the expression of four additional biallelically expressed genes, GGCX (2p11.2), BCL2L11 (2q13), TCF7L1 (2p11.2) and RALB (2q14.2). We detected the expression of all four genes in chromosome 2 hybrid cells, regardless of the parental origin of the human chromosome 2 (Figure S2). No detection of these genes was obtained in RT- controls. We also used transcribed polymorphisms to show mono-allelic LRRTM1 expression in tissue samples from a minority of unrelated postmortem human brains (3 out of 18) by RT-PCR and sequencing or restriction digestion (not shown). However, biallelic expression was found in 15 out of 18 brains, and LRRTM1 was expressed at similar levels from both alleles in the adult human cerebral cortex of five individuals showing biallelic expression (analyzed by quantitative PCR). All brain tissue was obtained from normal controls that were unaffected by major neurological or psychiatric disease. These results suggest that imprinting of LRRTM1 is either variable between individuals (as for the imprinted genes IMPT1 or IGF226), and/or variable between different brain regions or cell/tissue types (as for UBE3A27). We also found mono-allelic paternal expression of LRRTM1 in four out of four unrelated EBV-transformed human lymphoblastoid cell lines that were heterozygous for a transcribed SNP (Figure 2).
Figure 2.
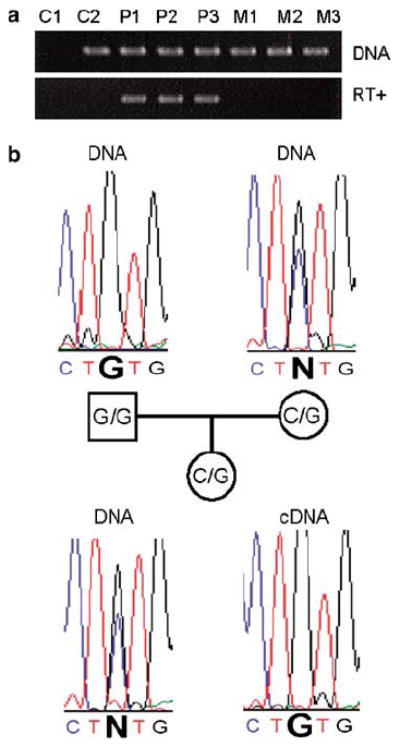
LRRTM1 expression is downregulated maternally in humans. (a) Data are shown from A9 cells that each contains a single human chromosome 2 of known parental origin. Products are shown from PCR using primers specific for human LRRTM1. Upper panel: the human gene is present in genomic DNA (labeled ‘DNA’) from all cell lines tested, apart from C1 (mouse A9 cell line containing no human chromosome). Lower panel: the human gene is only expressed (detected by RT-PCR) in three cell lines containing paternally derived human chromosome 2s (P1, P2, P3), and not in three cell lines containing maternally derived human chromosome 2s (M1, M2, M3). (C2 is a human fibroblast cell line). (b) Monoallelic paternal expression of LRRTM1 in a human EBV-transformed lymphoblastoid cell line. Sequence traces derived from genomic DNA (labeled ‘DNA’) surrounding a C/G SNP are shown for a father, mother and child. The child is heterozygous, but the cDNA prepared from mRNA showed expression of only the paternally inherited G allele of LRRTM1.
By use of in situ hybridization in the mouse, we found that Lrrtm1 is expressed predominantly in the nervous system by postmitotic neurons, but also in some nonneuronal tissues (Figure 3; Figure S3). Expression is upregulated in the mouse brain during embryonic development and early postnatally. In adult brain, Lrrtm1 expression is most prominent in the forebrain, particularly in the thalamus (in most or all nuclei), and in cortical areas including hippocampus, piriform and posterior cingulate (Figure 3; Figure S3).
Figure 3.
