

Abstract
Background & Aims
Strictures occur in ~30% of patients with Crohn’s disease and are characterized by intestinal smooth muscle hyperplasia, hypertrophy, and fibrosis due to excess extracellular matrix production including collagen. Insulin-like Growth Factor-I (IGF-I) expression is increased in smooth muscle cells of the muscularis propria in Crohn’s disease and in animal models of Crohn’s disease, including TNBS-induced colitis. While upregulated IGF-I is conjectured to cause smooth muscle cell growth and collagen production in the inflamed intestine, its role in the development of fibrosis has not been directly demonstrated.
Methods
Colitis was induced in IGF-I(+/−) or wildtype C57BL/6J mice by rectal administration of TNBS or ethanol vehicle. After 7 days colonic smooth muscle cells were isolated and used to prepare RNA or protein lysates. Transcript levels of IGF-IEa, IGF binding protein (IGFBP)-3, IGFBP-5, TGF-β1 and collagen IαI were measured by quantitative RT-PCR. Corresponding protein levels were measured by western blot or ELISA. Fibrosis was measured using digital image analysis of Masson’s-trichrome stained histologic sections.
Results
In IGF-I(+/−) mice, which express significantly lower levels of IGF-I than wildtype, the response to TNBS-induced colitis: upregulation of IGF-I, IGFBP-3, IGFBP-5, and collagen IαI expression, the resulting collagen deposition, and fibrosis are all significantly diminished compared to C57BL/6J wildtype controls. TGF-β1 expression and its increase following TNBS administration are not altered in IGF-I(+/−) mice compared to wildtype.
Conclusions
The findings indicate IGF-I is a key regulator in intestinal smooth muscle of smooth hyperplasia and excess collagen production that leads to fibrosis and longterm to stricture formation.
Keywords: Stricture, Colitis, Crohn’s disease, smooth muscle
INTRODUCTION
Trinitrobenzene sulfonic acid (TNBS) induces chronic inflammation in murine colon that all portions of the wall, including muscularis propria, and is utilized as a model of Crohn’s disease 1. The response of smooth muscle cells of muscularis propria to inflammation, whether from TNBS or Crohn’s Disease, cellular hyperplasia and hypertrophy and increased extracellular matrix production, including collagen I, results in fibrosis 2, 3. In Crohn’s disease, expression of Insulin-like Growth Factor-I (IGF-I) is increased in the intestine including muscle cells of the muscularis propria 3, 4. Similar events occur in animal models of inflammation: TNBS-induced and dextran sulfate-induced colitis 5, 6 and peptidoglycan-polysaccharide-induced ileocolitis 7. IGF-I is an endogenous growth factor playing a central role in the growth and development of visceral and vascular smooth muscle. In human intestine, IGF-I endogenous to smooth muscle cells regulates muscle cell growth of the muscularis propria in normal intestine by simultaneously stimulating proliferation and inhibiting apoptosis, and also stimulates collagen-I production 8, 9. While increased expression of IGF-I in the muscularis propria in Crohn’s Disease and in animal models of colitis is conjectured to contribute to the resulting muscle hyperplasia and fibrosis that contributes to stricture formation, this paper is the first to demonstrate this directly 3, 4, 7, 10, 11.
IGF-I is produced by the liver, with the main circulating form encoded by the IGF-IEa splice variant in humans, rats and mice, and also by target tissues including intestinal smooth muscle 8, 12. Two lines of investigation suggest that the effects of IGF-I on the smooth muscle cells of the intestine are largely governed by endogenous IGF-I production. First, in mice with hepatic-deleted IGF-I, intestinal muscle develops normally 13. Second, mice over-expressing IGF-I develop hyperplasia of smooth muscle tissues including the intestinal muscularis propria 14, 15.
The effects of endogenous IGF-I in intestinal smooth muscle is mediated by the cognate IGF-I receptor tyrosine kinase coupled to activation of Erk1/2 and p70S6 kinase that jointly stimulate proliferation, and collagen I gene expression, and GSK-3β that inhibits apoptosis, 9, 16, 17. IGF-I stimulates collagen I expression via Erk1/2 in rat colon 18. We recently showed that αVβ3 integrin (the vitronectin receptor) and its ligands, vitronectin and fibronectin, are expressed by intestinal smooth muscle cells in humans and upregulated in Crohn’s disease strictures 19. Occupancy of αVβ3 integrin by these ligands increases the intensity and duration of IGF-I stimulated IGF-I receptor activation and mediates excess smooth muscle growth in patients with stricturing Crohn’s disease 4.
This study examines the role of endogenous IGF-I in the response of the smooth muscle cells of the murine muscularis propria to TNBS-induced colitis. Deletion of IGF-I or of its receptor is a lethal mutation 20, thus the current study was performed in IGF(+/−) heterozygous mice which have a ~70% reduction in IGF-I, and in C57BL/6J littermate controls. The results show that the effects of TNBS-induced colitis on the smooth muscle cells of the muscularis propria, upregulation of gene expression and protein levels of IGF-I, IGFBP-5 and IGFBP-3 and collagen IαI, smooth muscle cell growth and increased fibrosis are all significantly diminished in IGF-I(+/−) mice compared to C57BL/6J wildtype controls. The results delineate for the first time the crucial role that IGF-I has in mediating the responses of intestinal smooth muscle to inflammation, muscle cell hyperplasia and fibrosis, and suggest that that these two features that characterize the response of the murine colon to TNBS may also be relevant to stricturing Crohn’s disease which is characterized by similar pathophysiologic events.
METHODS
Induction of Intestinal Inflammation with TNBS in the Mouse Colon
Colonic smooth muscle cells were isolated from the circular muscle layer of IGF(+/−) mice or C57BL/6J wildtype littermate controls (Jackson Laboratories, Bar Harbor, ME) 7 days following induction of colitis with 6 mg 2,4,6 trinitrobenzene sulfonic acid (TNBS, Sigma, St Louis, MO) in 50% ethanol (v/v) or vehicle alone 2, 21–24. All animal treatments were approved by the Institutional Animal Care and Use Committee of VCU.
The severity of inflammation was assessed using two validated protocols 25–27: scoring of macroscopic damage and histological evaluation. Macroscopic damage scores measured the presence and severity of adhesions (score 0–2), the maximum thickness of the bowel wall (in mm), and the absence or presence of diarrhea (0–1). Histological scoring evaluated the extent of destruction of normal mucosal architecture (0–3), the presence and degree of cellular infiltration (0–3), the extent of muscle thickening (0–3), presence or absence of crypt abscesses (0–1) and the presence or absence of goblet cell mucus (0–1). The average thickness of the muscularis propria was measured using image scanning micrometry at 5 villous bases per section by blinded reviewers and reported in µm’s.
Isolation of Colonic Smooth Muscle Cells
Muscle cells were isolated from the circular muscle layer of colon as reported previously after dissection of the layer and enzymatic digestion in Dulbecco’s modification of Eagle’s medium with 0.0375% collagenase (type II), and 0.1% soybean trypsin inhibitor 8, 28, 29. Total RNA or whole cell lysates were prepared from isolated muscle cells. Cells isolated in this way possess a smooth muscle phenotype: immunostaining for smooth muscle markers but not fibroblast markers, expression of γ-enteric actin, and the physiologic characteristics of contractile intestinal smooth muscle cells 30. Other cell types including epithelial cells, endothelial cells, neurons, and interstitial cells of Cajal are not detected in these cultures 30.
