

Abstract
Axonal loss is the principal cause of chronic disability in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). In C57BL/6 mice with EAE induced by immunization with myelin oligodendrocyte glycoprotein peptide 35–55, the first evidences of axonal damage in spinal cord were in acute subpial and perivascular foci of infiltrating neutrophils and lymphocytes and included intra-axonal accumulations of the endovesicular Toll-like receptor TLR8, and the inflammasome protein NAcht leucine-rich repeat protein 1 (NALP1). Later in the course of this illness, focal inflammatory infiltrates disappeared from the spinal cord, but there was persistent activation of spinal cord innate immunity and progressive, bilaterally symmetric loss of small-diameter corticospinal tract axons. These results support the hypothesis that both contact-dependent and paracrine interactions of systemic inflammatory cells with axons and an innate immune-mediated neurodegenerative process contribute to axonal loss in this multiple sclerosis model.
Introduction
Therapies that inhibit trafficking of pathogenic mononuclear cells into the CNS diminish the incidence of new demyelinative lesions in relapsing–remitting multiple sclerosis (Rudick et al., 2007; Goodin et al., 2008; Lopez-Diego and Weiner, 2008) but do not prevent progressive CNS axonal loss (Miller and Leary, 2007; Trapp and Nave, 2008). These observations, and evidences of sustained astroglial and microglial activation in progressive multiple sclerosis (Prineas et al., 2001; Moll et al., 2009), have given rise to the hypothesis that persistent activation of CNS innate immunity contributes to axonal loss in multiple sclerosis (Kutzelnigg et al., 2005; Tanuma et al., 2006; Marik et al., 2007; O'Brien et al., 2008; Weiner, 2009).
Experimental autoimmune encephalomyelitis (EAE) is a primarily T-cell-mediated disorder that has provided important insights into the pathogenesis and treatment of multiple sclerosis (Gold et al., 2006; Steinman and Zamvil, 2006). Axon loss is primarily responsible for permanent disability in EAE (Wujek et al., 2002). EAE elicited in C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG peptide) causes axonal vacuolization and fragmentation and eventual spinal cord atrophy (Bannerman et al., 2005; Wang et al., 2005; Bannerman and Hahn, 2007; Herrero-Herranz et al., 2008; Jones et al., 2008), all features also seen in multiple sclerosis (Kornek et al., 2001; Evangelou et al., 2005; Miller and Leary, 2007). Thus, MOG peptide EAE provides a good model with which to explore the interplay between the CNS immune milieu and axonal loss in multiple sclerosis.
We found that genes involved in neutrophil trafficking were induced in spinal cord soon after the administration of complete Freund's adjuvant (CFA), even in mice in which adaptive immunity had been genetically inactivated (Mombaerts et al., 1992). These very early inductions were followed, in mice with intact adaptive immunity that received MOG peptide in CFA, by the transient formation of intense subpial and perivascular infiltrates in spinal cord, comprised principally of neutrophils and lymphocytes. Intra-axonal accumulations of amyloid precursor protein (APP), hypophosphorylated neurofilament heavy chains (NF-H), Toll-like receptor-8 (TLR8), the inflammasome protein NAcht leucine-rich repeat protein 1 (NALP1) (Tschopp et al., 2003; Lamkanfi and Dixit, 2009), and Rab7, an endolysosomal vesicle-associated protein (Rink et al., 2005; Saxena et al., 2005; Deinhardt et al., 2006), first became detectable within these inflammatory foci. Since APP and NF-H are transported centrifugally and Rab7 centripetally in axons (Koo et al., 1990; Roy et al., 2000; Kamal et al., 2001; Lazarov et al., 2005; Deinhardt et al., 2006; Satpute-Krishnan et al., 2006; Shea and Chan, 2008), these observations indicate that axonal transport was impaired within these inflammatory infiltrates. Later in the course of MOG peptide EAE, neutrophils and most effector T cells were cleared from CNS. However, evidences of CNS innate immune activation persisted, and there was a progressive, symmetrical loss of small-diameter corticospinal axons, a pattern of corticospinal tract degeneration resembling that in patients with chronic multiple sclerosis (Ganter et al., 1999; DeLuca et al., 2004).
Materials and Methods
Mice.
All mice described in this study were adult C57BL/6 males and were housed in a pathogen-free facility. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of California Davis. RAG1−/− mice (B6.129S7-Rag1tm1Mom/J, Jackson Laboratory #002216) were used to examine innate immune responses in the absence of adaptive immunity; these mice lack the capacity to produce mature T or B lymphocytes (Mombaerts et al., 1992). Thy1-STOP-EYFP mice (B6.Cg-Tg(Thy1-EYFP)15Jrs/J, Jackson Laboratory #005630), in which Thy1 promoter-driven expression of enhanced yellow fluorescent protein (EYFP) in neurons was prevented by a loxP-flanked STOP sequence, were bred with Emx1Cre mice (B6.129S2-Emx1tm1(Cre)Krj/J, Jackson Laboratory #005628), in which Cre is constitutively expressed in forebrain neurons, to generate offspring which, in spinal cord corticospinal axons but not other spinal cord axons, were selectively labeled with EYFP (Bareyre et al., 2005).
MOG peptide-EAE.
MOG peptide-EAE was induced in 3-month-postnatal C57BL/6 mice by subcutaneous flank administration of 300 μg of rodent MOG peptide (amino acids 35–55, New England Peptides) in CFA containing 5 mg/ml killed Mycobacterium tuberculosis (Difco) on day 0, with intraperitoneal administration of 75 ng of pertussis toxin on days 0 and 2. “CFA control mice” received CFA and pertussis toxin, but no MOG peptide, and normal control mice received no injections. The mice were weighed and examined daily. Neurological deficits were graded on a five-point scale (limp tail or waddling gait = 1; limp tail and waddling gait = 2; single limb paresis and ataxia 2.5; double limb paresis = 3; single limb paralysis and paresis of second limb = 3.5; full paralysis of 2 limbs = 4; moribund = 4.5; and death = 5) (Zhang et al., 2003; Bannerman et al., 2005, 2007).
Isolation of leukocytes from mouse spleen/lymph nodes and CNS.
Mice killed by CO2 asphyxiation were perfused with ice-cold PBS. Spleens and draining lymph nodes were harvested, combined, minced in PBS, and pushed through a 40 μm mesh. Red blood cells were lysed with ACK solution (Quality Biologicals). Brains and spinal cords were minced and digested at 37°C for 30 min in PBS containing 0.04 units of Liberase R1 (Roche) and 10 μg of DNase I (Roche) per ml. Softened fragments were pushed through a 100 μm mesh. Mononuclear cells from spleen/lymph nodes and from CNS were isolated via a discontinuous 40/70% (v/v) Percoll gradient.
Ex vivo T-cell responses.
Mixed splenocytes and lymph node cells were cultured in 200 μl of RPMI 1640 containing 10% FBS, 2 mm l-glutamine, 0.1 mm nonessential amino acids, 100 U penicillin-streptomycin, 50 μm 2-mercaptoethanol, and 1 mm sodium pyruvate with or without 50 μg/ml MOG peptide (amino acids 35–55) for 24 h. The cells were incubated with brefeldin A (GolgiPlug, BD Bioscience) or brefeldin A plus ionomycin (Calbiochem, 750 ng/ml) and phorbol 12-myristate 13-acetate (PMA; 50 ng/ml, Sigma-Aldrich) for the last 5 h (Park et al., 2005).
Flow cytometry.
Mixed splenocytes and lymph node cells were immunostained after the 24 h culture described above. CNS mononuclear cells were immunostained after incubation at 37°C for 3 h in RPMI 1640 containing 10% FBS, 2 mm l-glutamine, 0.1 mm nonessential amino acids, 100 U/ml penicillin-streptomycin, 50 μm 2-mercaptoethanol, and 1 mm sodium pyruvate in the presence of brefeldin A. Immediately before immunostaining, Fc receptors were blocked for 10 min with anti-CD16/32. For Th1/Th17 lymphocyte analysis, cells were stained with Pacific Blue-labeled anti-mouse CD4, fixed, permeabilized using a Cytofix/Cytoperm Plus Kit according to the manufacturer's protocol and stained with allophycocyanin (APC)-labeled anti-mouse IFN-γ and phycoerythrin-labeled anti-mouse IL-17 (all reagents from BD Bioscience). For regulatory T lymphocyte (Treg) analysis, cells were stained with Pacific Blue-labeled anti-mouse CD4 (BD Bioscience) and APC-labeled anti-mouse CD25 (eBioscience), fixed, and permeabilized using fixation and permeabilization kits (eBioscience) and then intracellularly stained with phycoerythrin-labeled anti-mouse/rat Foxp3 (eBioscience). Immunostained cells were analyzed using a Cyan FACS (Dako Cytomation).
RNA isolation and qRT/PCR.
Mice killed by CO2 asphyxiation were perfused with ice-cold PBS. Pooled spinal cords from three MOG peptide, CFA control, or normal control mice were homogenized and stored in RNAlater solution (Ambion). RNA was isolated using RNeasy Lipid Tissue Mini Kit (QIAGEN) and stored at −80°C. cDNA was prepared using Reaction Ready first-strand cDNA synthesis kits (SuperArray Bioscience). Real-time PCR was performed using mouse Toll-like receptor signaling pathway microarrays and real-time SYBR green PCR master mixes (SuperArray Bioscience). To rule out DNA contamination, non-reverse-transcribed (non-RT) controls were included for every RNA batch prepared. Additional primer sets were used to verify the microarray results and to examine expression levels of additional genes involved in innate immunity and not included in the arrays (supplemental Table 1, available at www.jneurosci.org as supplemental material). The mRNA levels of the assayed genes were normalized to mRNA levels of the housekeeping gene Hsp90ab1.
Spinal cord immunohistology.
Mice anesthetized by intraperitoneal administration of ketamine (150 mg/kg) and xylazine (16 mg/kg) were perfused with PBS, followed by 4% paraformaldehyde (v/v) in PBS. Lumbosacral spinal cords from at least three mice on each of days 3, 7, 10, 12, 14, 21, 35, and 98–101 postadministration of MOG peptide in CFA, or of CFA alone (both groups also received two injections of pertussis toxin as described above) were immunohistologically examined. Paraffin-embedded sections were subjected to antigen retrieval with either sodium citrate, pH 6.0, or citraconic anhydride, pH 7.4 (Alelú-Paz et al., 2008). The sources for the primary antibodies used for immunohistology, and the antigen retrieval methods (if any) used with each, are listed in supplemental Table 2, available at www.jneurosci.org as supplemental material. Bound antibodies were detected using species- and isotype-specific fluorescently conjugated or biotinylated secondary antibodies and visualized by laser scanning confocal microscopy.