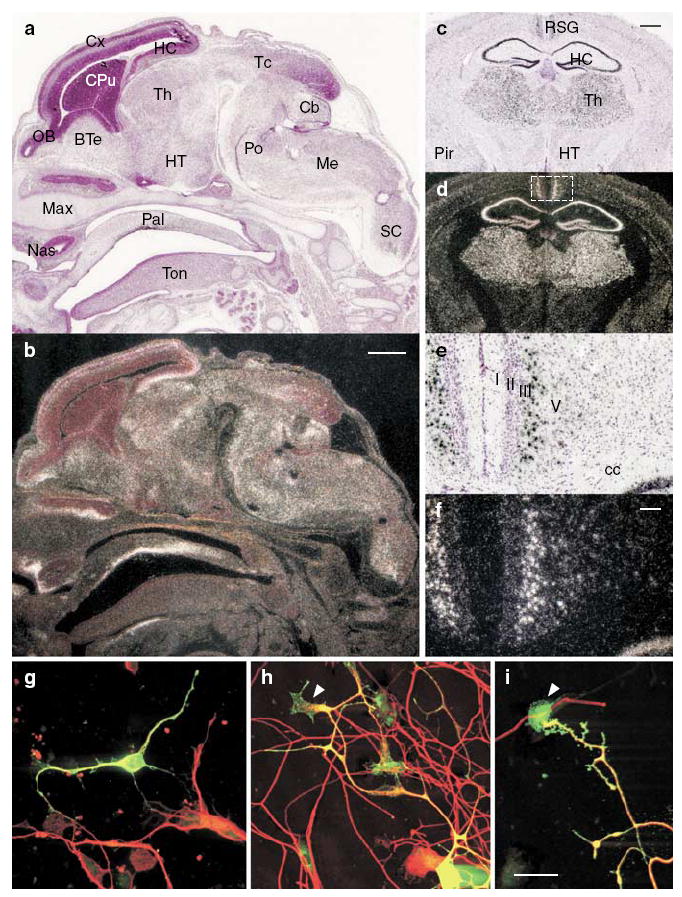
Developmental increase of Lrrtm1 mRNA expression in mouse thalamus, hippocampus and retrosplenial cortex, and localization of LRRTM1 in neurons. In situ hybridization analysis in sagittal sections of E15 mouse embryos (a and b) and coronal sections of adult mouse brain (c–f). Bright-field images counterstained with hematoxylin (a, c and e) and corresponding dark-field autoradiographs are shown (b, d and f). The white signal in the dark-field images indicates Lrrtm1 expression. (e and f) Higher magnification of the retrosplenial granular cortex (boxed area in d) shows a high level of Lrrtm1 expression in layer III–IV neurons. (g–i) Myc-LRRTM1 was transfected into E17 mouse cortical neurons (g) and nucleofected into rat P18 dorsal root ganglion neurons (h and i) and detected 40 h later with anti-myc antibody. Neurons were visualized with anti-β-tubulin antibody. LRRTM1 is localized to the cell soma and in the neurites; in neurites it is also localized to lamellipodia of the growth cones (arrowheads in h and i). BTe, basal telencephalon; Cb, cerebellum; cc, corpus callosum; CPu, caudate-putamen; Cx, cerebral cortex; HC, hippocampus; HT, hypothalamus; Max, maxilla, Me, medulla; Nas, nasal cavity; OB, olfactory bulb; Pal, palatine; Pir, piriform cortex; Po, pons; RSG, retrosplenial granular cortex; Sc, Spinal cord; Tc, tectum; Th, thalamus; Ton, tongue. Scale bar is 500 μm in (a–d), 100 μm in (e–f) and 20 μm in (g–i).
In northern blot analysis of adult human brain, LRRTM1 also showed predominant expression in forebrain regions including thalamus and cerebral cortex (Figure S4). By in situ hybridization in coronal sections of the post-mortem developing human brain (14–16 weeks gestation; Figure 4), strong expression was observed in anterior sections throughout the cortical plate and in septum, caudate and putamen. The absence of signal in the subventricular zone argues against a direct involvement in neurogenesis (Figure 4). Transcript distribution was similar in more caudal sections with the addition of signal in dorsolateral thalamus (Figure 4). More caudal still, thalamic signal shifted ventrally to a structure consistent with the lateral geniculate body (Figure 4). The striking signal in caudate and putamen in human (Figure 4) was not present in mouse, at least not at E15 and in adult. In addition, expression within the human thalamus was more restricted as compared to the mouse, with staining relatively limited to dorsomedial regions.
Figure 4.
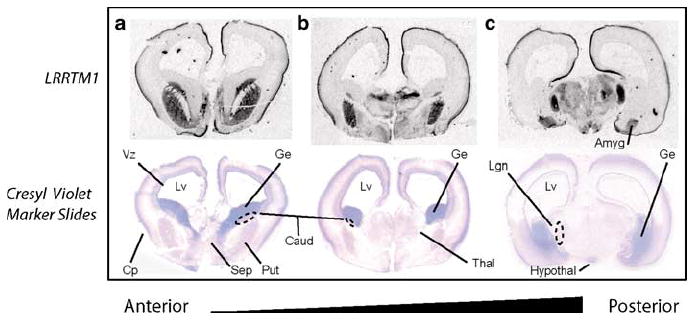
In situ hybridization analysis of LRRTM1 expression in the developing human brain at 15 weeks’ gestation. In coronal sections of anterior brain (a), expression is strong throughout the cortical plate and otherwise restricted to septum, caudate, and putamen. Transcript distribution is similar in more caudal sections (b) with the addition of signal in dorsolateral thalamus. More caudal still (c), thalamic signal shifts ventrally to a structure consistent with the lateral geniculate body. Abbreviations are as follows: Amyg (Amygdala), Caud (Caudate), Cp (Cortical), Ge (Ganglionic Eminence), Hypothal (Hypothalamus), Lgn (Lateral Geniculate Nucleus), Lv (Lateral Ventricle), Thal (Thalamus), Vz (Ventricular Zone).
No consistent asymmetric expression was observed in any of three human developing brains examined by in situ hybridization (14–16 weeks’ gestation), regardless of whether cerebral hemispheres were analyzed in aggregate or cortical sub-regions were examined in isolation (dorsolateral, temporal, ventrolateral or cingulated). Similarly to embryonic brain, LRRTM1 was expressed at similar levels (that is, symmetrically) in all analyzed regions of left and right adult human cerebral cortex (several different cortical regions from five individuals were analyzed by quantitative PCR). We also quantified left- and right-brain Lrrtm1 mRNA expression levels in rats and embryonic mice (see Materials and methods), but did not detect evidence for asymmetrical expression in rodents. In addition, we tested cerebral cortex, cerebellum, brain stem, olfactory bulb, thymus, heart, lung, liver, intestine, pancreas, spleen, kidney, muscle and testis of two reciprocal crossed F1 mice between C57BL/6J and JF1 strains for allele-specific expression. Each of the F1 mice was 30 weeks old. Expression of Lrrtm1 was detected in cerebral cortex, cerebellum and brain stem, but it was biallelic (Figure S5).