RNA preparation and quantitative RT-PCR
Total RNA was prepared using RNAqueous™ prep kits (Ambion, Austin, TX). Quantitative real-time PCR (qRT-PCR) was used to measure the levels of expression of: IGF-IEa (NM_0184052), IGFBP-5 (NM_010518), IGFBP-3 (NM_008343), TGF-β1 (NM_011577) and Collagen Iα1 (NM_007742). First stand cDNA synthesis was performed using qScript™ cDNA preps kits (Quanta, Gaithersburg, MD). Quantitative RT-PCR was performed using TAQman master mix, 2.5 pmol each of the forward and reverse primers (Applied Biosystems, Foster City, CA), and 2 µl of cDNA in a GeneAmp 5700 Sequence Detection System (Applied Biosystems, Foster City, CA). Fluorescence values for amplicons were recorded and the cycle threshold determined. Results were reported using the 2−ΔΔCt method based on β-actin amplification which remained constant between control and TNBS-treated mice.
Preparation of whole cell lysates
Cell lysates were prepared from freshly isolated muscle cells as described previously in an immunoprecipitation buffer consisting of (in mM): 50 Tris-HCl (pH 7.5), 150 NaCl, 50 NaF, 1 Na orthovanadate, 1 dithiothreitol, 1 phenylmethylsulfonyl fluoride and 0.5% NP-40 to which was added 1 µg/ml leupeptin, 1 µg/ml pepstatin A, and 1 µg/ml aprotinin prior to immunoblot or ELISA 19, 31, 32.
Immnoblot Analysis
Collagen IαI, cleaved and total caspase-3, and PCNA were measured by immunoblot analysis using standard methods 17, 19. Proteins in lysates containing equal protein (BioRad DC kit, Hercules, CA) were separated with SDS-PAGE and electrotransferred to nitrocellulose prior to incubation with antibodies recognizing the protein of interest: Collagen IαI (C-18, 1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), cleaved caspase-3(Asp175) (1:1000 dilution, Cell Signaling Technologies, Beverly, MA), total caspase-3 (1:1000 dilution, Cell Signaling Technologies, Beverly, MA), PCNA (PC10, 1:2000 dilution, Cell Signaling Technologies, Beverly, MA), or β-actin (1:1000 dilution, Cell Signaling Technologies, Beverly, MA). Bands were visualized with enhanced chemiluminescence using a FluoChem 8800 (Alpha Innotech, San Leandro, CA) and the digital images quantified using AlphaEaseFC version 3.1.2 software. Densitometric values for protein bands of were reported in arbitrary units above background values after normalization to total protein levels or β-actin.
Measurement of apoptosis
Apoptosis in smooth muscle cells was measured as described previously4. Briefly, levels of cleaved caspase-3(Asp-175) and of total caspase-3 were measured by immunoblot analysis as above and normalized to β-actin levels. The ratio of cleaved caspase-3/total caspase-3 was used as a measure of apoptosis in cells.
Measurement of IGF-I, IGFBP-3 and TGF-β1 by ELISA
IGF-I, IGFBP-5, IGFBP-3 and TGF-β1 levels were measured in lysates prepared from colonic muscle cells isolated from TNBS-treated or vehicle treated IGF-I(+/−) mice or C57BL/6J wildtype mice 32. Proteins were measured in samples containing equal protein using specific ELISAs for murine IGF-I, IGFBP-3, IGFBP-5 or TGF-β1 (R&D Systems, Minneapolis, MN) that have no appreciable cross-reactivity, for IGF-I: with IGF-II, insulin or IGF binding proteins; for IGFBPs: with IGF-I, IGF-II, insulin or other IGF binding proteins; and for TGF-β1: with TGF-β2, TGF-β3, activins, inhibins or BMP’s, respectively. Results were calculated as ng peptide/mg protein.
Masson’s Trichrome Staining
Masson’s trichrome staining was used as a measure of collagen deposition and fibrosis. Briefly, 7 µm sections were fixed in 4% fresh paraformaldehyde and then 20% sucrose prior to staining with Masson’s-trichrome (Sigma-Aldrich, St Louis, MO). Microscopic images were digitized, and the area positive for collagen staining determined by Red-Green-Blue (RGB) segmentation using fixed thresholding values according to Ortolan et al 33. Digital images were analyzed using AlphaEaseFC version 3.1.2 software. Results were calculated as collagen area within the muscularis propria in each of three consecutive microscopic fields per section measured by a blinded reviewer and reported as percent of the total muscularis propria area.
Statisical analysis
Values represent means ± SE of n experiments, where n represents the number of experiments on separate animals or on cells derived from separate animals. Statistical significance was tested by Student’s t-test for either paired or unpaired data as appropriate.
RESULTS
TNBS-induced colitis
Mean macroscopic (Fig 1A) and histologic scores (Fig 1B and C) on day 7 were similar between vehicle treated wildtype C57BL/6J mice and IGF+/− mice. The increase in both macroscopic (Fig 1A) and histologic scores in response to TNBS was significantly lower in IGF-I(+/−) mice compared to wildtype mice (Fig 1B and C). The thickness of the muscularis propria in vehicle treated mice was unchanged between wildtype mice and IGF-I(+/−) mice, 59 ± 2 µM and 58 ± 2 µm, respectively. The thickness of the muscularis propria layer following TNBS treatment was significantly different between wildtype mice, 155 ± 3 µm, and IGF-I(+/−) mice, 93 ± 3 µm (Fig 1C and 1D).
Figure 1. Responses to TNBS-induced colitis are decreased in IGF(+/−) mice.
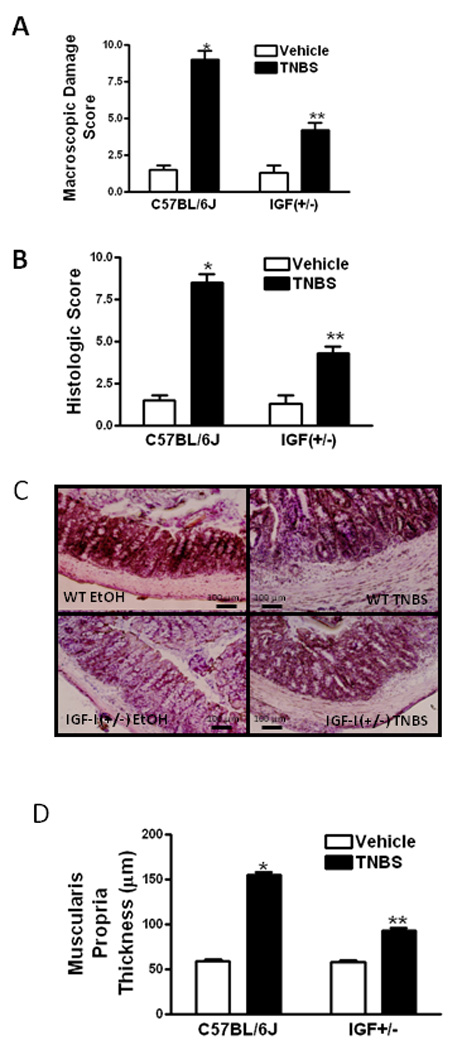
Macroscopic damage scores (Panel A) and histologic scores (Panel B) in TNBS-induced colitis were diminished in IGF-I(+/−) mice compared to wildtype mice. Macroscopic damage scores and histologic scores were measured 7 days following TNBS administration using validated measures 25, 27. Panel C: Representative hematoxylin and eosin (H&E) stained sections of colons from IGF-I(+/−) and wildtype (WT) mice 7 days after TNBS or vehicle administration. Panel D: The increase in muscularis propria thickness in response to TNBS was diminished in IGF-I(+/−) mice compare to wildtype mice as measured by image scanning micrometry. Values represent means ± SE of 8–11 animals in each group. * denotes P < 0.05 vs. vehicle-treated animals, ** denotes P < 0.05 vs wildtype mice.