In Thy1-STOP-EYFP/EmxCre double transgenic mice, EYFP expression was visualized in 10 μm cryostat cross-sections of L3 spinal cord by immunostaining with a fluorescein-conjugated anti-green fluorescent protein antibody (Rockland). Microscopic fields encompassing both dorsal corticospinal tracts were photographed using a 100× objective mounted on a Nikon laser scanning confocal microscope, and images were tiled together using Nikon NIS-Elements software. All EYFP-labeled axons in the lumbar dorsal corticospinal tract cross-sections prepared from the MOG peptide EAE and CFA control mice were counted with the aid of NIH ImageJ software.
Data analysis and statistics.
Molecular data were derived from at least three independent experiments at each time point. Results of spinal cord qRT/PCR assays were analyzed using the Mann–Whitney U test. Differences between qRT/PCR results obtained from MOG peptide or CFA control spinal cord extracts and those obtained from normal mice of the same age were classified as significant if p < 0.05 and the fold difference from normal mice was ≥1.5. Immunohistological data were obtained from at least four MOG peptide and four CFA control mice at each time point.
Results
Mice immunized with MOG peptide demonstrate the subacute onset and chronic persistence of clinical neurological deficits
Mice immunized with MOG peptide in CFA usually began to show neurological deficits between days 12 and 13 (rarely as early as day 10), reached peak severity between days 17 and 21, and continued to have substantial deficits through day 100 (Fig. 1). CFA control mice showed no clinical neurological deficits.
Figure 1.

Clinical course of MOG peptide-immunized mice. Mice were examined daily and scored as described in Materials and Methods. Clinical scores are shown for mice immunized with MOG peptide in CFA. CFA control mice showed no clinical neurological deficits at any time point. Data are means ± SEMs (n = 8).
MOG peptide-specific Th1, Th17, and Th17/Th1 lymphocytes are generated in peripheral lymphoid tissues by day 7 post-MOG peptide immunization
MOG peptide-specific Th1 (CD4+ IFNγ+), Th17 (CD4+ IL17+) and Th17/Th1 (CD4+ IFNγ+ IL17+) (Park et al., 2005; Annunziato et al., 2007; Nakae et al., 2007) lymphocytes became detectable by flow cytometry in pooled spleen/lymph nodes by day 7 post-MOG peptide (Fig. 2A). The functional properties of mouse Th17/Th1 lymphocytes have not yet been established, but human Th17/Th1 lymphocytes, like Th17 lymphocytes, are far less susceptible than Th1 lymphocytes to suppression by Tregs (Annunziato et al., 2007). Numbers of MOG peptide-specific Th1, Th17, and Th17/Th1 lymphocytes in peripheral lymphoid tissues peaked by day 12, then fell progressively over ensuing weeks. MOG peptide-specific T lymphocytes were not detected at any time point in pooled spleen/lymph nodes of CFA control mice.
Figure 2.

Flow cytometric analysis of CNS homing by systemic mononuclear cells. A–D, MOG-specific Th1, Th17, and Th17/Th1 (CD4+ IFN-γ+ IL-17+) cells in pooled spleen/lymph nodes (A), CNS (pooled brain and spinal cord) immune mononuclear cells (B), CNS Th1, Th17 and Th17/Th1 cells (C), and CNS Teff (Th1, Th17 and Th17/Th1) and Tregs (D). Data shown in A–D are means ± SEMs of at least three independent experiments at each time point. E, Example of the flow cytometric analysis of CNS CD4-gated mononuclear cells prepared from a day 14 post-CFA/MOG peptide mouse for intracellular expression of IFN-γ and IL-17, showing 3.4% Th1 cells (IFN-γ high, IL-17 low), 13.64% Th17 cells (IFN-γ low, IL-17 high) and 1.86% Th17/Th1 cells (IFN-γ high, IL-17 high).
Substantial accumulation of systemic immune cells in CNS commences between days 14 and 21 post-MOG peptide
Mononuclear cell yields from Percoll-fractionated collagenase/DNase digests of pooled brain and spinal cord increased sharply between days 14 and 21 post-MOG peptide, then dropped progressively over ensuing months, but remained consistently at baseline levels in CFA control mice (Fig. 2B). CD4-gated flow cytometry (see Fig. 2E for an example) demonstrated small numbers of Th1, Th17, and Th17/Th1 lymphocytes in CNS by day 12 post-MOG peptide, with maximal numbers of each of these CD4+ lymphocyte subsets attained between days 14 and 21 postimmunization (Fig. 2C).
At the peak of CNS lymphocyte infiltration in the MOG peptide-immunized mice, the ratio of Th17 to Th1 cells in CNS was >2 (Fig. 2C). This contrasted with spleen/lymph node ratios of both MOG-specific Th17 to Th1 cells (Fig. 2A) and of total Th17 to Th1 cells (data not shown) <0.5 in these mice. These results are in accord with those reported by Bailey et al. (2007), who showed that, both in vivo and in vitro, CNS myeloid dendritic cells preferentially polarize CD4+ T cells to Th17, while in the periphery, Th1 is the predominant CD4+ T-cell phenotype.
CNS levels of CD4+ CD25+ Foxp3+ Tregs initially rose at a rate similar to that of total CNS Th1+ Th17+ Th17/Th1 effector cells (Teffs). From day 21 onward, by which time the mice had reached peak clinical severity, the CNS Treg/Teff ratio was always >2 (Fig. 2D). This result was presumably a consequence of the activation and rapid proliferation of Tregs in the inflamed CNS (O'Connor et al., 2007), and is likely to have prevented further worsening of the disease (McGeachy et al., 2005).
To supplement these flow cytometric analyses of lymphocyte subsets in whole CNS, spinal cord extracts from MOG peptide-immunized and CFA control mice were assayed for mRNAs encoding lymphocyte transcription factors and other immune cell subtype-specific constituents by qRT/PCR. Both granzyme A mRNA and Itga2 mRNA rose early after immunization in mice that received MOG peptide in CFA. Granzyme A is a proapoptotic protease expressed by natural killer (NK), NKT, and a subset of cytotoxic T cells (Metkar et al., 2008; Trapani and Bird, 2008). Itga2 mRNA encodes CD49b (also known as CD49b, VLA-2 α chain and integrin α2 chain), which is expressed by NK and NKT cells, a subset of CD8+ T cells, and possibly by microglial/monocyte-derived macrophages (Slifka et al., 2000; Pellicci et al., 2005; Schleicher et al., 2005). Granzyme A and Itga2 mRNAs also rose early after administration of CFA to control mice, but, by day 14 postimmunization and thereafter, these mRNAs were markedly higher in MOG peptide EAE than in CFA control mice (Table 1). By day 14 postinjection, there were significant accumulations of transcription factor mRNAs associated with Th17 cells (ROR-γt), Th1 cells (T-bet), and Tregs (Foxp3) in the MOG peptide-immunized mice, but not in CFA control mice (Fig. 3). While levels of CD4 mRNA in MOG peptide spinal cords were not significantly higher than in CFA control spinal cords until day 14 postinjection, the abundance of CD3 mRNA, a pan-T lymphocyte marker, had already risen significantly in MOG peptide mice by day 10 postimmunization (Fig. 3). Quantitative RT-PCR also showed increased levels of mRNAs encoding proteins characteristic of microglia and macrophages (CD11b) and of B lineage cells (CD19 and IgM). Among these immune cell marker mRNAs, only IgM mRNA continued to rise throughout the course of the disease (Fig. 3).
Table 1.
qRT/PCR assays of immune-related mRNAs in spinal cords of mice given MOG peptide in CFA (MOG) and in CFA control (CFA) mice
| Day 3 |
Day 7 |
Day 10 |
Day 14 |
Day 21 |
Day 35 |
Day 98 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CFA | MOG | CFA | MOG | CFA | MOG | CFA | MOG | CFA | MOG | CFA | MOG | CFA | MOG | |
| Mincle | 4.0 | 4.4 | 5.4 | 19.8 | 6.6 | 9.1 | 4.3 | 185.1 | 2.4 | 116.7 | 4.8 | 185.5 | 0.6 | 3.7 |
| Dectin1 | 0.7 | 1.6 | 1.5 | 6.6 | 1.9 | 2.6 | 2.0 | 265.1 | 1.6 | 167.3 | 1.4 | 144.2 | 1.0 | 18.1 |
| COX2 | 5.1 | 6.0 | 3.7 | 6.4 | 2.5 | 17.3 | 1.9 | 9.9 | 1.3 | 6.0 | 2.9 | 11.6 | 1.2 | 1.3 |
| IL1β | 2.2 | 1.6 | 2.8 | 13.8 | 2.4 | 5.9 | 1.2 | 76.8 | 1.0 | 53.9 | 2.6 | 125.5 | 0.6 | 2.2 |
| IL1R1 | 1.8 | 2.1 | 1.5 | 2.0 | 1.5 | 2.2 | 1.2 | 3.1 | 1.3 | 3.3 | 2.3 | 6.5 | 1.7 | 2.4 |
| ASC | 1.0 | 0.6 | 1.2 | 1.2 | 1.3 | 1.2 | 0.9 | 56.0 | 0.8 | 14.0 | 1.0 | 10.7 | 1.0 | 4.1 |