In rodent primary sensory (DRG) and cortical neurons (Figure 3), and in cerebellar granular neurons (data not shown), overexpressed LRRTM1 localized to the cell soma, neurites and lamellipodia of growth cones, suggesting a function in axon guidance and/or synaptogenesis. Unexpectedly, in transfected MRC5, Cos-7 and Neuro-2a cells, LRRTM1 colocalized with endoplasmic reticulum (ER) markers (Figure 5 and Figure S6). Live-cell staining for overexpressed LRRTM1 in DRG neurons revealed that the protein is not accessible on the plasma membrane under conditions that allowed surface detection of a related member of the LRR protein superfamily, Lingo1 (Figure 5). These results suggest that endogenous LRRTM1 may have a role in intracellular trafficking within axons. However, it remains possible that LRRTM1 is localized to plasma membrane in cells expressing an unidentified LRRTM1 chaperone or coreceptor protein that promotes its processing and/or transport.
Figure 5.
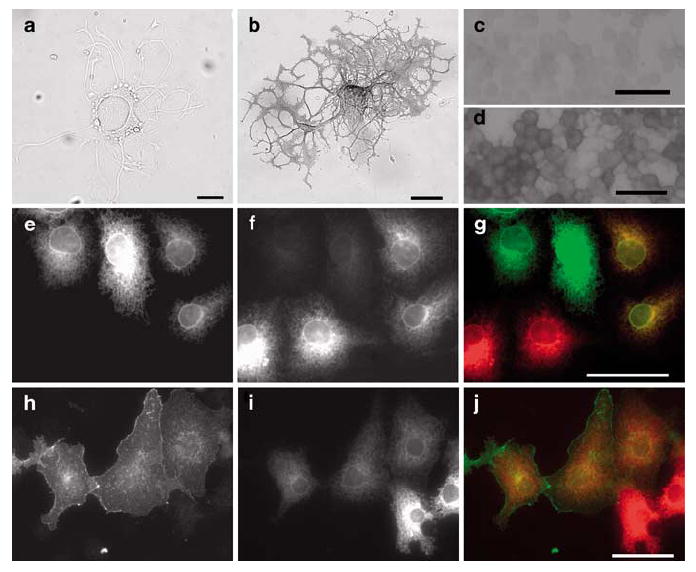
LRRTM1 is not localized on the plasma membrane. No cell-surface expression of LRRTM1 is seen in primary sensory neurons electroporated with myc-LRRTM1 construct and detected by live-cell staining with alkaline phosphatase-conjugated anti-myc antibody (a); strong cell-surface staining of myc-Lingo1 expressing neuron is seen (b). Live-cell staining resulted in 20–30 strongly stained DRG neurons per well for myc-Lingo1 but none for myc-LRRTM1. Similarly, in neuro-2a neuroblastoma cells myc-LRRTM1 (c) is not, but myc-lingo1 is (d) localized to plasma membrane as detected by live-cell staining. In permeabilized Cos-7 cells LRRTM1 (anti-myc antibody; e) is colocalized (merge picture; g) with ER-marked dsRED2-ER (f), whereas lingo1 (anti-myc antibody; h) is only partially colocalized with dsRED2-ER, and it is partially localized on the plasma membrane (i; merge picture: j). Scale bars are 50 μm.
We analyzed methylation within 2 CpG islands that correspond to the predicted promoter and coding exon of LRRTM1, and a third island roughly 18 kb upstream of LRRTM1, in 17 lymphoblastoid cell lines and 17 human post-mortem brain samples, but we did not find evidence that these CpG islands are differentially methylated regions (DMRs) (data not shown).
We tested CTNNA2 for mono-allelic expression in 10 post-mortem brain samples from normal, unrelated individuals, by use of a transcribed SNP in exon 12, but expression was always biallelic, in contrast to LRRTM1 (data not shown). We did not detect CTNNA2 expression in the A9 hybrids, or in human lymphoblastoid cell lines, by RT-PCR. The first CTNNA2 exon is over 750 kb from LRRTM1 (Figure 1), and several LD blocks away, further suggesting that the regulation of these genes is distinct. There are no other genes within 1.13 Mb of LRRTM1, and the next closest gene expressed in the forebrain (expression data available via the UCSC genome browser) is over 2.75 Mb distally (LRRTM4; Figure 1).
In order to analyze the evolution of LRRTM1, we compared it between humans and chimpanzees. There is no fixed amino acid difference between human LRRTM1 and chimpanzee LRRTM1, and it is not among the genes that are differently expressed between adult humans and adult chimpanzees in brain, liver, kidney, heart or testis.28 Furthermore, biallelic expression of LRRTM1 was detected in two out of two chimpanzee brain samples (see Materials and methods).