Diminished IGF-I levels in IGF-I(+/−) mice
Smooth muscle cells of the mouse intestine express the IGF-IEa gene from which IGF-I is produced by alternative splicing. Expression of IGF-IEa increased 3.4 ± 0.3 fold in colonic smooth muscle cells of wildtype mice 7 days following TNBS administration compared with vehicle treated mice. IGF-IEa transcript levels were 0.60 ± 0.08 fold lower in vehicle treated IGF-I(+/−) mice than in wildtype mice. Following TNBS administration, IGF-IEa expression increased only 1.42 ± 0.10 fold (Fig 2A).
Figure 2. TNBS-induced upregulated IGF-I expression and production is decreased in IGF-I(+/−) mice.
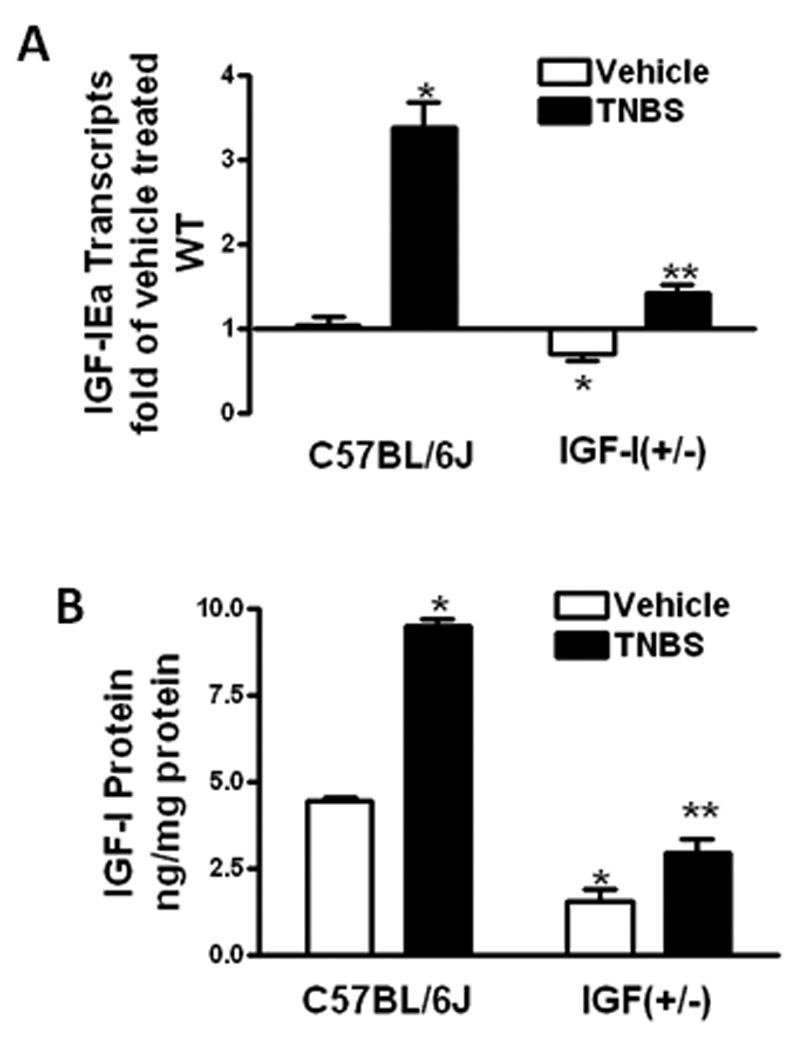
Panel A: IGF-IEa transcripts in muscle cells of vehicle treated IGF-I(+/−) mice were lower than wildtype mice. The increase IGF-IEa transcript levels with TNBS administration in IGF-I(+/−) mice was lower than in wildtype mice. IGF-IEa transcripts were measured by qRT-PCR by the 2−ΔΔCt method using β-actin as control. Panel B: IGF-I protein levels in muscle cells of vehicle treated IGF-I(+/−) mice were lower than in wildtype mice. The increase IGF-I in IGF-I(+/−) mice with TNBS treatment was lower than in wildtype mice. IGF-I was measured by ELISA. Results were expressed as ng/mg protein. Values represent the mean ± SE of 8–11 animals in each group. * denotes P < 0.05 vs vehicle treated wildtype mice, ** denotes P < 0.05 vs TNBS treated wildtype mice.
IGF-I protein levels in vehicle treated wildtype mice, 4.48 ± 0.10 ng/mg protein, also increased, to 9.52 ± 0.21 ng/mg protein, with TNBS treatment. IGF-I protein levels were lower in vehicle treated IGF-I(+/−) mice, 1.36 ± 0.36 ng/mg protein, than in wildtype mice and increased only to 2.97 ± 0.37 ng/mg protein after TNBS administration (Fig 2B).
TNBS-induced upregulation of IGFBP-5 and IGFBP-3
Intestinal smooth muscle cells express IGFBP-5 which exerts both IGF-I-dependent effects on smooth muscle cell hyperplasia and IGF-I-independent effects on smooth muscle cell hyperplasia and collagen production 34. IGFBP-5 expression in colonic smooth muscle cells of vehicle treated wildtype mice increased by 2.6 ± 0.2 fold with TNBS treatment. IGFBP-5 expression was 0.8 ± 0.3 fold lower in vehicle treated IGF-I(+/−) mice than in wildtype vehicle treated controls and increased only 0.9 ± 0.2 fold following treatment with TNBS (Fig 3A).
Figure 3. TNBS-induced upregulated IGFBP-5 and IGFBP-3 expression and production is decreased in IGF-I(+/−) mice.