| NALP1 | 1.9 | 0.7 | 2.3 | 1.5 | 2.2 | 1.3 | 1.6 | 22.2 | 1.2 | 12.5 | 1.3 | 9.6 | 1.1 | 4.5 |
| Caspase1 | 0.6 | 0.5 | 0.9 | 0.6 | 0.6 | 0.6 | 0.5 | 10.3 | 0.4 | 2.7 | 0.6 | 2.4 | 0.5 | 2.2 |
| Caspase3 | 0.8 | 1.2 | 1.5 | 0.6 | 0.9 | 0.8 | 0.7 | 2.3 | 0.7 | 1.8 | 0.9 | 1.0 | 1.0 | 1.3 |
| Caspase11 | 1.3 | 1.1 | 2.6 | 3.4 | 1.1 | 1.7 | 0.9 | 33.8 | 0.5 | 9.0 | 0.7 | 9.5 | 0.7 | 6.9 |
| MPO | 6.0 | 3.8 | 1.8 | 7.7 | 3.2 | 6.0 | 2.5 | 16.2 | 1.5 | 9.0 | 1.5 | 5.6 | 1.8 | 2.9 |
| Ela2 | 34.4 | 5.6 | 8.3 | 6.6 | 17.7 | 7.2 | 8.0 | 26.9 | 3.1 | 9.3 | 6.9 | 1.3 | 0.4 | 0.7 |
| CXCL1 | 4.2 | 3.8 | 3.4 | 17.8 | 3.0 | 10.8 | 2.0 | 47.5 | 0.7 | 35.1 | 1.2 | 13.4 | 1.3 | 7.1 |
| CXCR1/IL8Ra | 3.1 | 1.3 | 2.1 | 5.4 | 5.9 | 3.9 | 2.7 | 5.9 | 1.3 | 6.8 | 0.9 | 6.4 | 1.0 | 1.2 |
| CXCR2/IL8Rb | 14.0 | 7.8 | 9.5 | 25.2 | 23.4 | 16.8 | 14.7 | 43.1 | 4.7 | 30.6 | 7.3 | 19.8 | 1.6 | 2.6 |
| G-CSF | 1.6 | 0.8 | 0.7 | 5.0 | 1.0 | 3.7 | 1.1 | 4.0 | 0.8 | 8.1 | 1.3 | 6.9 | 0.8 | 1.1 |
| TLR1 | 1.5 | 1.4 | 1.5 | 2.4 | 1.8 | 2.0 | 1.3 | 15.7 | 1.2 | 16.3 | 1.8 | 19.7 | 0.8 | 3.5 |
| TLR2 | 0.6 | 0.9 | 0.7 | 1.8 | 0.9 | 1.5 | 0.8 | 18.1 | 0.6 | 8.4 | 0.6 | 5.8 | 0.9 | 5.7 |
| TLR3 | 1.4 | 0.9 | 1.0 | 1.3 | 1.3 | 1.1 | 1.4 | 2.4 | 1.4 | 2.9 | 0.8 | 1.2 | 1.1 | 1.4 |
| TLR4 | 1.1 | 1.3 | 1.2 | 1.6 | 1.0 | 1.5 | 1.2 | 8.1 | 1.2 | 5.4 | 1.7 | 10.0 | 1.1 | 2.2 |
| TLR6 | 0.5 | 0.6 | 0.7 | 1.1 | 0.7 | 0.8 | 0.9 | 4.1 | 0.4 | 4.4 | 1.3 | 7.3 | 0.7 | 1.3 |
| TLR7 | 1.0 | 1.3 | 1.1 | 1.1 | 1.2 | 1.0 | 0.7 | 7.6 | 1.0 | 9.4 | 0.9 | 5.9 | 0.5 | 1.6 |
| TLR8 | 0.9 | 1.3 | 1.8 | 2.7 | 1.7 | 2.0 | 1.2 | 20.5 | 1.3 | 13.0 | 2.3 | 19.2 | 1.0 | 3.8 |
| TLR9 | 1.1 | 1.1 | 1.1 | 1.4 | 1.2 | 1.0 | 1.1 | 5.0 | 1.1 | 5.2 | 2.0 | 11.4 | 1.0 | 2.0 |
| TLR11 | 0.6 | 1.0 | 1.1 | 1.5 | 2.1 | 2.0 | 1.9 | 25.6 | 1.2 | 21.8 | 1.7 | 15.0 | 1.2 | 7.8 |
| TLR12 | 1.0 | 1.1 | 1.6 | 1.3 | 1.6 | 1.3 | 1.1 | 19.5 | 1.3 | 15.3 | 1.2 | 6.4 | 1.5 | 5.0 |
| TLR13 | 1.0 | 1.3 | 1.7 | 1.5 | 2.0 | 1.3 | 1.0 | 22.2 | 0.8 | 9.7 | 1.0 | 7.7 | 1.1 | 4.2 |
| Rab7 | 1.1 | 1.3 | 1.2 | 1.0 | 1.7 | 1.1 | 0.8 | 1.8 | 1.3 | 1.1 | 1.5 | 1.1 | 1.3 | 1.7 |
| APP | 1.0 | 1.0 | 1.1 | 0.6 | 1.2 | 1.1 | 0.8 | 0.7 | 0.8 | 0.6 | 1.0 | 0.6 | 1.5 | 1.5 |
| MyD88 | 1.2 | 1.2 | 0.9 | 1.3 | 1.5 | 1.5 | 1.3 | 4.4 | 1.2 | 3.6 | 1.8 | 10.5 | 1.3 | 1.8 |
| CD45 | 1.5 | 1.4 | 1.5 | 3.3 | 2.1 | 2.0 | 1.3 | 17.2 | 0.9 | 15.5 | 1.4 | 10.0 | 0.8 | 4.2 |
| Grzm a | 1.6 | 1.1 | 3.9 | 4.6 | 3.2 | 3.0 | 2.5 | 18.0 | 1.7 | 25.0 | 2.3 | 4.2 | 1.9 | 1.7 |
| Itga2 | 2.3 | 1.2 | 3.0 | 1.8 | 1.9 | 1.4 | 2.4 | 4.0 | 1.7 | 5.8 | 1.8 | 6.1 | 1.6 | 3.2 |
Mice were sacrificed on days −1 (normal control), 3, 7, 10, 14, 21, 35, and 98 postinjection with MOG peptide in CFA or with CFA alone. Each determination employed three mice, and three determinations were made at each time point. Data are presented as means of fold difference between normal control and the MOG or CFA control group. Boldface indicates that mRNA levels were at least 1.5-fold higher, and significantly different, in the MOG or CFA control group than in the day −1 control spinal cords at that time point (p < 0.05 Mann–Whitney U test). Additional spinal cord innate immune qRT/PCR assays, not specifically mentioned in the text, are listed in supplemental Table 3, available at www.jneurosci.org as supplemental material.
Figure 3.

qRT/PCR for immune marker mRNAs in MOG peptide and CFA control spinal cords harvested on days −1 (normal control), 3, 7, 10, 14, 21, 35, and 98. The x axis shows days post-CFA/MOG (circles/solid line) or CFA (squares/dotted line). The y axis shows the ratio of abundance of each mRNA in MOG or CFA to that in normal controls. Fold changes are provided for T-cell surface markers (CD3, CD4, CD8), transcription factors associated with Th1 (T-bet), Th17 (ROR-γt), or Treg (Foxp3) T cells; and markers for B cells (IgM and CD19) and macrophages/microglia (CD11b). Data are means ± SEMs of at least three independent experiments. *Results in CFA/MOG peptide spinal cords were significantly higher than in CFA control spinal cords (p < 0.05); θ, results in CFA control spinal cords were significantly higher than in CFA/MOG peptide spinal cords (p < 0.05, Mann–Whitney U test).
Activation of CNS innate immunity commences before substantial accumulation in CNS of systemic immune cells, and involves genes that participate in neutrophil trafficking
Toll-like receptors (TLRs) and other pattern recognition receptors are the principal afferent arms of innate immunity, and are coupled to effector mechanisms which include phagocytosis, secretion of cytokines and chemokines, assembly of inflammasomes, and trafficking of circulating immune cells to affected tissues (Creagh and O'Neill, 2006; Kawai and Akira, 2007; Martinon and Tschopp, 2007; O'Neill and Bowie, 2007). We used qRT-PCR to assay levels of expression of mRNAs encoding TLRs, selected inflammasome components and cytokines, and other proteins involved in innate immune responses during the course of MOG peptide EAE. Within 3 d after administration of MOG peptide in CFA, there were significant spinal cord inductions of mRNAs encoding the microglial/macrophage transcript, CD11b (Santambrogio et al., 2001; Herber et al., 2006) (Table 1, Fig. 3) and of two genes implicated in neutrophil trafficking, CXCL1 and CLEC4E, the latter of which encodes the macrophage inducible C-type lectin, or Mincle (Table 1) (Anthony et al., 1998; Balch et al., 2002; Carlson et al., 2008; van Vliet et al., 2008; Wells et al., 2008; Yamasaki et al., 2008). Levels of these mRNAs rose to a comparable degree in spinal cords of CFA control mice (Table 1), and also in spinal cords of Rag1−/− mice given MOG peptide in CFA (Table 2), despite the lack of mature B and T cells in this mutant strain (Mombaerts et al., 1992). Hence, these gene inductions must have taken place in the Rag1−/− mice independent of any adaptive immune response, and in the CFA control mice in the absence of an adaptive immune response directed specifically against MOG peptide. It is likely that transient activation of microglia, demonstrated by increases in spinal cord levels of CD11b and Mincle mRNAs, was largely responsible for the early induction of spinal cord cytokine and chemokine mRNAs (Raghavendra et al., 2004). Further experiments demonstrated that CFA, rather than pertussis toxin, was largely responsible for these mRNA inductions (Table 3). The mechanism by which CFA entrained these early CNS innate immune responses in wild-type and immunocompromised mice may involve interleukin-1β (IL-1β) (Roy et al., 2006); subcutaneous CFA elicits massive production of IL-1β at the injection site, and a >10-fold increase in CSF IL-1β content (Samad et al., 2001), and manipulations that elevate CNS IL-1β increase CNS CXCL1 mRNA and induce CNS neutrophil infiltration (Campbell et al., 2002; Shaftel et al., 2007).
Table 2.
mRNA fold changes in spinal cords of RAG1−/− mice induced by injection of MOG peptide in CFA
| Day 7 | Day 10 | Day 14 | |
|---|---|---|---|
| MPO | 2.6 | 4.2 | 16.8 |
| COX2 | 2.1 | 2.3 | 1.2 |
| Mincle | 5.8 | 6.7 | 8.8 |
| IL1β | 3.3 | 4.2 | 1.6 |
| CXCR1 | 3.0 | 2.1 | 3.1 |
| CXCR2 | 5.7 | 3.9 | 10.9 |
| CXCL1 | 4.1 | 5.6 | 2.1 |
| CCL5 | 1.8 | 1.5 | 2.8 |
| IL1R1 | 3.2 | 1.7 | 1.8 |
| IL6R | 2.4 | 1.6 | 1.6 |
| Ela2 | 5.0 | 7.1 | 18.9 |
| CD45 | 1.6 | 1.2 | 2.1 |
| CD11b | 1.5 | 0.9 | 1.5 |
mRNAs tested but showing no change in spinal cords of RAG1−/− mice that had been injected with MOG peptide in CFA: eNOS, TNFα, C3, NFKB, C5, HMGB1, IL6, NFIL6, HSPA1A, C5aR, CD3e, CD3g, CCR2, CCL2, CCL6, CCL8, Granzyme A, IL27p28, FOXp3, CD4, CD8, DX5, CXCL9, CXCL10, CXCL12, CX3CL1, CX3CR1, IFNα, and CCR3. Mice were sacrificed on day −1 (RAG1−/− control), 7, 10, or 14 postinjection. Each determination employed three mice, and three determinations were made at each time point. Data are presented as means of fold differences of CFA-MOG over the day −1 RAG1−/− controls. Boldface indicates that mRNA levels were at least 1.5-fold higher, and significantly different, in the CFA-MOG spinal cords than in the day −1 RAG1−/− control spinal cords at that time point, with p < 0.05 (Mann–Whitney U test).