Discussion
Crow proposed that handedness, brain asymmetry and schizophrenia share an underlying genetic relationship.14 However, no genes have previously been shown to associate across these traits. We carried out SNP-based association screening for human relative hand skill, under a parent-of-origin model, within four candidate genes close to or within 6 mb of a peak of paternal-specific linkage on 2p12, in a sample of sib pairs recruited for the presence of RD. We found strong paternal-specific association with relative hand skill implicating the gene LRRTM1 (leucine-rich repeat transmembrane neuronal 1) and a region of 80 kb upstream of this gene (which is located within an intron of CTNNA2). We did not find this association in a sample of normal twin-based sibships from Australia. However, consistent with our parent-of-origin association data, we went on to show that LRRTM1 is downregulated on the maternally inherited chromosome, and is therefore a newly identified imprinted locus in the human genome. We also found that the same haplotype that drove the paternal association with handedness was overtransmitted paternally, but not maternally, to schizophrenic/schizoaffective individuals in 1002 affected families of European descent (P = 0.0014). This was a specific hypothesis test that requires no statistical adjustment, and indicates that LRRTM1 is a novel susceptibility factor for schizophrenia. We also found consistent evidence for association in a large case–control sample of European descent that was collected in Scotland and Germany.
We did not find that the ‘risk’ 2-2 haplotype that we identified in European family-based samples was associated with schizophrenia in a Han Chinese family-based sample. The frequency of the 2-2 risk haplotype was somewhat higher in the Han Chinese, at 18%. The failure to replicate may nonetheless be related to modest power in the Chinese sample of 270 families. Alternatively, the nonreplication in the Chinese may indicate that the 2-2 haplotype does not carry risk variation in this population (it may also be relevant that native Chinese speakers show some morphological differences to English speakers in language-related areas of the brain29). A further study in a larger Chinese sample will be required to distinguish these possibilities, together with identification of the functional variants in European populations.
Of a total of approximately 35 000 human genes, only roughly 40 were previously known to be imprinted, and the total number is unlikely to exceed this greatly.30-32 The finding that LRRTM1 is imprinted, and maternally suppressed, was a confirmation of the parent-of-origin genetic data that led to this locus, because the a priori probability that LRRTM1 would be imprinted was very low (roughly 1 in 1000). Our association data suggest that allelic variation upstream of LRRTM1, that may affect the gene’s expression, is relevant functionally for brain asymmetry when inherited on the active paternal chromosome, but not on the relatively inactive maternal chromosome, at certain locations and time points during human brain development. However, the ‘risk’ haplotype that we identified at LRRTM1 cannot explain the majority of the paternal-specific linkage across the genomic region of 2p12 (Figure 1), and therefore additional genetic or epigenetic variability at LRRTM1, or neighboring genes, may also be responsible partly for the linkages of handedness and schizophrenia to 2p12.9 Although we found evidence that LRRTM1 is variably imprinted in the post-mortem adult human brain (as is the imprinted gene 5HT2A),33 we cannot conclude directly, on the basis of our data, that variability in imprinting of LRRTM1 is involved in individual differences in human handedness and schizophrenia liability. Our data indicate a role for the 2-2 haplotype in handedness variability and susceptibility to schizophrenia, in European populations, while the data on imprinting are consistent with the paternal-specific nature of this effect.
Our data suggest that a subtype of schizophrenia, linked to misregulation of human LRRTM1, may have its origins in fetal neurodevelopment. Since LRRTM1 appears to underlie the strongest linkage to schizophrenia in the genome, as identified by a meta-analysis of 20 genomewide linkage scans,16 it is possible that LRRTM1 dysfunction causes a major, common subtype of schizophrenia. Assessing the frequency of this subtype will require studies in further clinical and epidemiological samples, together with a better definition of the functional genetic and epigenetic variation at the LRRTM1 locus. This information may have a substantial impact on pharmacogenetic studies and the development of new treatments for schizophrenia, by allowing patient heterogeneity to be accounted for in clinical trial studies. The receptor-like structure of LRRTM1 also suggests that it may be a drug-tractable target in its own right.
LRRTM1 is one of a four-member family of type I transmembrane proteins containing leucine-rich repeat (LRR) domains, which are commonly involved in protein–protein interactions (LRRs are present in the Slits and Nogo-receptor,34,35 involved in axonal pathfinding). Each LRRTM member has a specific, dynamically regulated regional brain-expression distribution.35 Using several gene-functional approaches, we found that LRRTM1 is likely to play a role during the development of specific forebrain structures by influencing neuronal differentiation and connectivity, with a possible role in intracellular trafficking in axons. Thus, LRRTM1 is an ideal candidate gene for having an involvement in subtle developmental abnormalities of the central nervous system.
When comparing expression between human and mouse, it seems that there is a striking absence of signal in caudate and putamen in mouse, at least at e15 and in adult. Also, expression within the fetal human thalamus is more restricted as compared to the mouse, with staining relatively limited to dorsomedial regions that have been implicated in schizophrenia.36,37 We found no evidence for overtly asymmetrical expression of Lrrtm1 in the developing or adult brain of rodents or humans, using in situ hybridization and/or quantitative PCR. However, we cannot rule out a subtle asymmetry of function or expression at some restricted time point during human brain development, as for the transcription factor LMO4.38 It remains possible that LRRTM1 is important in the establishment, consolidation or elaboration of the left–right axis during human brain growth and development, particularly before 14 weeks’ gestation, which was the earliest stage that we were able to analyze in human. (Population-level morphological asymmetries of the cerebral cortex are already noticeable shortly after this time, and are visible by ultrasound in normal fetuses at 20–22 weeks’ gestation.39) Therefore, detailed studies of the roles of LRRTM1 and the pathways in which it functions in mammalian brain development are warranted, particularly at developmental time points earlier than 14 weeks in humans, that may reveal critical new insights into the establishment and/or maintenance of normal and abnormal human brain function and asymmetry.