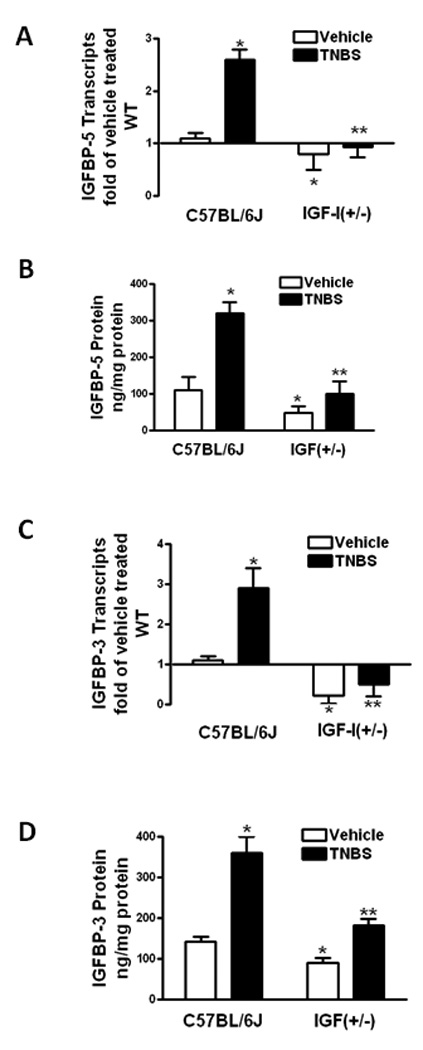
Panel A: IGFBP-5 transcripts in muscle cells of vehicle treated IGF-I(+/−) mice were lower than in wildtype mice. The increase in IGFBP-5 transcripts with TNBS treatment in IGF-I(+/−) mice was lower than wildtype mice. IGFBP-5 transcripts were measured by qRT-PCR by the 2−ΔΔCt method using β-actin as control. Panel B: IGFBP-5 protein levels in muscle cells of vehicle treated IGF-I(+/−) mice were lower than in wildtype mice. The increase in IGFBP-5 levels with TNBS treatment in IGF-I(+/−) mice was lower than in wildtype mice. IGFBP-5 was measured by ELISA. Panel C: IGFBP-3 transcripts in muscle cells of vehicle treated IGF-I(+/) mice were lower than in C57BL/6J wildtype mice. The increase in IGFBP-3 transcripts with TNBS treatment in IGF-I(+/−) mice was lower than in wildtype mice. IGFBP-3 transcripts were measured by qRT-PCR by the 2−ΔΔCt method using β-actin as control. Panel D: IGFBP-3 protein levels in muscle cells of vehicle treated IGF-I(+/−) mice were lower than in C57BL/6J wildtype mice. The increase in IGFBP-3 protein with TNBS treatment in IGF-I(+/−) mice was lower than in wildtype mice. IGFBP-3 was measured by ELISA. Results were expressed as ng/mg protein. Results were expressed as ng/mg protein. Values represent the mean ± SE of 8–11 animals in each group. * denotes P < 0.05 vs vehicle treated wildtype mice, ** denotes P < 0.05 vs TNBS treated wildtype mice.
IGFBP-5 protein levels in vehicle treated wildtype mice, 110 ± 36 ng/mg protein, were increased to 320 ± 30 ng/mg protein with TNBS treatment. In IGF-I(+/−) mice levels of IGFBP-5 protein were lower in vehicle treated mice, 49 ± 18 ng/mg protein, than in wildtype and increased to 100 ± 35 ng/mg protein with TNBS treatment (Fig 3B).
Intestinal smooth muscle cells express IGFBP-3 which exerts both IGF-I-dependent effects on smooth muscle cell hyperplasia and IGF-I-independent effects on collagen production 35. IGFBP-3 expression in colonic smooth muscle cells of vehicle treated wildtype increased by 2.9 ± 0.5 fold with TNBS treatment. IGFBP-3 expression was 2.1 ± 0.4 fold lower in vehicle treated IGF-I(+/−) mice than in wildtype vehicle treated controls and increased only 1.5 ± 0.3 fold with TNBS treatment (Fig 3C).
IGFBP-3 protein levels in vehicle treated wildtype mice, 142 ± 13 ng/mg protein, were increased to 360 ± 40 ng/mg protein with TNBS treatment. In IGF-I(+/−) mice levels of IGFBP-3 protein were lower in vehicle treated mice, 91 ± 12 ng/mg protein, than in wildtype and increased to 183 ± 15 ng/mg protein with TNBS treatment (Fig 3D).
TNBS-induced upregulation of TGF-β1
Intestinal smooth muscle cells express TGF-β1 which stimulates expression and production of collagen IαI and IGFBP-3 in these cells. TGF-β1 expression levels were the same in vehicle treated IGF-I(+/−) mice and wildtype controls (Fig 5A). Treatment with TNBS induced a similar increase in TGF-β1 expression in IGF(+/−) mice, 2.84 ± 0.17 fold increase, and wildtype mice, 2.95 ± 0.21 fold increase (Fig 4A).
Figure 5. TNBS-induced collagen IαI expression and fibrosis are decreased in muscle cells of IGF-I(+/−) mice.
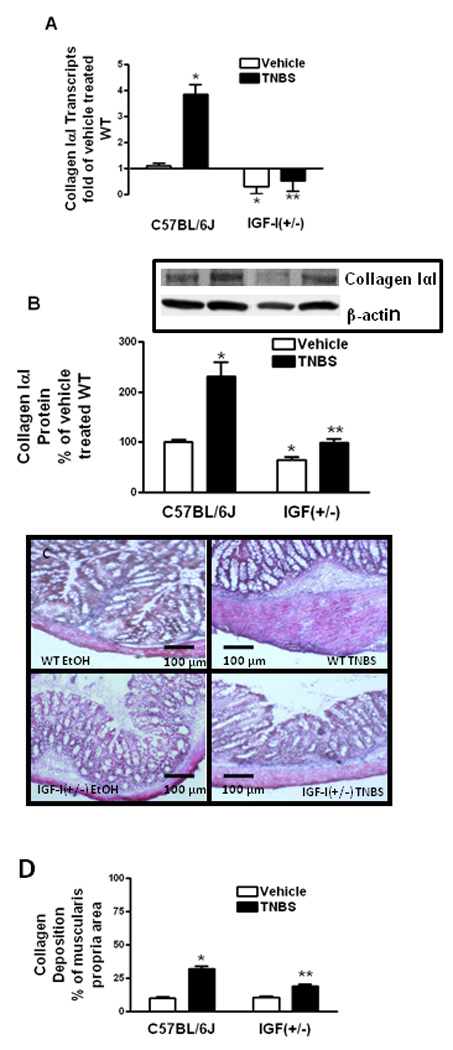
Panel A: Collagen IαI transcripts in muscle cells of vehicle treated IGF-I(+/−) mice and the increase in response to TNBS were lower than in wildtype mice. Collagen IαI transcripts were measured by qRT-PCR by the 2−ΔΔCt method using β-actin as control. Panel B: Collagen IαI protein levels in muscle cells of vehicle treated IGF-I(+/−) mice and the increase in response to TNBS were lower than in wildtype mice. Inset: Representative immunoblots of collagen IαI and β-actin. Results were expressed as relative levels after normalization to β-actin levels. Panel C: Representative Masson’s-trichrome stained sections of colons from IGF-I(+/−) and wildtype (WT) mice 7 days after rectal administration of TNBS or vehicle. Panel D: Collagen levels are in the muscularis propria of vehicle treated IGF(+/−) mice are similar to wildtype mice but significantly lower amounts of collagen deposition occur in IGF(+/−) mice with TNBS treatment. Results were expressed as percent of muscularis propria area. Values represent the mean ± SE of 8–11 animals in each group. * denotes P < 0.05 vs vehicle treated wildtype mice, ** denotes P < 0.05 vs TNBS treated wildtype mice.
Figure 4. TNBS-induced TGF-β1 expression and production is unchanged in muscle cells of IGF-I(+/−) mice.