Table 3.
mRNA fold changes in spinal cord of C57BL/6 mice injected with CFA, CFA plus pertussis toxin (PTx), or PTx alone
| 3 h postinjection |
24 h postinjection |
48 h postinjection |
72 h postinjection |
96 h postinjection |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CFA | CFA + PTx | PTx | CFA | CFA + PTx | PTx | CFA | CFA + PTx | PTx | CFA | CFA + PTx | PTx | CFA | CFA + PTx | PTx | |
| MPO | 2.1 | 0.7 | 0.5 | 4.1 | 0.9 | 0.5 | 1.3 | 0.5 | 0.7 | 1.3 | 1.1 | 0.4 | 15.9 | 1.0 | 0.9 |
| COX2 | 2.6 | 5.9 | 0.9 | 3.5 | 3.9 | 0.7 | 5.5 | 3.8 | 1.0 | 2.9 | 2.1 | 0.9 | 5.5 | 4.4 | 0.8 |
| Mincle | 1.0 | 2.2 | 0.4 | 1.8 | 2.1 | 0.4 | 2.4 | 1.6 | 0.6 | 1.4 | 1.5 | 0.3 | 4.0 | 4.0 | 0.6 |
| IL1β | 1.1 | 1.2 | 0.4 | 0.9 | 0.8 | 0.4 | 0.8 | 1.1 | 0.4 | 1.1 | 1.1 | 0.4 | 2.3 | 3.1 | 0.8 |
| CXCL1 | 4.3 | 34.9 | 0.4 | 3.2 | 2.3 | 0.6 | 15.4 | 4.4 | 1.5 | 5.0 | 1.7 | 0.7 | 8.3 | 10.0 | 0.8 |
| CD45 | 1.5 | 1.5 | 1.2 | 2.4 | 1.9 | 1.3 | 2.1 | 1.7 | 1.3 | 1.6 | 1.8 | 1.1 | 2.7 | 2.1 | 1.3 |
mRNAs tested but showing no change in spinal cords of mice given CFA, CFA plus PTx, or PTx alone: HMGB1, NF-kb1, CX3CR1, CX3CL1, C5, NF-IL6, and CXCL9. Mice were sacrificed at the indicated times postinjection with CFA, CFA plus pertussis toxin, or pertussis toxin alone. Each determination employed three mice, and three determinations were made at each time point. Data are presented as means of fold differences over uninjected control mice. Boldface indicates that mRNA levels were at least 1.5-fold higher, and significantly different, than in the control samples at that time-point, with p < 0.05 (Mann–Whitney U test).
Among the immune system-related mRNAs we assayed by qRT/PCR in spinal cord extracts from the MOG peptide-immunized and CFA control mice (Table 1, Fig. 3; supplemental Table 3, available at www.jneurosci.org as supplemental material), the earliest substantial alterations specific to MOG peptide-immunized mice were greater than fourfold increases in mRNAs encoding the innate immune cell activator, dectin-1 (Shah et al., 2008), and granulocyte-colony stimulating factor (G-CSF, also known as CSF-3), the latter a protein that enhances neutrophil trafficking and survival (Eyles et al., 2006; Wengner et al., 2008) by day 7 post-MOG peptide (Table 1). Three days later (day 10 post-MOG peptide), immunoreactive G-CSF had accumulated on the spinal meninges of both MOG peptide-immunized and CFA control mice (Fig. 4A).
Figure 4.

Early perivascular and subpial neutrophil- and lymphocyte-rich inflammatory infiltrates in MOG peptide EAE mice. A, Expression of immunoreactive granulocyte colony-stimulating factor (G-CSF) on the spinal meninges on day 10 post-CFA/MOG peptide; this was no longer evident by day 12 post-CFA/MOG peptide, when a subpial infiltrate of Ly6G+ neutrophils had formed. B, Perivascular infiltrate of Ly6G+ neutrophils and CD3ε+ lymphocytes on day 12 post-CFA/MOG peptide. C, Another perivascular inflammatory infiltrate, containing Ly6G+/MMP8+ neutrophils, but without substantial recruitment to the infiltrate of IBA1+ microglia or monocyte-derived macrophages.
Subpial and perivascular neutrophil-rich inflammatory infiltrates are evanescently present in spinal cord shortly before the onset of clinical neurological deficits
Immunohistological evaluation of spinal cord on days 3 and 7 postadministration of MOG peptide in CFA, or of CFA alone, revealed occasional Ly6G+ neutrophils (Mildner et al., 2008) adhering to the meninges and surfaces of the nerve roots, but no intraparenchymal neutrophil infiltrates (data not shown). By day 12 post-MOG peptide, spinal cord perivascular and subpial infiltrates of Ly6G+ neutrophils containing matrix metalloproteinase-8 (MMP-8), an enzyme that facilitates neutrophil tissue penetration, had appeared in the MOG peptide-immunized mice (Fig. 4). These inflammatory infiltrates, which were most often located in ventral and lateral spinal cord white matter, and rarely in the dorsal funiculi, also contained substantial numbers of CD3ε+ T lymphocytes (Fig. 4B), but, by day 12 post-MOG peptide, contained only sparse numbers of Iba1+ microglia (Fig. 4C). By day 14 post-MOG peptide and at later time points, however, Ly6G+ neutrophils had become a much less prominent component of the spinal cord inflammatory infiltrates, which were increasingly dominated by Iba1+ microglial and monocyte-derived macrophages (data not shown).
Morphological evidences of acute spinal cord axonopathy become detectable by day 12 post-MOG peptide
Hypophosphorylated NF-H immunoreactivity, detectable by immunostaining with the SMI32 monoclonal antibody, is normally expressed in perikarya of spinal cord motor neurons, but not in axons in spinal cord white matter (Bannerman et al., 2005). In EAE and multiple sclerosis, damaged axons become SMI32+ (Trapp et al., 1998; Pitt et al., 2000; Wang et al., 2005; Petzold et al., 2008). We first detected SMI32+ axons in lumbosacral spinal cord white matter on day 12 post-MOG peptide; at this time point, these SMI32+ axons were almost always situated within inflammatory infiltrates (Fig. 5). As has previously been noted in multiple sclerosis autopsy specimens (Trapp et al., 1998; Bjartmar et al., 2001) and in a rat adoptive transfer EAE model (Aboul-Enein et al., 2006), some of these damaged axons, viewed in cross-section, were still encircled by myelin basic protein-positive (MBP+) immunoreactivity on day 12 post-MOG peptide (Fig. 5C).
Figure 5.

Axons are damaged within spinal cord inflammatory infiltrates in MOG peptide EAE mice. A, Ly6G+ neutrophil-rich inflammatory infiltrate containing APP+ damaged axons, from a day 14 post-CFA/MOG peptide mouse. B, By day 21 post-CFA/MOG peptide, SMI32+ and APP+ damaged axons are seen outside zones of overt infiltration by DAPI+ inflammatory cells; note the low overlap between SMI32 and APP immunoreactivities in this field. C, This field was in the ventrolateral lumbar spinal cord of a mouse given CFA/MOG peptide 12 d previously. Within a zone of DAPI nuclear-stained inflammatory cells, an SMI32+ axon still encircled by myelin basic protein (MBP) immunoreactivity is indicated by the arrow, a demyelinated SMI32+ axon by an arrowhead, and an apparently vacuolated SMI32+ axon surrounded by a ring of MBP immunoreactivity by an asterisk.
Axonal hypophosphorylated NF-H immunoreactivity was often ring-like in sections that had been subjected to sodium citrate antigen retrieval before immunostaining with SMI32 (Figs. 5, 6, 7A). This ring-like distribution of immunoreactivity was much less prominent, however, when citraconic anhydride, rather than sodium citrate, was used for antigen retrieval (Fig. 7B).
Figure 6.

Accumulations of the endovesicular Toll-like receptor TLR8 in SMI32+ axons. Left, SMI32+ axons in ventrolateral spinal cord at days 12, 35, and 98 post-CFA/MOG peptide, respectively. Center, Same fields immunostained for TLR8. Right, Merged panels, with DAPI nuclear staining as well. Note that the ring-like pattern of SMI32 immunostaining is more prominent on day 12 post-CFA/MOG peptide, and that the SMI32 mAb and the anti-TLR8 antibody often bind to the same axons.
Figure 7.

SMI32+ axons in MOG peptide EAE mice contain endolysosome-associated Rab7 and express inflammasome protein NALP1, which contributes to the processing of IL-1β. A, Ventrolateral spinal cord of a day 14 post-CFA/MOG peptide mouse immunostained (from left to right) for Rab7, SMI32, and the two markers together. The white arrowhead indicates an SMI32+ axon containing Rab7. B, Ventrolateral spinal cord of a day 21 post-CFA/MOG peptide mouse immunostained (from left to right) for NALP1, SMI32, and both markers. By 21 d post-CFA/MOG peptide, many SMI32+ axons expressed NALP1. C, Western blot demonstrating induction of IL-1β precursor (pro-IL-1β) and IL-1 β in spinal cords of day 14 post-CFA/MOG peptide mice. The asterisk indicates a nonspecific band.
APP accumulates in axons in acute multiple sclerosis lesions (Bitsch et al., 2000; Frischer et al., 2009), and anti-APP, like SMI32, has proven useful for identifying damaged axons in EAE (Einstein et al., 2006; Moreno et al., 2006; Herrero-Herranz et al., 2008). Though there were no consistent changes in spinal cord expression of APP mRNA during the course of MOG peptide EAE (Table 1), accumulations of immunoreactive APP in spinal cord axons became evident at approximately the time of onset of clinical neurological deficits in MOG peptide-immunized mice. Interestingly, colabeling of spinal cord sections with SMI32 and anti-APP demonstrated that only occasional axons, viewed in cross-section, simultaneously expressed both markers, with many being only SMI32+ or APP+ (Fig. 5).
Activation of TLR8, an endolysosomal TLR, has been reported to cause neuronal death and inhibit neurite formation (Ma et al., 2006, 2007). We observed induction of TLR8 mRNA in MOG peptide-EAE spinal cord by day 14 postimmunization (Table 1). Accumulations of immunoreactive TLR8 were already present in some SMI32+ axons within spinal cord inflammatory foci by day 12 post-MOG peptide (Fig. 6). TLR8 immunoreactivity was not detected in dorsal corticospinal tract axons (data not shown). Levels of mRNAs encoding most other TLRs were also elevated in spinal cords of MOG peptide-immunized mice from day 14 onwards, in conjunction with elevations of mRNAs encoding the TLR downstream signaling molecule, MyD88 (Gorden et al., 2006; O'Neill and Bowie, 2007). With the exception of rare axons that contained accumulations of immunoreactive TLR3, TLRs other than TLR8 were not detected in axons (data not shown). Bone marrow chimera experiments have shown that ablation of MyD88 expression in CNS radiation-resistant cells delays the onset and diminishes the clinical severity and acute axonopathy in MOG peptide EAE (Prinz et al., 2006), possibly by inhibiting CNS MyD88-mediated canonical TLR8 signal transduction (Gorden et al., 2006).
Some SMI32+ axons in spinal cord white matter of the MOG peptide EAE mice contained accumulations of immunoreactive Rab7 (Fig. 7A). Rab7 is an endolysosome-associated protein that participates in fast retrograde vesicular protein transport in axons (Bucci et al., 2000; Harrison et al., 2003; Saxena et al., 2005; Deinhardt et al., 2006).