Humans have the strongest population-level bias in handedness of any primate,1,2 and LRRTM1 is a candidate for having had a role in the evolution of this trait. As the human and mouse predicted LRRTM1 proteins are 96% identical, and the human and chimpanzee proteins are 100% identical, any potential human-specific properties of LRRTM1 may involve the spatiotemporal control of its imprinted expression. (It is interesting that recent analysis of conserved noncoding elements demonstrated that a sequence element 130 kb downstream of LRRTM1 displays accelerated evolution in the human lineage, which may be important for the gene’s regulation).40 We detected only biallelic, nonimprinted expression of Lrrtm1 in cerebral cortex, cerebellum, and brain stem from two 30-week-old reciprocal crossed F1 mice between C57BL/6J and JF1 strains (data not shown), and only biallelic expression in the postmortem brains of two adult chimpanzees. Larger sample sizes will be needed to test whether imprinting of LRRTM1 is found only in humans. (There is a high level of discordance in imprinting between humans and mice;32 the gene DLX5 is an example.41) It will also be interesting to compare the regulation of LRRTM1 longitudinally during the development of humans and other species, as the imprinted regulation may be restricted to certain developmental periods.38 In addition, human genetic variation at this locus in nonclinical populations could be analyzed with dense marker genotyping, to test for evidence of selection, particularly with regard to the risk haplotype.
Genomic imprinting can arise when the optimal level of maternal resource investment in offspring differs between the two parental sexes, in polygamous mating systems.31,42 Paternally inherited alleles of imprinted genes often sequester more maternal resources than do maternally inherited alleles, especially when influencing growth in utero.31,42 The imprinting of LRRTM1 in humans, therefore, suggests an intriguing evolutionary scenario, in which the parental sexes have conflicting interests in relation to the outcome of lateralized brain development in their offspring, which underlies much of human cognition and behavior. Further study of the neural systems and behaviors that LRRTM1 influences will be required to understand the selective forces that drove LRRTM1 to be imprinted.
We did not detect evidence for differential methylation of the paternal and maternal alleles at the CpG islands associated directly with LRRTM1, and therefore the mechanism underlying imprinted expression of LRRTM1 remains unclear. However, many other imprinted genes are not regulated by differential methylation within their own promoters, but are regulated instead by imprinted control centers which may be located many kilobases distant.31 Also, allele-specific histone modifications are emerging currently as equally important determinants of imprinted gene expression.43 The lack of methylation does not, therefore, refute the imprinted expression of LRRTM1, which we observed in three different tissues and experimental settings. We recommend that the mechanism of imprinted regulation at this locus should be investigated as a priority, as epigenetic misregulation of LRRTM1 may have clinical relevance and could underlie much of the paternal linkage of 2p12 to schizophrenia.
We analyzed schizophrenia and schizoaffective disorder jointly in the New York/Oxford sample to maximize the numbers of informative paternal transmissions for the risk haplotype (Table 1). However, this sample was not sufficiently large to test for heterogeneity of the association effect between these two patient groups. Our previous linkage analysis9 of 2p12 had indicated that the linkage was stronger for the subset of sibling pairs with schizophrenia only (paternal LOD 4.72), but this remains to be investigated more thoroughly by targeted association analysis in additional samples.
LRRTM1 is the first genetic influence on human handedness to be identified. Handedness, brain asymmetry and schizophrenia are likely to be etiologically complex traits with several, or many, genetic and environmental influences. As we detected an effect of LRRTM1 on handedness in dyslexic siblings, but not in normal twin-based sibships, it is possible that LRRTM1 influences behavioral lateralization more strongly in clinically selected populations for psychiatric/neurodevelopmental dysfunctions, than in the general population. This would suggest an interactive, nonadditive effect of LRRTM1 with other genetic and/or environmental effects that predispose to neuropsychiatric dysfunction, such that the effect of LRRTM1 depends on other risk factors in order to manifest fully. However, it is also possible that the failure to replicate the association with handedness was due to low power and a relatively small replication sample in our study. It is a well-described statistical feature of genetic association analysis that follow-up samples should usually be much larger than initial samples to provide adequate power, as the genetic effect in the initial sample is typically inflated.44 Unfortunately, we are unaware of other family-based collections with accurate quantitative assessments of handedness, that are suitable for genetic studies.