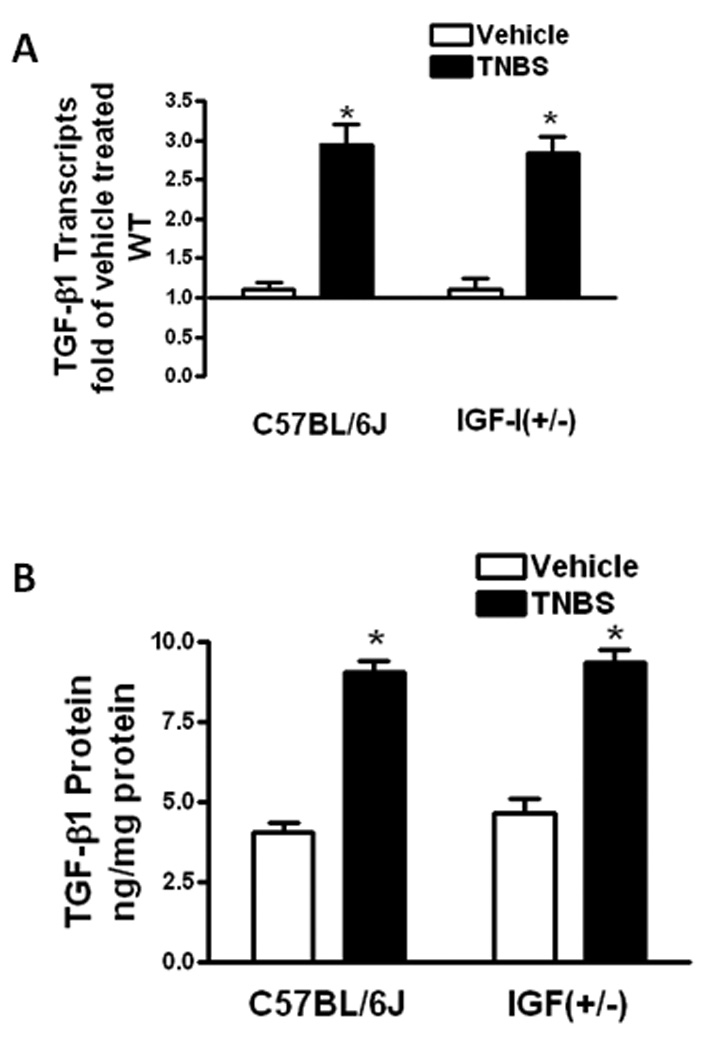
Panel A: TGF-β1 transcripts in muscle cells of vehicle treated IGF-I(+/−) mice and wildtype mice were similar. TGF-β1 transcript levels were increased to a similar extent in wildtype and IGF-I(+/−) mice with TNBS treatment. TGF-β1 transcripts were measured by qRT-PCR by the 2−ΔΔCt method using β-actin as control. Panel B: TGF-β1 protein levels in muscle cells of vehicle treated IGF-I(+/−) mice and wildtype mice were similar. TGF-β1 protein levels increased to a similar extent with TNBS treatment in wildtype and IGF-I(+/−) mice. TGF-β1 was measured by ELISA. Results were expressed as ng/mg protein. Values represent the mean ± SE of 8–11 animals in each group. * denotes P < 0.05 vs vehicle treated wildtype mice.
TGF-β1 protein levels in vehicle treated wildtype mice, 4.08 ± 0.29 ng/mg protein, increased to 9.06 ± 0.34 ng/mg protein with TNBS treatment. In IGF-I(+/−) mice levels of TGF-β1 protein were similar to vehicle treated wildtype, 4.65 ± 0.45 ng/mg protein, as was the increase in TGF-β1 levels to 9.35 ± 0.41 ng/mg protein with TNBS treatment (Fig 4B).
TNBS-induced collagen IαI expression and fibrosis
Collagen I is the main isoform in the intestine. In intestinal smooth muscle cells in vitro collagen IαI expression is positively regulated by IGF-I, IGFBP-5 and IGFBP-3 and by TGF-β112, 34–36. Collagen IαI expression in wildtype mice increased 3.86 ± 0.36 fold after TNBS treatment compared to vehicle treatment. Collagen IαI expression was 1.4 ± 0.4 fold lower in vehicle treated IGF-I(+/−) mice than in wildtype vehicle treated mice, and increased only 1.8 ± 0.4 fold with TNBS treatment (Fig 5A).
Collagen IαI protein levels in vehicle treated wildtype mice increased 131 ± 29% with TNBS treatment (Fig 6B). In IGF-I(+/−) mice collagen IαI protein was 65 ± 5% of that in vehicle treated wildtype mice, and increased only 52 ± 6% 7 days with TNBS treatment (Fig 5B).
Figure 6. TNBS-induced smooth muscle cell proliferation is decreased in IGF-I(+/−) mice.
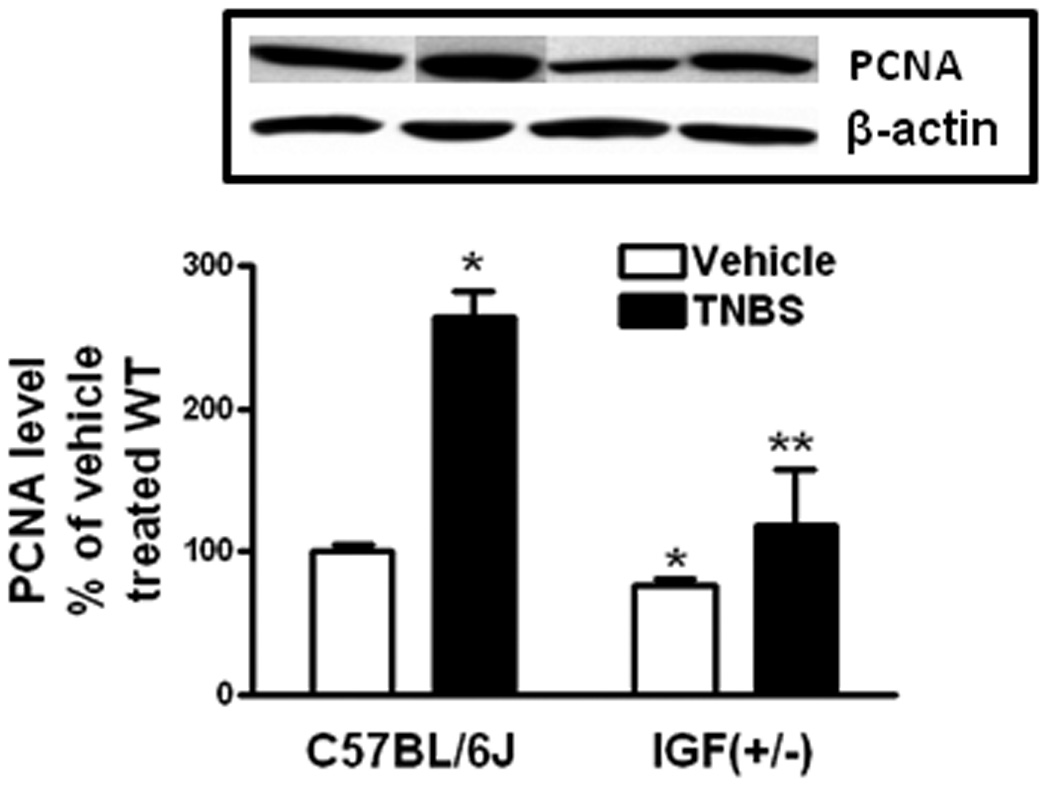
Levels of PCNA in muscle cells of vehicle treated IGF-I(+/−) were lower than in vehicle treated wildtype mice. The increase in proliferating muscle cells with TNBS treatment was lower in IGF-I(+/−) mice. Inset: representative immunoblots of PCNA and β-actin levels. Results were expressed as levels relative to vehicle treated wildtype mice after normalization of β-actin levels. Values represent the mean ± SE of 11 animals in each group. * denotes P < 0.05 vs vehicle treated wildtype mice, ** denotes P < 0.05 vs TNBS treated wildtype mice.