Inflammasomes process pro-interleukin-1β (pro-IL1β) to the proinflammatory cytokine IL1β (Tschopp et al., 2003; Faustin et al., 2007; Franchi et al., 2009; Lamkanfi and Dixit, 2009). Spinal cord levels of mRNAs encoding multiple inflammasome-associated proteins, including NALP1, caspase 1, caspase 3, apoptosis-associated speck-like protein containing a caspase-activating recruitment domain (ASC), and pro-IL1β (Faustin et al., 2007; Martinon and Tschopp, 2007; de Rivero Vaccari et al., 2008; Franchi et al., 2009), were increased by day 14 post-MOG peptide (Table 1). Immunoreactive pro-IL1β and IL-1β were substantially elevated in spinal cord by day 14 post-MOG peptide (Fig. 7C), and by day 21 post-MOG peptide, many SMI32+ axons in spinal cord white matter contained immunoreactive NALP1. These results suggest the existence of an intrinsic neuronal innate immune contribution to proinflammatory cytokine production in MOG peptide EAE. Prior studies have shown that neuronal expression of NALP1 is also induced by trauma or ischemia (Liu et al., 2004; de Rivero Vaccari et al., 2008), and that transfection-induced overexpression of NALP1 in cultured neurons results in their apoptosis (Liu et al., 2004).
Progressive, bilaterally symmetric loss of dorsal corticospinal tract axon numbers in MOG peptide EAE
Selective loss of small-diameter corticospinal tract axons has been documented in multiple sclerosis (Ganter et al., 1999; DeLuca et al., 2004). Both immunohistological and cortical motor evoked potential studies have suggested there is a substantial diminution in corticospinal axon numbers in mice with long-standing MOG peptide EAE (Amadio et al., 2006; Black et al., 2006). Enumeration of specifically labeled corticospinal axons would be desirable to strengthen this conclusion. Corticospinal axons are unique in extending from the forebrain into the spinal cord without an intervening synapse, and can therefore be identified in spinal cord by labeling constituents of forebrain neuronal perikarya that undergo centrifugal axonal transport. Bareyre et al. (2005) devised a genetic approach to accomplish this, using mice bred to express Thy1-STOP-EYFP, in which the STOP sequence that prevents neuronal Thy1 promoter-driven EYFP expression is flanked by loxP sites, and Cre, driven by the forebrain-specific Emx promoter, which excises the STOP sequence and activates forebrain neuronal EYFP expression. By this means, they established that all adult mouse EYFP+ dorsal corticospinal axons were <1.5 μm in diameter, most having diameters between 0.4 and 0.6 μm. Using this genetic labeling procedure, we documented progressive, bilaterally symmetrical loss of dorsal corticospinal axons in the lumbar spinal cords of MOG peptide EAE EmxCre/Thy1-STOP-EYFP mice, to 56% and 26% of control levels on days 35 and 101 post-MOG peptide, respectively (Fig. 8, Table 4). Immunostaining with SMI312, an antibody mixture that recognizes multiple phosphorylated neurofilament epitopes and heavily immunostains rodent dorsal corticospinal tracts (Szaro et al., 1990; Frischer et al., 2009), confirmed the severe loss of dorsal corticospinal tract axons in day 101 post-MOG peptide mice (Fig. 8B). Focal inflammatory infiltrates involving the dorsal corticospinal tracts were rarely seen, but, by day 35 post-MOG peptide, there was extensive infiltration of these tracts by Iba1+ and CD68+ microglial/monocyte-derived macrophages (Fig. 8) (Ohsawa et al., 2000; Rodriguez et al., 2007; Santos et al., 2008).
Figure 8.
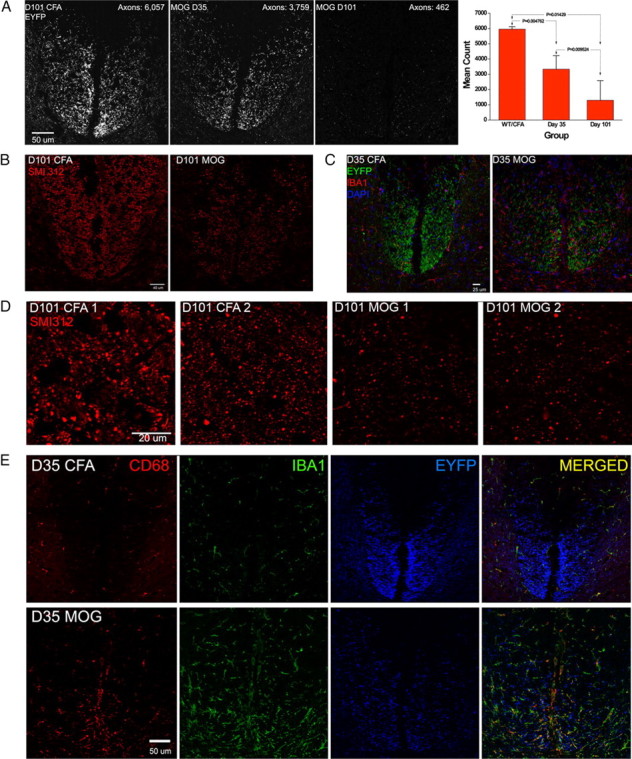
Progressive loss of EYFP+ dorsal corticospinal axons in MOG peptide EAE. A, Optical sections (0.13 mm) of the L3 dorsal corticospinal tracts (dCSTs) of day 101 CFA control; day 35 CFA/MOG peptide; and day 101 CFA/MOG peptide EmxCre/Rosa-STOP-EYFP transgenic mice. The far right panel shows the total (right plus left) EYFP+ axon counts in the combined right plus left dorsal dCSTs at L3 of four normal or CFA controls; six d 35 CFA/MOG peptide mice; and four d 101 CFA/MOG peptide mice (means ± SDs). B, dCSTs of a day 101 post-CFA control and a day 101 post-CFA/MOG peptide EAE mouse, immunostained with SMI312, which recognizes multiple phosphorylated neurofilament epitopes; note the severe depletion of SMI312+ axons in the EAE specimen and that dCST axons are shown in green. C, dCSTs of day 35 post-CFA control and day 35 post-CFA/MOG peptide EAE EmxCre/Rosa-STOP-EYFP mice immunostained for the microglial/macrophage marker Iba1. Note the prominence of Iba1+ cells in the EAE specimen. D, SMI312 immunostained axons within the dCSTs of two day 101 post-CFA control mice (on the left) and two day 101 post-CFA/MOG peptide mice (on the right) in 0.13 mm optical sections, confirming severe dCST axonal depletion in chronic MOG peptide EAE. E, Greater prominence of both Iba1+ and CD68+ microglial/monocyte-derived macrophages in day 35 post-CFA/MOG peptide than day 35 post-CFA control EmxCre/Rosa-STOP-EYFP lumbar spinal cord dorsal funiculi. EYFP+ dCST axons are pseudocolored blue in this panel.
Table 4.
Loss of dorsal CST axons in the lumbar spinal cords of MOG peptide EAE mice is bilaterally symmetric
| Days post-MOG peptide | Treatment | Axon count |
r2 | |
|---|---|---|---|---|
| Right | Left | |||
| Normal | Untreated | 2823 | 2945 | |
| 35 | CFA 1 | 3116 | 3010 | |
| 101 | CFA 1 | 2830 | 3069 | |
| 101 | CFA 2 | 2937 | 3120 | |
| 0.997 | ||||
| 35 | MOG peptide 1 | 1982 | 2329 | |
| 35 | MOG peptide 2 | 1804 | 1955 | |
| 35 | MOG peptide 3 | 1819 | 1489 | |
| 35 | MOG peptide 4 | 1593 | 1937 | |
| 35 | MOG peptide 5 | 702 | 979 | |
| 35 | MOG peptide 6 | 1776 | 1659 | |
| 0.974 | ||||
| 101 | MOG peptide 7 | 275 | 307 | |
| 101 | MOG peptide 8 | 247 | 215 | |
| 101 | MOG peptide 9 | 397 | 536 | |
| 101 | MOG peptide 10 | 1870 | 1335 | |
| 0.956 | ||||
EYFP+ axons were counted in the right and left dorsal corticospinal tracts in L3 spinal cord cross-sections from normal, CFA control, and MOG peptide EAE EmxCre/RosaSTOP-EYFP mice. The regression coefficient r2 was close to 1 in each group, indicating little variance between the axon counts on the right and left side in either the control or MOG peptide EAE mice (Snedecor and Cochran, 1989).
Discussion
Acute parenchymal inflammatory infiltrates in MOG peptide EAE
Early evidence for an interplay in spinal cord between adaptive and CNS innate immunity in MOG peptide EAE was the induction, on day 7 postinjection, of G-CSF mRNA in the spinal cords of mice given MOG peptide plus CFA, but not in CFA control mice. A small “first wave” of leptomeningeal and choroid plexus Th17 cells (Kivisäkk et al., 2009; Reboldi et al., 2009) may have contributed to this induction of spinal cord G-CSF synthesis (Smith et al., 2007, 2008). Because G-CSF augments neutrophil survival, mobilization, and adhesion (Eyles et al., 2006; Wengner et al., 2008), these early leptomeningeal and choroid plexus Th17 cells may also have contributed to the formation of neutrophil-rich infiltrates in spinal cord by day 12 post-MOG peptide (Smith et al., 2007, 2008; Kroenke et al., 2008; von Vietinghoff and Ley, 2008).
Participation by neutrophils in CNS inflammatory infiltrates has previously been reported in EAE (Nygårdas and Hinkkanen, 2002; Zehntner et al., 2005; Carlson et al., 2008; Kroenke et al., 2008). Neutrophils secrete proteases that permeabilize the blood–brain barrier (Leib et al., 2000; Gidday et al., 2005; Gurney et al., 2006; Folgueras et al., 2008), and proinflammatory cytokines and complement components (Määttä et al., 1998; Nguyen et al., 2008). Inhibiting CNS neutrophil infiltration in proteolipid peptide-induced EAE by antibody-mediated depletion of blood neutrophils, or constitutive disruption of CXCR2, the receptor for the neutrophil-attracting chemokine, CXCL1, prevented blood–brain barrier disruption and attenuated clinical severity of EAE, and this attenuation was reversed in CXCR2−/− mice by systemic administration of wild-type (CXCR2+/+) neutrophils (Carlson et al., 2008). In vitro, neutrophils are neuronotoxic via a contact-dependent, protease-mediated mechanism (Dinkel et al., 2004). These observations, together with the temporal and spatial coordination between the neutrophil-rich inflammatory infiltrates and the onset of axonopathy in the spinal cords of MOG peptide-immunized mice, support the hypothesis that these early inflammatory infiltrates exerted contact-dependent or paracrine axonal toxicity. While neutrophil-rich perivascular and subpial inflammatory infiltrates have not been recognized as features of multiple sclerosis (Raine, 1994), and Japanese patients with selective demyelination of the optic nerves and spinal cord in association with neutrophil-rich inflammatory infiltrates (Ishizu et al., 2005; Matsuoka et al., 2007) may have had neuromyelitis optica (Devic's disease) rather than multiple sclerosis (Wingerchuk et al., 2007; Kira, 2008), the brief tenure of neutrophils in spinal cord in EAE suggests that their presence in occasional early multiple sclerosis lesions may have been missed.