In addition to schizophrenia, other neurodevelopmental disorders including bipolar disorder, autism and language impairment have shown evidence for associations with left-handedness and/or abnormal asymmetrical brain structure/function.2,10,11,45-49 LRRTM1 is, therefore, also a candidate for involvement in these traits. The LRRTMs are a four-member gene family first described in 2003, and it is interesting to note that LRRTM3 has recently been proposed as a susceptibility factor for late-onset Alzheimer’s disease.50 We recommend, therefore, that the whole LRRTM gene family is investigated in relation to psychiatric and neurological disorders.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the families who took part in this study; David Smith, Catherine Joachim and OPTIMA (Oxford Project to Investigate Memory and Ageing) for access to post-mortem brain tissue; Xiayi Ke for testing his screening algorithm for imprinted genes; Phil Burnet for plasmids and samples; Dianne Gerelli and the Human Developmental Brain Resource (London, UK) for pilot in situ hybridization studies; Jordana Tsenova for ideas on genetic analysis; Eris Duro for help with mutation screening; Abigail Woodroffe for handling genotype data; Julie V Perederiy for help with in situ experiments; Drs Dermot Walsh and F Anthony O’Neill made critical contributions to the collection of the Irish Study of High Density Schizophrenia Families (ISHDSF). Drs Enn Jõeste and Liina Kiho from North Estonian Regional Hospital, Tallinn, helped in collecting human brain tissue. David Goldstein, Kevin Shianna and Dongliang Ge provided genotype data. Han Chinese Schizophrenia Sample data and biomaterials were collected as a part of the National Institute of Mental Health (NIMH) Schizophrenia Genetics Initiative. A complete list of the people who contributed on the NIMH project is in the Supplementary material. CF was a NARSAD Young Investigator (National Alliance for Research on Schizophrenia and Depression). APM is a Wellcome Principal Research Fellow. RW-M is a Wellcome Trust Research Career Development Fellow. SEF is a Royal Society Research Fellow. TT is a Wellcome Trust International Senior Research Fellow in Biomedical Science in Central Europe. JN is supported by the Swiss National Science Foundation and by a Marie Curie Intra-European Fellowship. BSA was supported by a Fellowship from the Tourette Syndrome Association. This research was funded additionally by The Wellcome Trust (UK); the Schizophrenia Research Fund (UK); the Academy of Finland and the Sigrid Juselius Foundation (Finland); the Graham Boeckh Chair Program in Schizophrenia to GAR at McGill University; Warner-Lambert, Parke-Davis Pharmaceuticals Company, and National Institute of Mental Health grant R01 MH-44245 (LED); National Institute of Health grants MH41953 (collection and analysis of the ISHDSF) and MH61399 (MK); and by grant ‘Research on Psychiatric and Neurological disorders and Mental Health’ from the ministry of Health, Labor and Welfare of Japan (H14-Kokoro-002), and Grant-in-Aid for Scientific Research on Priority Areas (C) Medical Genome Science from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Clyde Francks conceived and directed this research.
Footnotes
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Rogers LJ, Andrew R, editors. Comparative Vertebrate Lateralization. Cambridge University Press; Cambridge, UK: 2002. [Google Scholar]
- 2.Hugdahl K, Davidson RJ, editors. The Asymmetrical Brain. MIT Press; Cambridge, MA: 2003. [Google Scholar]
- 3.Mevorach C, Humphreys GW, Shalev L. Attending to local form while ignoring global aspects depends on handedness: evidence from TMS. Nat Neurosci. 2005;8:276–277. doi: 10.1038/nn1400. [DOI] [PubMed] [Google Scholar]
- 4.Geschwind DH, Miller BL, DeCarli C, Carmelli D. Heritability of lobar brain volumes in twins supports genetic models of cerebral laterality and handedness. Proc Natl Acad Sci USA. 2002;99:3176–3181. doi: 10.1073/pnas.052494999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Francks C, Fisher SE, MacPhie IL, Richardson AJ, Marlow AJ, Stein JF, et al. A genomewide linkage screen for relative hand skill in sibling pairs. Am J Hum Genet. 2002;70:800–805. doi: 10.1086/339249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher SE, Francks C, Marlow AJ, MacPhie IL, Newbury DF, Cardon LR, et al. Independent genome-wide scans identify a chromosome 18 quantitative-trait locus influencing dyslexia. Nat Genet. 2002;30:86–91. doi: 10.1038/ng792. [DOI] [PubMed] [Google Scholar]
- 7.Francks C, Fisher SE, Marlow AJ, MacPhie IL, Taylor KE, Richardson AJ, et al. Familial and genetic effects on motor coordination, laterality, and reading-related cognition. Am J Psychiatry. 2003;160:1970–1977. doi: 10.1176/appi.ajp.160.11.1970. [DOI] [PubMed] [Google Scholar]