Since the expression of the pro-fibrotic factors: IGF-I, IGFBP-5, IGFBP-3, was decreased in IGF-I(+/−) mice although expression of another major pro-fibrotic factor, TGF-β1, was not altered, we hypothesized that diminished collagen IαI production in response to TNBS would be accompanied by diminished fibrosis. This was tested using Masson’s-trichrome staining and measurement of collagen deposition and fibrosis within the muscularis. In vehicle-treated wildtype and IGF-I(+/−) mice, fibrosis measurements were similar (Fig. 5C). Increased fibrosis in wildtype mice with TNBS treatment was lower in IGF-I(+/−) mice treated with TNBS (Fig. 5C). Collagen deposition within the muscularis propria of vehicle treated IGF-I(+/−) mice, 10.6 ± 0.7% of total area, was similar to wildtype mice, 10.1 ± 0.6% of total area (Fig. 5D). Less collagen deposition and fibrosis occurred in IGF-I(+/−) mice with TNBS treatment: 19.1 ± 1.7% of total area, than in wildtype mice: 32.1 ± 2.3% of total area. It is worth noting that the lower levels of fractional collagen deposition occurring in IGF-I(+/−) mice per cross-sectional area occurred in addition to the diminished expansion of the muscularis propria in TNBS-treated IGF-I(+/−) mice compared to wildtype (Fig 1C and D).
Muscle cell proliferation following TNBS-induced colitis
Proliferation of muscle cells was measured from the levels of proliferating cell nuclear antigen (PCNA) present. In wildtype mice proliferation increased 264 ± 18% with TNBS treatment over that in vehicle treated wildtype mice. In contrast, PCNA levels in smooth muscle cells of IGF-I(+/−) mice treated with TNBS increased by only 156 ± 40% over that observed in vehicle treated IGF-I(+/−) mice (Fig. 6).
Muscle cell apoptosis following TNBS-induced colitis
In strictured intestinal muscle of Crohn’s disease when IGF-I expression increases, muscle cell apoptosis decreases 4. We hypothesized that endogenous IGF-I regulated smooth muscle growth in TNBS-induced colitis in part by regulating apoptosis.
Relative cleaved caspase-3/caspase-3 levels in vehicle treated wildtype mice, 4.13 ± 0.47, decreased to 0.67 ± 0.25 with TNBS treatment (Fig 7). In vehicle treated IGF-I(+/−) mice relative cleaved caspase-3/caspase-3 levels, 1.25 ± 0.33, were lower than wildtype mice and decreased further to 0.45 ± 0.2 with TNBS treatment (Fig. 7).
Figure 7. TNBS-induced decreased apoptosis in smooth muscle cells is accentuated in IGF-I(+/−) mice.
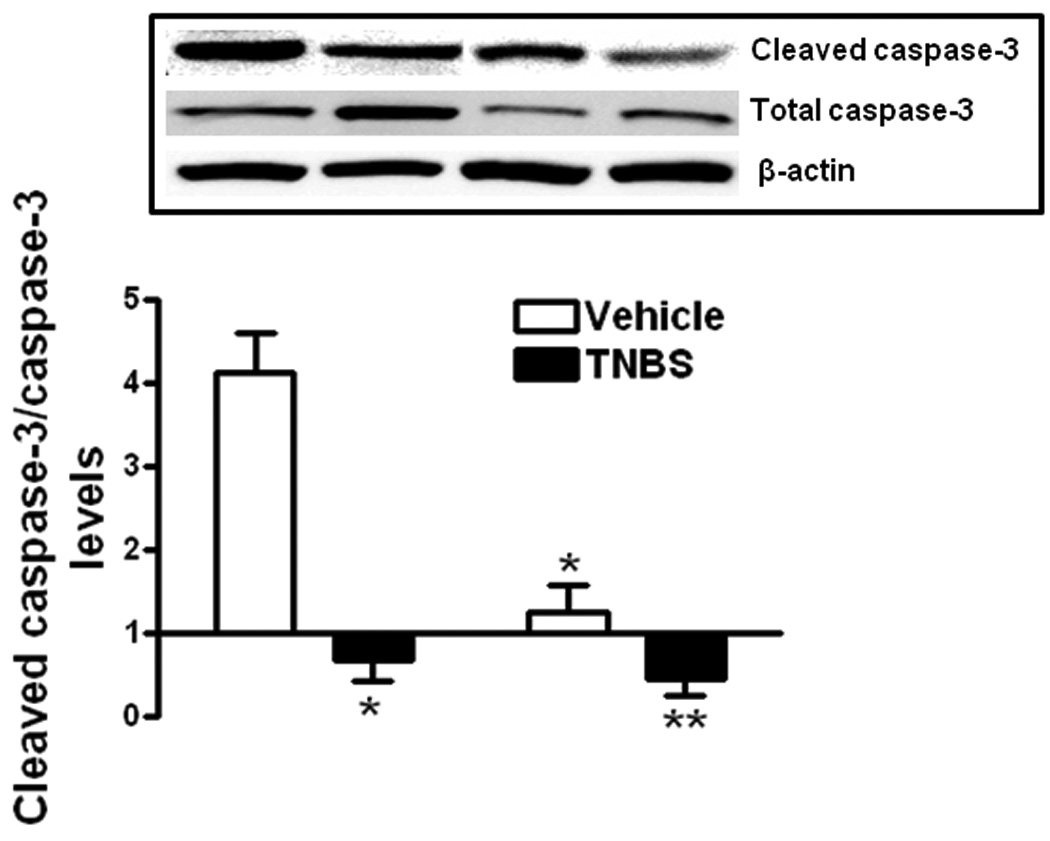
Levels of apoptosis in muscle cells of vehicle treated IGF-I(+/−) mice were lower than in vehicle treated wildtype mice. Apoptosis was decreased further in wildtype and IGF-I(+/−) mice with TNBS treatment compared to vehicle treated mice. The decrease with TNBS treatment in IGF-I(+/−) mice was lower than in wildtype mice. Inset: Representative immunoblots of cleaved caspase-3(Asp-175), total caspase-3 and β-actin. Apoptosis was measured cleaved caspase-3(Asp-175)/total caspase-3 levels and results expressed as the relative levels after normalization to β-actin levels. Values represent the mean ± SE of 8 animals in each group. * denotes P < 0.05 vs vehicle treated wildtype mice, ** denotes P < 0.05 vs TNBS treated wildtype mice.
DISCUSSION
TNBS-induced colitis is a model for Crohn’s disease sharing similar pathophysiologic processes including a close parallelism in the responses of the muscularis propria in TNBS-induced colitis and of the muscularis propria in stricturing Crohn’s disease. In both Crohn’s disease and TNBS-induced colitis, three pathophysiologic events contribute to stricture formation: smooth muscle hyperplasia, smooth muscle cell hypertrophy and excess net extracellular matrix production including collagen I that results in fibrosis. Common to both are upregulation of IGF-I, IGFBP-5 and IGFBP-3 and of TGF-β1 specifically by smooth muscle cells of the muscularis propria 3, 4, 27. This paper demonstrates for the first time the functional role of IGF-I in regulating the responses of muscularis propria smooth muscle to TNBS-induced colitis including: increased muscle hyperplasia, excess collagen IαI production and increased fibrosis.