Axonal abnormalities within inflammatory foci
Mechanisms previously proposed to injure axons in EAE and multiple sclerosis include attack on axolemma by antibodies and complement (Mead et al., 2002, 2004; Jégou et al., 2007; Mathey et al., 2007; Barnett et al., 2009); proinflammatory mediators generated by macrophages and astroglia (Ahmed et al., 2002; Storch et al., 2002; Ayers et al., 2004; Wang et al., 2005); and axonal mitochondrial dysfunction induced by influx of Na+ and Ca2+ (Kornek et al., 2001; Craner et al., 2004; Dutta et al., 2006; Waxman, 2006; Forte et al., 2007; Friese et al., 2007; Mahad et al., 2009; O'Malley et al., 2009; Ouardouz et al., 2009a,b; Trapp and Stys, 2009). Whatever the cause of axonal injury, early axonal abnormalities that we observed in MOG peptide EAE were accumulations of APP and Rab7 within axons in spinal cord inflammatory foci. We interpret these findings as indicative of disturbances in both anterograde and retrograde axonal transport (Kamal et al., 2001; Lazarov et al., 2005; Deinhardt et al., 2006; Satpute-Krishnan et al., 2006; Shriver and Dittel, 2006; Rasmussen et al., 2007; Shindler et al., 2007; Frischer et al., 2009).
While APP and Rab7 are not known to be neurotoxic, affected axons in MOG peptide EAE also contained accumulations of TLR8 and NALP1. TLR8, an endovesicular TLR, is activated by oligonucleotides derived either from viruses or dying cells (Gibbard et al., 2006; Baccala et al., 2007; Krieg and Vollmer, 2007). TLR8 was of particular interest to us because its' activation in cultured neurons by a synthetic ligand was reported to inhibit neurite outgrowth and induce neuronal apoptosis (Ma et al., 2006, 2007), and because we observed TLR8 in damaged axons in acute inflammatory infiltrates. NALP1, a member of the inflammasome complex responsible for activation of proinflammatory caspases and interleukin-1β (Tschopp et al., 2003; Lamkanfi and Dixit, 2009), was present in damaged axons from day 21 through 35 post-MOG peptide. Messenger RNAs encoding NALP1 and multiple other members of the inflammasome complex were induced in spinal cord by day 14 post-MOG peptide, at which time point we documented substantial elevations in spinal cord pro-IL-1β and IL-1β. Prior studies demonstrated that NALP1 is induced in neurons by physical trauma or ischemia, that prolonged CNS overexpression of IL-1β damages axons (Campbell et al., 2007), and that death of traumatized neurons is diminished by inhibiting inflammasome function (Liu et al., 2004; de Rivero Vaccari et al., 2008).
Progressive loss of small-diameter corticospinal axons in MOG peptide EAE
Corticospinal tract atrophy, selectively affecting small-diameter axons, has been documented in autopsy specimens from multiple sclerosis patients (Ganter et al., 1999; DeLuca et al., 2004), and in mice with chronic and relapsing forms of EAE (Black et al., 2006; Liu et al., 2008). We have now shown, by both a genetic labeling procedure that permits specific enumeration of corticospinal axons (Bareyre et al., 2005) and confirmed by phosphorylated pan-neurofilament immunostaining (Szaro et al., 1990; Frischer et al., 2009), that small-diameter dorsal corticospinal tract axons are depleted in mice with MOG peptide EAE. We think it likely that a “dying back central axonopathy” (Kremenchutzky et al., 2006) was largely responsible for this axonal loss, because, in the lumbar spinal cords of each of the EAE mice we examined, the right and left dorsal corticospinal tracts were affected equally, and because the axonal loss progressed during the chronic phase of their illness, during which no new focal spinal cord inflammatory lesions were demonstrable. Activation of microglia and astroglial hypertrophy are prominent in white matter tracts in progressive forms of multiple sclerosis and in chronic EAE (De Keyser et al., 1999; Bannerman et al., 2007; Rasmussen et al., 2007). In these regions, extracellular glutamate homeostasis is impaired (Werner et al., 2001; Shijie et al., 2009) and production of potentially neurotoxic inflammatory mediators is elevated (Diestel et al., 2003).
We have not ruled out the possibility, however, that death of pyramidal neurons within cerebral cortical lesions also contributed to dorsal corticospinal axonal loss in the MOG peptide EAE mice. Cerebral cortical lesions in which neurons are lost by apoptosis are increasingly recognized in patients with multiple sclerosis and in chronic EAE; these lesions, which contribute to progressive gray matter atrophy and disability (Peterson et al., 2001; Vercellino et al., 2005, 2007; Fisher et al., 2008; Fisniku et al., 2008), lack overt inflammatory infiltrates but contain abundant activated microglia (Peterson et al., 2001; Rasmussen et al., 2007; Vercellino et al., 2007; Stadelmann et al., 2008; Trapp and Nave, 2008).
In conclusion, the immunological milieu in which axonal loss occurs in MOG peptide EAE is heterogeneous. At the time of onset of clinical deficits, there are immunohistological evidences of axonal damage within acute subpial and perivascular inflammatory infiltrates. Neutrophils, as well as T lymphocytes, are prominent components of these very early spinal cord inflammatory infiltrates; whether this evanescent infiltration by neutrophils is restricted to EAE, or occurs in occasional acute multiple sclerosis lesions, is worthy of further study. In addition to expressing well recognized markers for axons under attack (hypophosphorylated NF-H and APP), these damaged axons also accumulate the endovesicular Toll-like receptor TLR8 and the inflammasome protein NALP1, both of which are known to participate in neuronal injury in other contexts (Ma et al., 2006, 2007; de Rivero Vaccari et al., 2008). Over ensuing months, during which the focal inflammatory infiltrates subside, but elevated levels of expression of genes involved in innate immunity persist, the mice demonstrate progressive, symmetric and severe loss of small-diameter dorsal corticospinal axons and accumulations of macrophages in the dorsal corticospinal tracts. This finding strengthens support for the hypothesis that corticospinal tract axonal loss, a leading cause of persistent disability in multiple sclerosis (Ganter et al., 1999; DeLuca et al., 2004; Kremenchutzky et al., 2006), is a consequence of sustained activation of CNS innate immunity (Kutzelnigg et al., 2005; Tanuma et al., 2006; Trapp and Nave, 2008; Weiner, 2009). Finally, MOG peptide EAE will provide a robust model system with which to quantitatively evaluate the efficacy of interventions to prevent progressive axonal loss and chronic neurological disability in autoimmune CNS demyelinative disorders.
Footnotes
This work was supported by National Institutes of Health RO1 NS025044 and by Shriners Hospitals for Children.
References
- Aboul-Enein F, Weiser P, Höftberger R, Lassmann H, Bradl M. Transient axonal injury in the absence of demyelination: a correlate of clinical disease in acute experimental autoimmune encephalomyelitis. Acta Neuropathol. 2006;111:539–547. doi: 10.1007/s00401-006-0047-y. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Doward AI, Pryce G, Taylor DL, Pocock JM, Leonard JP, Baker D, Cuzner ML. A role for caspase-1 and -3 in the pathology of experimental allergic encephalomyelitis: inflammation versus degeneration. Am J Pathol. 2002;161:1577–1586. doi: 10.1016/S0002-9440(10)64436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alelú-Paz R, Iturrieta-Zuazo I, Byne W, Haroutunian V, García-Villanueva M, Rábano A, García-Amado M, Prensa L, Giménez-Amaya JM. A new antigen retrieval technique for human brain tissue. PLoS One. 2008;3:e3378. doi: 10.1371/journal.pone.0003378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadio S, Pluchino S, Brini E, Morana P, Guerriero R, Boneschi FM, Comi G, Zaratin P, Muzio V, del Carro U. Motor evoked potentials in a mouse model of chronic multiple sclerosis. Muscle Nerve. 2006;33:265–273. doi: 10.1002/mus.20463. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Filì L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol. 1998;8:923–926. doi: 10.1016/s0960-9822(07)00373-9. [DOI] [PubMed] [Google Scholar]
- Ayers MM, Hazelwood LJ, Catmull DV, Wang D, McKormack Q, Bernard CC, Orian JM. Early glial responses in murine models of multiple sclerosis. Neurochem Int. 2004;45:409–419. doi: 10.1016/j.neuint.2003.08.018. [DOI] [PubMed] [Google Scholar]
- Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ plarize CD4+TH-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Balch SG, Greaves DR, Gordon S, McKnight AJ. Organization of the mouse macrophage C-type lectin (Mcl) gene and identification of a subgroup of related lectin molecules. Eur J Immunogenet. 2002;29:61–64. doi: 10.1046/j.1365-2370.2002.00266.x. [DOI] [PubMed] [Google Scholar]
- Bannerman P, Hahn A, Soulika A, Gallo V, Pleasure D. Astrogliosis in EAE spinal cord: derivation from radial glia, and relationships to oligodendroglia. Glia. 2007;55:57–64. doi: 10.1002/glia.20437. [DOI] [PubMed] [Google Scholar]
- Bannerman PG, Hahn A, Ramirez S, Morley M, Bönnemann C, Yu S, Zhang GX, Rostami A, Pleasure D. Motor neuron pathology in experimental autoimmune encephalomyelitis: studies in THY1-YFP transgenic mice. Brain. 2005;128:1877–1886. doi: 10.1093/brain/awh550. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Parratt JD, Cho ES, Prineas JW. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann Neurol. 2009;65:32–46. doi: 10.1002/ana.21524. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kinkel RP, Kidd G, Rudick RA, Trapp BD. Axonal loss in normal-appearing white matter in a patient with acute MS. Neurology. 2001;57:1248–1252. doi: 10.1212/wnl.57.7.1248. [DOI] [PubMed] [Google Scholar]
- Black JA, Liu S, Hains BC, Saab CY, Waxman SG. Long-term protection of central axons with phenytoin in monophasic and chronic-relapsing EAE. Brain. 2006;129:3196–3208. doi: 10.1093/brain/awl216. [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SJ, Wilcockson DC, Butchart AG, Perry VH, Anthony DC. Altered chemokine expression in the spinal cord and brain contributes to differential interleukin-1β-induced neutrophil recruitment. J Neurochem. 2002;83:432–441. doi: 10.1046/j.1471-4159.2002.01166.x. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Deacon RM, Jiang Y, Ferrari C, Pitossi FJ, Anthony DC. Overexpression of IL-1β by adenoviral-mediated gene transfer in the rat brain causes a prolonged hepatic chemokine response, axonal injury and the suppression of spontaneous behaviour. Neurobiol Dis. 2007;27:151–163. doi: 10.1016/j.nbd.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Carlson T, Kroenke M, Rao P, Lane TE, Segal B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med. 2008;205:811–823. doi: 10.1084/jem.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127:294–303. doi: 10.1093/brain/awh032. [DOI] [PubMed] [Google Scholar]
- Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Wilczak N, Leta R, Streetland C. Astrocytes in multiple sclerosis lack beta-2 adrenergic receptors. Neurology. 1999;53:1628–1633. doi: 10.1212/wnl.53.8.1628. [DOI] [PubMed] [Google Scholar]
- DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 2004;127:1009–1018. doi: 10.1093/brain/awh118. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diestel A, Aktas O, Hackel D, Hake I, Meier S, Raine CS, Nitsch R, Zipp F, Ullrich O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med. 2003;198:1729–1740. doi: 10.1084/jem.20030975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel K, Dhabhar FS, Sapolsky RM. Neurotoxic effects of polymorphonuclear granulocytes on hippocampal primary cultures. Proc Natl Acad Sci U S A. 2004;101:331–336. doi: 10.1073/pnas.0303510101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- Einstein O, Grigoriadis N, Mizrachi-Kol R, Reinhartz E, Polyzoidou E, Lavon I, Milonas I, Karussis D, Abramsky O, Ben-Hur T. Transplanted neural precursor cells reduce brain inflammation to attenuate chronic experimental autoimmune encephalomyelitis. Exp Neurol. 2006;198:275–284. doi: 10.1016/j.expneurol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Evangelou N, DeLuca GC, Owens T, Esiri MM. Pathological study of spinal cord atrophy in multiple sclerosis suggests limited role of local lesions. Brain. 2005;128:29–34. doi: 10.1093/brain/awh323. [DOI] [PubMed] [Google Scholar]
- Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony-stimulating factor and neutrophils—forgotten mediators of inflammatory disease. Nat Clin Pract Rheumatol. 2006;2:500–510. doi: 10.1038/ncprheum0291. [DOI] [PubMed] [Google Scholar]
- Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol. 2008;64:255–265. doi: 10.1002/ana.21436. [DOI] [PubMed] [Google Scholar]
- Fisniku LK, Chard DT, Jackson JS, Anderson VM, Altmann DR, Miszkiel KA, Thompson AJ, Miller DH. Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann Neurol. 2008;64:247–254. doi: 10.1002/ana.21423. [DOI] [PubMed] [Google Scholar]
- Folgueras AR, Fueyo A, García-Suárez O, Cox J, Astudillo A, Tortorella P, Campestre C, Gutiérrez-Fernández A, Fanjul-Fernández M, Pennington CJ, Edwards DR, Overall CM, López-Otín C. Collagenase-2 deficiency or inhibition impairs experimental autoimmune encephalomyelitis in mice. J Biol Chem. 2008;283:9465–9474. doi: 10.1074/jbc.M709522200. [DOI] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci U S A. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganter P, Prince C, Esiri MM. Spinal cord axonal loss in multiple sclerosis: a post-mortem study. Neuropathol Appl Neurobiol. 1999;25:459–467. doi: 10.1046/j.1365-2990.1999.00205.x. [DOI] [PubMed] [Google Scholar]
- Gibbard RJ, Morley PJ, Gay NJ. TLR8 endosomal conserved features in the extracellular domain of human toll-like receptor 8 are essential for pH-dependent signaling. J Biol Chem. 2006;281:27503–27511. doi: 10.1074/jbc.M605003200. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Goodin DS, Cohen BA, O'Connor P, Kappos L, Stevens JC, Stevens JC. Assessment: the use of natalizumab (Tysabri) for the treatment of multiple sclerosis (an evidence-based review) Neurology. 2008;71:766–773. doi: 10.1212/01.wnl.0000320512.21919.d2. [DOI] [PubMed] [Google Scholar]
- Gorden KK, Qiu XX, Binsfeld CC, Vasilakos JP, Alkan SS. Cutting edge: activation of murine TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol. 2006;177:6584–6587. doi: 10.4049/jimmunol.177.10.6584. [DOI] [PubMed] [Google Scholar]
- Gurney KJ, Estrada EY, Rosenberg GA. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003;23:6494–6506. doi: 10.1128/MCB.23.18.6494-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Maloney JL, Roth LM, Freeman MJ, Morgan D, Gordon MN. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia. 2006;53:382–391. doi: 10.1002/glia.20272. [DOI] [PubMed] [Google Scholar]
- Herrero-Herranz E, Pardo LA, Gold R, Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. 2008;30:162–173. doi: 10.1016/j.nbd.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Ishizu T, Osoegawa M, Mei FJ, Kikuchi H, Tanaka M, Takakura Y, Minohara M, Murai H, Mihara F, Taniwaki T, Kira J. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain. 2005;128:988–1002. doi: 10.1093/brain/awh453. [DOI] [PubMed] [Google Scholar]
- Jégou JF, Chan P, Schouft MT, Griffiths MR, Neal JW, Gasque P, Vaudry H, Fontaine M. C3d binding to the myelin oligodendrocyte glycoprotein results in an exacerbated experimental autoimmune encephalomyelitis. J Immunol. 2007;178:3323–3331. doi: 10.4049/jimmunol.178.5.3323. [DOI] [PubMed] [Google Scholar]
- Jones MV, Nguyen TT, Deboy CA, Griffin JW, Whartenby KA, Kerr DA, Calabresi PA. Behavioral and pathological outcomes in MOG 35–55 experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;199:83–93. doi: 10.1016/j.jneuroim.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Kira J. Neuromyelitis optica and asian phenotype of multiple sclerosis. Ann NY Acad Sci. 2008;1142:58–71. doi: 10.1196/annals.1444.002. [DOI] [PubMed] [Google Scholar]
- Kivisäkk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, Khoury SJ. Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol. 2009;65:457–469. doi: 10.1002/ana.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Bauer J, Djamshidian A, Weissert R, Wallstroem E, Stefferl A, Zimprich F, Olsson T, Linington C, Schmidbauer M, Lassmann H. Distribution of a calcium channel subunit in dystrophic axons in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain. 2001;124:1114–1124. doi: 10.1093/brain/124.6.1114. [DOI] [PubMed] [Google Scholar]
- Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, Ebers GC. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129:584–594. doi: 10.1093/brain/awh721. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007;220:251–269. doi: 10.1111/j.1600-065X.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–2712. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Inflammasomes: guardians of cytosolic sanctity. Immunol Rev. 2009;227:95–105. doi: 10.1111/j.1600-065X.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25:2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib SL, Leppert D, Clements J, Täuber MG. Matrix metalloproteinases contribute to brain damage in experimental pneumococcal meningitis. Infect Immun. 2000;68:615–620. doi: 10.1128/iai.68.2.615-620.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lo CF, Ning X, et al. Expression of NALP1 in cerebellar granule neurons stimulates apoptosis. Cell Signal. 2004;16:1013–1021. doi: 10.1016/j.cellsig.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Liu Z, Li Y, Zhang J, Elias S, Chopp M. Evaluation of corticospinal axon loss by fluorescent dye tracing in mice with experimental autoimmune encephalomyelitis. J Neurosci Methods. 2008;167:191–197. doi: 10.1016/j.jneumeth.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Diego RS, Weiner HL. Novel therapeutic strategies for multiple sclerosis—a multifaceted adversary. Nat Rev Drug Discov. 2008;7:909–925. doi: 10.1038/nrd2358. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lü J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Haynes RL, Sidman RL, Vartanian T. TLR8: an innate immune receptor in brain, neurons and axons. Cell Cycle. 2007;6:2859–2868. doi: 10.4161/cc.6.23.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Määttä JA, Sjöholm UR, Nygårdas PT, Salmi AA, Hinkkanen AE. Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998;90:162–175. doi: 10.1016/s0165-5728(98)00135-0. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, Lassmann H, Turnbull DM. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132:1161–1174. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, Al-Hayani A, Davies SN, Rasband MN, Olsson T, Moldenhauer A, Velhin S, Hohlfeld R, Meinl E, Linington C. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204:2363–2372. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T, Matsushita T, Kawano Y, Osoegawa M, Ochi H, Ishizu T, Minohara M, Kikuchi H, Mihara F, Ohyagi Y, Kira J. Heterogeneity of aquaporin-4 autoimmunity and spinal cord lesions in multiple sclerosis in Japanese. Brain. 2007;130:1206–1223. doi: 10.1093/brain/awm027. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- Mead RJ, Singhrao SK, Neal JW, et al. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol. 2002;18:458–465. doi: 10.4049/jimmunol.168.1.458. [DOI] [PubMed] [Google Scholar]
- Mead RJ, Neal JW, Griffiths MR, Linington C, Botto M, Lassmann H, Morgan BP. Deficiency of the complement regulator CD59a enhances disease severity, demyelination and axonal injury in murine acute experimental allergic encephalomyelitis. Lab Invest. 2004;84:21–28. doi: 10.1038/labinvest.3700015. [DOI] [PubMed] [Google Scholar]
- Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, Kim S, Raja SM, Shi L, Simon MM, Froelich CJ. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. 2008;29:720–733. doi: 10.1016/j.immuni.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Mildner A, Djukic M, Garbe D, Wellmer A, Kuziel WA, Mack M, Nau R, Prinz M. Ly-6G+CCR2− myeloid cells rather than Ly-6ChighCCR2+ monocytes are required for the control of bacterial infection in the central nervous system. J Immunol. 2008;181:2713–2722. doi: 10.4049/jimmunol.181.4.2713. [DOI] [PubMed] [Google Scholar]
- Miller DH, Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol. 2007;6:903–912. doi: 10.1016/S1474-4422(07)70243-0. [DOI] [PubMed] [Google Scholar]
- Moll NM, Cossoy MB, Fisher E, Staugaitis SM, Tucky BH, Rietsch AM, Chang A, Fox RJ, Trapp BD, Ransohoff RM. Imaging correlates of leukocyte accumulation and CXCR4/CXCL12 in multiple sclerosis. Arch Neurol. 2009;66:44–53. doi: 10.1001/archneurol.2008.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature. 1992;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- Moreno B, Hevia H, Santamaria M, Sepulcre J, Muñoz J, García-Trevijano ER, Berasain C, Corrales FJ, Avila MA, Villoslada P. Methylthioadenosine reverses brain autoimmune disease. Ann Neurol. 2006;60:323–334. doi: 10.1002/ana.20895. [DOI] [PubMed] [Google Scholar]
- Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, Galvan MD, Anderson AJ. Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes in vitro and in vivo after spinal cord injury. J Neuroinflamm. 2008;5:1–13. doi: 10.1186/1742-2094-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygårdas PT, Hinkkanen AE. Up-regulation of MMP-8 and MMP-9 activity in the BALB/c mouse spinal cord correlates with the severity of experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2002;128:245–254. doi: 10.1046/j.1365-2249.2002.01855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien K, Fitzgerald DC, Naiken K, Alugupalli KR, Rostami AM, Gran B. Role of the innate immune system in autoimmune inflammatory demyelination. Curr Med Chem. 2008;15:1105–1115. doi: 10.2174/092986708784221458. [DOI] [PubMed] [Google Scholar]