- 8.Francks C, DeLisi LE, Fisher SE, Laval SH, Rue JE, Stein JF, et al. Confirmatory evidence for linkage of relative hand skill to 2p12-q11. Am J Hum Genet. 2003;72:499–502. doi: 10.1086/367548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francks C, DeLisi LE, Shaw SH, Fisher SE, Richardson AJ, Stein JF, et al. Parent-of-origin effects on handedness and schizophrenia susceptibility on chromosome 2p12-q11. Hum Mol Genet. 2003;12:3225–3230. doi: 10.1093/hmg/ddg362. [DOI] [PubMed] [Google Scholar]
- 10.DeLisi LE, Sakuma M, Kushner M, Finer DL, Hoff AL, Crow TJ. Anomalous cerebral asymmetry and language processing in schizophrenia. Schizophr Bull. 1997;23:255–271. doi: 10.1093/schbul/23.2.255. [DOI] [PubMed] [Google Scholar]
- 11.Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeLisi LE, Svetina C, Razi K, Shields G, Wellman N, Crow TJ. Hand preference and hand skill in families with schizophrenia. Laterality. 2002;7:321–332. doi: 10.1080/13576500143000294. [DOI] [PubMed] [Google Scholar]
- 13.Orr KG, Cannon M, Gilvarry CM, Jones PB, Murray RM. Schizophrenic patients and their first-degree relatives show an excess of mixed-handedness. Schizophr Res. 1999;39:167–176. doi: 10.1016/s0920-9964(99)00071-7. [DOI] [PubMed] [Google Scholar]
- 14.Berlim MT, Mattevi BS, Belmonte-de-Abreu P, Crow TJ. The etiology of schizophrenia and the origin of language: overview of a theory. Compr Psychiatry. 2003;44:7–14. doi: 10.1053/comp.2003.50003. [DOI] [PubMed] [Google Scholar]
- 15.Williams NA, Close JP, Giouzeli M, Crow TJ. Accelerated evolution of Protocadherin11X/Y: a candidate gene-pair for cerebral asymmetry and language. Am J Med Genet B Neuropsychiatr Genet. 2006;141:623–633. doi: 10.1002/ajmg.b.30357. [DOI] [PubMed] [Google Scholar]
- 16.Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeLisi LE, Shaw SH, Crow TJ, Shields G, Smith AB, Larach VW, et al. A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry. 2002;159:803–812. doi: 10.1176/appi.ajp.159.5.803. [DOI] [PubMed] [Google Scholar]
- 18.Abecasis GR, Cookson WO, Cardon LR. Pedigree tests of transmission disequilibrium. Eur J Hum Genet. 2000;8:545–551. doi: 10.1038/sj.ejhg.5200494. [DOI] [PubMed] [Google Scholar]
- 19.Thiselton DL, Webb BT, Neale BM, Ribble RC, O’Neill FA, Walsh D, et al. No evidence for linkage or association of neuregulin-1 (NRG1) with disease in the Irish study of high-density schizophrenia families (ISHDSF) Mol Psychiatry. 2004;9:777–783. doi: 10.1038/sj.mp.4001530. [DOI] [PubMed] [Google Scholar]
- 20.Xiong L, Rouleau GA, DeLisi LE, St Onge J, Najafee R, Riviere JB, et al. CAA insertion polymorphism in the 3’UTR of Nogo gene on 2p14 is not associated with schizophrenia. Mol Brain Res. 2005;133:153–156. doi: 10.1016/j.molbrainres.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 21.Abecasis GR, Burt RA, Hall D, Bochum S, Doheny KF, Lundy SL, et al. Genomewide scan in families with schizophrenia from the founder population of Afrikaners reveals evidence for linkage and uniparental disomy on chromosome 1. Am J Hum Genet. 2004;74:403–417. doi: 10.1086/381713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van den Oord EJCG, Rujescu D, Robles JR, Giegling I, Birrell C, Bukszar J, et al. Factor structure and external validity of the PANSS revisited. Schizophr Res. 2006;82:213–223. doi: 10.1016/j.schres.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi S, Cui YH, Kojima T, Han YH, Zhou RL, Kamioka M, et al. Family-based association study of markers on chromosome 22 in schizophrenia using African-American, European-American, and Chinese families. Am J Med Genet B Neuropsychiatr Genet. 2003;120:11–17. doi: 10.1002/ajmg.b.20031. [DOI] [PubMed] [Google Scholar]
- 24.Kugoh H, Mitsuya K, Meguro M, Shigenami K, Schulz TC, Oshimura M. Mouse A9 cells containing single human chromosomes for analysis of genomic imprinting. DNA Res. 1999;6:165–172. doi: 10.1093/dnares/6.3.165. [DOI] [PubMed] [Google Scholar]
- 25.Mergenthaler S, Hitchins MP, Blagitko-Dorfs N, Monk D, Woll-mann HA, Ranke MB, et al. Conflicting reports of imprinting status of human GRB10 in developing brain: how reliable are somatic cell hybrids for predicting allelic origin of expression? Am J Hum Genet. 2001;68:543–545. doi: 10.1086/318192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakatani T, Wei M, Katoh M, Okita C, Wada D, Mitsuya K, et al. Epigenetic heterogeneity at imprinted loci in normal populations. Biochem Biophys Res Commun. 2001;283:1124–1130. doi: 10.1006/bbrc.2001.4916. [DOI] [PubMed] [Google Scholar]
- 27.Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, et al. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–847. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- 28.Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–1854. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- 29.Kochunov P, Fox P, Lancaster J, Tan LH, Amunts K, Zilles K, et al. Localized morphological brain differences between English-speaking Caucasians and Chinese-speaking Asians: new evidence of anatomical plasticity. Neuroreport. 2003;14:961–964. doi: 10.1097/01.wnr.0000075417.59944.00. [DOI] [PubMed] [Google Scholar]
- 30.Imprinted Gene Catalogue. Web. 2006 http://igc.otago.ac.nz/home.html.