Evidence supporting the role of IGF-I in the responses of smooth muscle to TNBS-induced colitis are as follows. 1. IGF-IEa expression and IGF-I protein levels were significantly lower in IGF-I(+/−) mice than wildtype mice and their upregulation in response to TNBS was lower than in wildtype mice; 2. Macroscopic damage scores and histologic scores including muscle thickness in TNBS-treated mice were lower in IGF-I(+/−) mice than in wildtype mice; 3. IGFBP-5 and IGFBP-3 expression and protein levels were lower in IGF-I(+/−) mice than in wildtype and their upregulation in response to TNBS was lower than in wildtype mice; 4. The numbers of proliferating muscle cells in the muscularis propria of TNBS-treated mice was lower in IGF-I(+/) mice than in wildtype mice; 5. The levels of apoptosis in the muscle cells of the muscularis propria of IGF-I(/+−) mice was lower in wildtype mice and were decreased further following TNBS treatment; 6. The levels of collagen IαI expression and production by muscle cells of the muscularis propria of IGF-I(+/−) mice was lower than in wildtype mice as was the increase in response to TNBS. 7. The increase in fibrosis within the muscularis propria in response to TNBS was diminished in IGF-I(+/−) mice compared to wildtype mice.
The importance of understanding the mechanisms operative within the intestine leading to fibrosis is underscored by the discrepancy between the levels of these mediators in the serum compared to the muscle layer of intestine. Serum levels of hepatic-derived IGF-I, IGFBP-5 and IGFBP-3, are lower in patients with active Crohn’s disease than in normal subjects, whereas the expression of IGF-I, IGFBP-5 and IGFBP-3 by smooth muscle cells in strictured intestine is increased compared to adjacent non-strictured intestinal muscle from the resection margin 4, 7, 35. The central role of IGF-I in this process is highlighted by the diminished smooth muscle growth and fibrosis in response to TNBS-induced colitis in IGF-I(+/−) mice compared to wildtype mice. These effects are mediated directly, via activation of the cognate IGF-I receptor, and likely indirectly via IGF-I-dependent upregulation of IGFBP-5, which stimulates muscle growth 32, 34, and IGFBP-3 which like IGF-I and IGFBP-5 increases collagen IαI production by muscle 2, 12, 34, 37. The effects of IGF-I revealed by using IGF-I(+/−) mice on levels of collagen IαI and fibrosis are specific since no alteration in TGF-β1 expression or protein levels was detected in vehicle or TNBS treated IGF-I(+/−) mice. The results indicate that while TGF-β1-dependent collagen IαI production in smooth muscle is one regulator of fibrosis in colitis, a significant component of the fibrosis that occurs in the intestine is due to IGF-I-dependent collagen IαI expression either directly or indirectly by the resultant upregulation of IGFBP-3 and IGFBP-5.
The response to specific mouse strains to TNBS colitis can be different. It is worth noting that the response of C57BL/6J wildtype mice and the IGF-I(+/−) mice on this genetic background to TNBS administration with upregulation of IGF-I and TGF-β1, IGF binding proteins and fibrosis are similar to the muscle cells of stricturing Crohn’s disease and dissimilar to other mouse strains such as BALB/c which respond in sequential fashion over time 38. Whereas some processes are similar, such as the progression of strictures after cessation of chronic TNBS administration in the absence of inflammation. TGF-β1 induces IGF-I expression in subepithelial myofibroblasts of TNBS-treated BALB/c mice and in an immortalized cell line of human intestinal fibroblasts 1, 38, 39. TGF-β1 also induces IGF-I expression in intestinal smooth muscle cells of C57BL/6J mice (unpublished observation). Therapeutic disruption of TGF-β, however, must be highly selective as targeted disruption of TGF-β1, or its signaling pathways: Smad2 or Smad4, are lethal mutations.
In summary, this paper demonstrates for the first time the key role that IGF-I plays in the development of two events that occur in intestinal muscle in response to inflammation. Directly or indirectly, via IGF binding proteins, IGF-I causes increased smooth muscle cell hyperplasia, increased collagen IαI production and fibrosis in TNBS-induced colitis. The clinical significance of these findings lie in the similar pathophysiologic processes involved in the muscle of the muscularis propria in stricturing Crohn’s disease. This study further suggests that given the crucial role of IGF-I in this process, inhibitors of IGF-I may be an effective pharmacologic strategy in patients with stricturing Crohn’s disease by virtue of their potential to impact two factors involved in stricture formation: muscle cell hyperplasia and fibrosis.
Acknowledgments
Grant support: This work was supported by DK-49691 from the National Institute of Diabetes, Digestive and Kidney Diseases
Footnotes
Disclosures: No conflicts of interest exist
REFERENCES
- 1.Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK, Schlitt HJ, Kitani A, Strober W. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007;178:5859–5870. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- 2.Zeeh JM, Riley NE, Hoffmann P, Reinshagen M, Goebell H, Gerken G. Expression of insulin-like growth factor binding proteins and collagen in experimental colitis in rats. Eur J Gastroenterol Hepatol. 2001;13:851–858. doi: 10.1097/00042737-200107000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Zimmermann EM, Li L, Hou YT, Mohapatra NK, Pucilowska JB. Insulin-like growth factor I and insulin-like growth factor binding protein 5 in Crohn's disease. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1022–G1029. doi: 10.1152/ajpgi.2001.280.5.G1022. [DOI] [PubMed] [Google Scholar]
- 4.Flynn RS, Murthy KS, Grider JR, Kellum JM, Kuemmerle JF. Endogenous IGF-I and [alpha]V[beta]3 Integrin Ligands Regulate Increased Smooth Muscle Hyperplasia in Stricturing Crohn's Disease. Gastroenterology. 2010;138:285–293. doi: 10.1053/j.gastro.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeeh JM, Hoffmann P, Sottili M, Eysselein VE, McRoberts JA. Up-regulation of insulinlike growth factor I binding sites in experimental colitis in rats. Gastroenterology. 1995;108:644–652. doi: 10.1016/0016-5085(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 6.Savendahl L, Underwood LE, Haldeman KM, Ulshen MH, Lund PK. Fasting prevents experimental murine colitis produced by dextran sulfate sodium and decreases interleukin-1 beta and insulin-like growth factor I messenger ribonucleic acid. Endocrinology. 1997;138:734–740. doi: 10.1210/endo.138.2.4941. [DOI] [PubMed] [Google Scholar]
- 7.Zimmermann EM, Sartor RB, McCall RD, Pardo M, Bender D, Lund PK. Insulinlike growth factor I and interleukin 1 beta messenger RNA in a rat model of granulomatous enterocolitis and hepatitis. Gastroenterology. 1993;105:399–409. doi: 10.1016/0016-5085(93)90713-m. [DOI] [PubMed] [Google Scholar]
- 8.Kuemmerle JF. Autocrine regulation of growth in cultured human intestinal muscle by growth factors. Gastroenterology. 1997;113:817–824. doi: 10.1016/s0016-5085(97)70176-8. [DOI] [PubMed] [Google Scholar]