- O'Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol. 2007;179:958–966. doi: 10.4049/jimmunol.179.2.958. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci. 2000;113:3073–3084. doi: 10.1242/jcs.113.17.3073. [DOI] [PubMed] [Google Scholar]
- O'Malley HA, Shreiner AB, Chen GH, Huffnagle GB, Isom LL. Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Mol Cell Neurosci. 2009;40:143–155. doi: 10.1016/j.mcn.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signaling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Coderre E, Basak A, Chen A, Zamponi GW, Hameed S, Rehak R, Yin X, Trapp BD, Stys PK. Glutamate receptors on myelinated spinal cord axons: I. GluR6 kainate receptors. Ann Neurol. 2009a;65:151–159. doi: 10.1002/ana.21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouardouz M, Coderre E, Zamponi GW, Hameed S, Yin X, Trapp BD, Stys PK. Glutamate receptors on myelinated spinal cord axons: II. AMPA and GluR5 receptors. Ann Neurol. 2009b;65:160–166. doi: 10.1002/ana.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicci DG, Hammond KJ, Coquet J, Kyparissoudis K, Brooks AG, Kedzierska K, Keating R, Turner S, Berzins S, Smyth MJ, Godfrey DI. DX5/CD49b-positive T cells are not synonymous with CD1d-dependent NKT cells. J Immunol. 2005;175:4416–4425. doi: 10.4049/jimmunol.175.7.4416. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Petzold A, Gveric D, Groves M, Schmierer K, Grant D, Chapman M, Keir G, Cuzner L, Thompson EJ. Phosphorylation and compactness of neurofilaments in multiple sclerosis: indicators of axonal pathology. Exp Neurol. 2008;213:326–335. doi: 10.1016/j.expneurol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, Hoffman B, Morgan BP. Immunopathology of secondar-progressive multiple sclerosis. Ann Neurol. 2001;50:646–657. doi: 10.1002/ana.1255. [DOI] [PubMed] [Google Scholar]
- Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, Rochford CD, Brück W, Becher B. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Raine CS. The Dale E. McFarlin Memorial Lecture: the immunology of the multiple sclerosis lesion. Ann Neurol. 1994;36:S61–S72. doi: 10.1002/ana.410360716. [DOI] [PubMed] [Google Scholar]
- Rasmussen S, Wang Y, Kivisäkk P, Bronson RT, Meyer M, Imitola J, Khoury SJ. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing-remitting experimental autoimmune encephalomyelitis. Brain. 2007;130:2816–2829. doi: 10.1093/brain/awm219. [DOI] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Alvarez-Erviti L, Blesa FJ, Rodríguez-Oroz MC, Arina A, Melero I, Ramos LI, Obeso JA. Bone-marrow-derived cell differentiation into microglia: a study in a progressive mouse model of Parkinson's disease. Neurobiol Dis. 2007;28:316–325. doi: 10.1016/j.nbd.2007.07.024. [DOI] [PubMed] [Google Scholar]
- Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem. 2006;281:14971–14980. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Coffee P, Smith G, Liem RK, Brady ST, Black MM. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci. 2000;20:6849–6861. doi: 10.1523/JNEUROSCI.20-18-06849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudick RA, Miller D, Hass S, Hutchinson M, Calabresi PA, Confavreux C, Galetta SL, Giovannoni G, Havrdova E, Kappos L, Lublin FD, Miller DH, O'Connor PW, Phillips JT, Polman CH, Radue EW, Stuart WH, Wajgt A, Weinstock-Guttman B, Wynn DR, et al. Health-related quality of life in multiple sclerosis: effects of natalizumab. Ann Neurol. 2007;62:335–346. doi: 10.1002/ana.21163. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. interleukin-1β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proc Natl Acad Sci U S A. 2001;98:6295–6300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AM, Calvente R, Tassi M, Carrasco MC, Martín-Oliva D, Marín-Teva JL, Navascués J, Cuadros MA. Embryonic and postnatal developmen of microglial cells in the mouse retina. J Comp Neurol. 2008;506:224–239. doi: 10.1002/cne.21538. [DOI] [PubMed] [Google Scholar]
- Satpute-Krishnan P, DeGiorgis JA, Conley MP, Jang M, Bearer EL. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci U S A. 2006;103:16532–16537. doi: 10.1073/pnas.0607527103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Bucci C, Weis J, Kruttgen A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J Neurosci. 2005;25:10930–10940. doi: 10.1523/JNEUROSCI.2029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher U, Hesse A, Bogdan C. Minute numbers of contaminant Cd8+ T cells or CD11b+CD11c+ NK cells are the source of IFN-γ in IL-12/IL-18-stimulated mouse macrophage populations. Blood. 2005;105:1319–1328. doi: 10.1182/blood-2004-05-1749. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1β expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood–brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9309. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah VB, Huang Y, Keshwara R, Ozment-Skelton T, Williams DL, Keshvara L. Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J Immunol. 2008;180:2777–2785. doi: 10.4049/jimmunol.180.5.2777. [DOI] [PubMed] [Google Scholar]
- Shea TB, Chan WK. Regulation of neurofilament dynamics by phosphorylation. Eur J Neurosci. 2008;27:1893–1901. doi: 10.1111/j.1460-9568.2008.06165.x. [DOI] [PubMed] [Google Scholar]
- Shijie J, Takeuchi H, Yawata I, Harada Y, Sonobe Y, Doi Y, Liang J, Hua L, Yasuoka S, Zhou Y, Noda M, Kawanokuchi J, Mizuno T, Suzumura A. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis. Tohoku J Exp Med. 2009;217:87–92. doi: 10.1620/tjem.217.87. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:3602–3609. doi: 10.1167/iovs.07-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver LP, Dittel BN. T-cell-mediated disruption of the neuronal microtubule network: correlation with early reversible axonal dysfunction in acute experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:999–1011. doi: 10.2353/ajpath.2006.050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka MK, Pagarigan RR, Whitton JL. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J Immunol. 2000;164:2009–2015. doi: 10.4049/jimmunol.164.4.2009. [DOI] [PubMed] [Google Scholar]
- Smith E, Zarbock A, Stark MA, Burcin TL, Bruce AC, Foley P, Ley K. IL-23 is required for neutrophil homeostasis in normal and neutrophilic mice. J Immunol. 2007;179:8274–8279. doi: 10.4049/jimmunol.179.12.8274. [DOI] [PubMed] [Google Scholar]
- Smith E, Stark MA, Zarbock A, Burcin TL, Bruce AC, Vaswani D, Foley P, Ley K. IL-17α inhibits the expansion of IL-17a-producing T cells in mice through a “short-loop” inhibition via IL-17 receptor. J Immunol. 2008;181:1357–1364. doi: 10.4049/jimmunol.181.2.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG. Statistical methods. Ed 8. Ames, Iowa: Blackwell; 1989. [Google Scholar]
- Stadelmann C, Albert M, Wegner C, Brück W. Cortical pathology in multiple sclerosis. Curr Opin Neurol. 2008;21:229–234. doi: 10.1097/01.wco.0000318863.65635.9a. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Storch MK, Weissert R, Steffer A, Birnbacher R, Wallström E, Dahlman I, Ostensson CG, Linington C, Olsson T, Lassmann H. MHC gene related effects on microglia and macrophages in experimental autoimmune encephalomyelitis determine the extent of axonal injury. Brain Pathol. 2002;12:287–299. doi: 10.1111/j.1750-3639.2002.tb00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaro BG, Whitnall MH, Gainer H. Phosphorylation-dependent epitopes on neurofilament proteins and neurofilament densities differs in axons in the corticospinal and primary sensory dorsal column tracts in the rat spinal cord. J Comp Neurol. 1990;302:220–235. doi: 10.1002/cne.903020204. [DOI] [PubMed] [Google Scholar]
- Tanuma N, Sakuma H, Sasaki A, Matsumoto Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006;112:195–204. doi: 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- Trapani JA, Bird PI. A renaissance in understanding the multiple and diverse functions of granzymes? Immunity. 2008;29:665–667. doi: 10.1016/j.immuni.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Stys PK, et al. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;5:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Martinon F, Burns K. NALPS: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- van Vliet SJ, García-Vallejo JJ, van Kooyk Y. Dendritic cells and C-type lectin receptors: coupling innate to adaptive immune responses. Immunol Cell Biol. 2008;86:580–587. doi: 10.1038/icb.2008.55. [DOI] [PubMed] [Google Scholar]
- Vercellino M, Plano F, Votta B, Mutani R, Giordana MT, Cavalla P. Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol. 2005;64:1101–1107. doi: 10.1097/01.jnen.0000190067.20935.42. [DOI] [PubMed] [Google Scholar]
- Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, Mutani R, Giordana MT, Cavalla P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol. 2007;66:732–739. doi: 10.1097/nen.0b013e31812571b0. [DOI] [PubMed] [Google Scholar]
- von Vietinghoff S, Ley K. Homeostatic regulation of blood neutrophil counts. J Immunol. 2008;181:5183–5188. doi: 10.4049/jimmunol.181.8.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Ayers MM, Catmull DV, Hazelwood LJ, Bernard CC, Orian JM. Astrocyte-associated axonal damage in pre-onset stages of experimental autoimmune encephalomyelitis. Glia. 2005;51:235–240. doi: 10.1002/glia.20199. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease. Ann Neurol. 2009;65:239–248. doi: 10.1002/ana.21640. [DOI] [PubMed] [Google Scholar]
- Wells CA, Salvage-Jones JA, Li X, Hitchens K, Butcher S, Murray RZ, Beckhouse AG, Lo YL, Manzanero S, Cobbold C, Schroder K, Ma B, Orr S, Stewart L, Lebus D, Sobieszczuk P, Hume DA, Stow J, Blanchard H, Ashman RB. The macrophage-inducible C-type lectin, Mincle, is an essential component of the innate immune response to Candida albicans. J Immunol. 2008;180:7404–7413. doi: 10.4049/jimmunol.180.11.7404. [DOI] [PubMed] [Google Scholar]
- Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood. 2008;111:42–49. doi: 10.1182/blood-2007-07-099648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating rector that senses damaged cells. Nat Immunol. 2008;9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- Zehntner SP, Brickman C, Bourbonnière L, Remington L, Caruso M, Owens T. Neutrophils that infiltrate the central nervous system regulate T cell responses. J Immunol. 2005;174:5124–5131. doi: 10.4049/jimmunol.174.8.5124. [DOI] [PubMed] [Google Scholar]
- Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]