- 31.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 32.Morison IM, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet. 2005;21:457–465. doi: 10.1016/j.tig.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Bunzel R, Blumcke I, Cichon S, Normann S, Schramm J, Propping P, et al. Polymorphic imprinting of the serotonin-2A (5-HT2A) receptor gene in human adult brain. Mol Brain Res. 1998;59:90–92. doi: 10.1016/s0169-328x(98)00146-6. [DOI] [PubMed] [Google Scholar]
- 34.Fournier AE, GrandPre T, Gould G, Wang X, Strittmatter SM. Nogo and the Nogo-66 receptor. Prog Brain Res. 2002;137:361–369. doi: 10.1016/s0079-6123(02)37027-4. [DOI] [PubMed] [Google Scholar]
- 35.Lauren J, Airaksinen MS, Saarma M, Timmusk T. A novel gene family encoding leucine-rich repeat transmembrane proteins differentially expressed in the nervous system. Genomics. 2003;81:411–421. doi: 10.1016/s0888-7543(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 36.Andrews J, Wang L, Csernansky JG, Gado MH, Barch DM. Abnormalities of thalamic activation and cognition in schizophrenia. Am J Psychiatry. 2006;163:463–469. doi: 10.1176/appi.ajp.163.3.463. [DOI] [PubMed] [Google Scholar]
- 37.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2004;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 38.Sun T, Patoine C, Abu-Khalil A, Visvader J, Sum E, Cherry TJ, et al. Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science. 2005;308:1794–1798. doi: 10.1126/science.1110324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hering-Hanit R, Achiron R, Lipitz S, Achiron A. Asymmetry of fetal cerebral hemispheres: in utero ultrasound study. Arch Dis Child Fetal Neonat Ed. 2001;85:F194–F196. doi: 10.1136/fn.85.3.F194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prabhakar S, Noonan JP, Paabo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- 41.Kimura MI, Kazuki Y, Kashiwagi A, Kai Y, Abe S, Barbieri O, et al. Dlx5, the mouse homologue of the human-imprinted DLX5 gene, is biallelically expressed in the mouse brain. J Hum Genet. 2004;49:273–277. doi: 10.1007/s10038-004-0139-2. [DOI] [PubMed] [Google Scholar]
- 42.Wilkins JF, Haig D. What good is genomic imprinting: the function of parent-specific gene expression. Nat Rev Genet. 2003;4:359–368. doi: 10.1038/nrg1062. [DOI] [PubMed] [Google Scholar]
- 43.Peters AH, Schubeler D. Methylation of histones: playing memory with DNA. Curr Opin Cell Biol. 2005;17:230–238. doi: 10.1016/j.ceb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Ioannidis JPA, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 45.Csernansky JG, Schindler MK, Splinter NR, Wang L, Gado M, Selemon LD, et al. Abnormalities of thalamic volume and shape in schizophrenia. Am J Psychiatry. 2004;161:896–902. doi: 10.1176/appi.ajp.161.5.896. [DOI] [PubMed] [Google Scholar]
- 46.De Fosse L, Hodge SM, Makris N, Kennedy DN, Caviness VS, Jr, McGrath L, et al. Language-association cortex asymmetry in autism and specific language impairment. Ann Neurol. 2004;56:757–766. doi: 10.1002/ana.20275. [DOI] [PubMed] [Google Scholar]
- 47.Herbert MR, Harris GJ, Adrien KT, Ziegler DA, Makris N, Kennedy DN, et al. Abnormal asymmetry in language association cortex in autism. Ann Neurol. 2002;52:588–596. doi: 10.1002/ana.10349. [DOI] [PubMed] [Google Scholar]
- 48.Paulesu E, Demonet JF, Fazio F, McCrory E, Chanoine V, Brunswick N, et al. Dyslexia: cultural diversity and biological unity. Science. 2001;291:2165–2167. doi: 10.1126/science.1057179. [DOI] [PubMed] [Google Scholar]
- 49.Sommer IE, Ramsey NF, Mandl RC, Kahn RS. Language lateralization in monozygotic twin pairs concordant and discordant for handedness. Brain. 2002;125:2710–2718. doi: 10.1093/brain/awf284. [DOI] [PubMed] [Google Scholar]
- 50.Majercak J, Ray WJ, Espeseth A, Simon A, Shi XP, Wolffe C, et al. LRRTM3 promotes processing of amyloid-precursor protein by BACE1 and is a positional candidate gene for late-onset Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:17967–17972. doi: 10.1073/pnas.0605461103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 52.Wright M, De Geus E, Ando J, Luciano M, Posthuma D, Ono Y, et al. Genetics of cognition: outline of a collaborative twin study. Twin Res. 2001;4:48–56. doi: 10.1375/1369052012146. [DOI] [PubMed] [Google Scholar]
- 53.Maegawa S, Yoshioka H, Itaba N, Kubota N, Nishihara S, Shirayoshi Y, et al. Epigenetic silencing of PEG3 gene expression in human glioma cell lines. Mol Carcinog. 2001;31:1–9. doi: 10.1002/mc.1034. [DOI] [PubMed] [Google Scholar]
- 54.Stanssens P, Zabeau M, Meersseman G, Remes G, Gansemans Y, Storm N, et al. High-throughput MALDI-TOF discovery of genomic sequence polymorphisms. Genome Res. 2004;14:126–133. doi: 10.1101/gr.1692304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102:15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klumperman J, Schweizer A, Clausen H, Tang BL, Hong W, Oorschot V, et al. The recycling pathway of protein ERGIC-53 and dynamics of the ER-Golgi intermediate compartment. J Cell Sci. 1998;111:3411–3425. doi: 10.1242/jcs.111.22.3411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.