- 9.Kuemmerle JF. Endogenous IGF-I protects human intestinal smooth muscle cells from apoptosis by regulation of GSK-3 beta activity. Am J Physiol Gastrointest Liver Physiol. 2005;288:G101–G110. doi: 10.1152/ajpgi.00032.2004. [DOI] [PubMed] [Google Scholar]
- 10.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 11.Louis E, Collard A, Oger AF, Degroote E, Aboul Nasr El Yafi FA, Belaiche J. Behaviour of Crohn's disease according to the Vienna classification: changing pattern over the course of the disease. Gut. 2001;49:777–782. doi: 10.1136/gut.49.6.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann EM, Li L, Hou YT, Cannon M, Christman GM, Bitar KN. IGF-I induces collagen and IGFBP-5 mRNA in rat intestinal smooth muscle. Am J Physiol. 1997;273:G875–G882. doi: 10.1152/ajpgi.1997.273.4.G875. [DOI] [PubMed] [Google Scholar]
- 13.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohneda K, Ulshen MH, Fuller CR, D'Ercole AJ, Lund PK. Enhanced growth of small bowel in transgenic mice expressing human insulin-like growth factor I. Gastroenterology. 1997;112:444–454. doi: 10.1053/gast.1997.v112.pm9024298. [DOI] [PubMed] [Google Scholar]
- 15.Williams KL, Fuller CR, Fagin J, Lund PK. Mesenchymal IGF-I overexpression: paracrine effects in the intestine, distinct from endocrine actions. Am J Physiol Gastrointest Liver Physiol. 2002;283:G875–G885. doi: 10.1152/ajpgi.00089.2002. [DOI] [PubMed] [Google Scholar]
- 16.Kuemmerle JF. Endogenous IGF-I promotes survival of human intestinal muscle cells by Akt-dependent inhibition of GSK-3β activity. Mol. Biol. Cell. 2002;13:165a. [Google Scholar]
- 17.Kuemmerle JF. IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase. Am J Physiol Gastrointest Liver Physiol. 2003;284:G411–G422. doi: 10.1152/ajpgi.00310.2002. [DOI] [PubMed] [Google Scholar]
- 18.Xin X, Hou YT, Li L, Schmiedlin-Ren P, Christman GM, Cheng HL, Bitar KN, Zimmermann EM. IGF-I increases IGFBP-5 and collagen alpha1(I) mRNAs by the MAPK pathway in rat intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2004;286:G777–G783. doi: 10.1152/ajpgi.00293.2003. [DOI] [PubMed] [Google Scholar]
- 19.Kuemmerle JF. Occupation of alphavbeta3-integrin by endogenous ligands modulates IGF-I receptor activation and proliferation of human intestinal smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1194–G1202. doi: 10.1152/ajpgi.00345.2005. [DOI] [PubMed] [Google Scholar]
- 20.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 21.Hosseini JM, Goldhill JM, Bossone C, Pineiro-Carrero V, Shea-Donohue T. Progressive alterations in circular smooth muscle contractility in TNBS-induced colitis in rats. Neurogastroenterol Motil. 1999;11:347–356. doi: 10.1046/j.1365-2982.1999.00165.x. [DOI] [PubMed] [Google Scholar]
- 22.Yamada Y, Marshall S, Specian RD, Grisham MB. A comparative analysis of two models of colitis in rats. Gastroenterology. 1992;102:1524–1534. doi: 10.1016/0016-5085(92)91710-l. [DOI] [PubMed] [Google Scholar]
- 23.Zeeh JM, Mohapatra N, Lund PK, Eysselein VE, McRoberts JA. Differential expression and localization of IGF-I and IGF binding proteins in inflamed rat colon. J Recept Signal Transduct Res. 1998;18:265–280. doi: 10.3109/10799899809047747. [DOI] [PubMed] [Google Scholar]
- 24.Qiao LY, Grider JR. Up-regulation of calcitonin gene-related peptide and receptor tyrosine kinase TrkB in rat bladder afferent neurons following TNBS colitis. Exp Neurol. 2007;204:667–679. doi: 10.1016/j.expneurol.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linden DR, Chen JX, Gershon MD, Sharkey KA, Mawe GM. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G207–G216. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- 26.Linden DR, Foley KF, McQuoid C, Simpson J, Sharkey KA, Mawe GM. Serotonin transporter function and expression are reduced in mice with TNBS-induced colitis. Neurogastroenterol Motil. 2005;17:565–574. doi: 10.1111/j.1365-2982.2005.00673.x. [DOI] [PubMed] [Google Scholar]
- 27.Hazelgrove KB, Flynn RS, Qiao L-Y, Grider JR, Kuemmerle JF. Endogenous IGF-I and {alpha}v{beta}3 integrin ligands regulate increased smooth muscle growth in TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1230–G1237. doi: 10.1152/ajpgi.90508.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuemmerle JF. Synergistic regulation of NOS II expression by IL-1 beta and TNF-alpha in cultured rat colonic smooth muscle cells. Am J Physiol. 1998;274:G178–G185. doi: 10.1152/ajpgi.1998.274.1.G178. [DOI] [PubMed] [Google Scholar]
- 29.Kuemmerle JF, Bushman TL. IGF-I stimulates intestinal muscle cell growth by activating distinct PI 3-kinase and MAP kinase pathways. Am J Physiol. 1998;275:G151–G158. doi: 10.1152/ajpgi.1998.275.1.G151. [DOI] [PubMed] [Google Scholar]
- 30.Teng B, Murthy KS, Kuemmerle JF, Grider JR, Sase K, Michel T, Makhlouf GM. Expression of endothelial nitric oxide synthase in human and rabbit gastrointestinal smooth muscle cells. Am J Physiol. 1998;275:G342–G351. doi: 10.1152/ajpgi.1998.275.2.G342. [DOI] [PubMed] [Google Scholar]
- 31.Kuemmerle JF, Murthy KS. Coupling of the insulin-like growth factor-I receptor tyrosine kinase to Gi2 in human intestinal smooth muscle: Gbetagamma -dependent mitogen-activated protein kinase activation and growth. J Biol Chem. 2001;276:7187–7194. doi: 10.1074/jbc.M011145200. [DOI] [PubMed] [Google Scholar]
- 32.Kuemmerle JF, Zhou H. Insulin-like growth factor-binding protein-5 (IGFBP-5) stimulates growth and IGF-I secretion in human intestinal smooth muscle by Ras-dependent activation of p38 MAP kinase and Erk1/2 pathways. J Biol Chem. 2002;277:20563–20571. doi: 10.1074/jbc.M200885200. [DOI] [PubMed] [Google Scholar]
- 33.Ortolan EVP, Spadella CT, Caramori C, Machado JLM, Gregorio EA, Rabello K. Microscopic, Morphometric and Ultrastructural Analysis of Anastomotic Healing in the Intestine of Normal and Diabetic Rats. Exp Clin Endocrinol Diabetes. 2008;116:198–202. doi: 10.1055/s-2007-993147. [DOI] [PubMed] [Google Scholar]
- 34.Flynn RS, Mahavadi S, Murthy KS, Kellum JM, Kuemmerle JF. Insulin-like growth factor-binding protein-5 stimulates growth of human intestinal muscle cells by activation of G{alpha}i3. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1232–G1238. doi: 10.1152/ajpgi.00323.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flynn RS, Mahavadi S, Murthy KS, Grider JR, Kellum JM, Akbari M, Kuemmerle JF. Endogenous IGFBP-3 Regulates Excess Collagen Expression in Intestinal Smooth Muscle Cells of Crohn's Disease Strictures. Inflammatory Bowel Diseases. 2010 doi: 10.1002/ibd.21351. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham MF, Bryson GR, Diegelmann RF. Transforming growth factor beta 1 selectively augments collagen synthesis by human intestinal smooth muscle cells. Gastroenterology. 1990;99:447–453. doi: 10.1016/0016-5085(90)91028-5. [DOI] [PubMed] [Google Scholar]
- 37.Kuemmerle JF. Endogenous IGF-I regulates IGF binding protein production in human intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2000;278:G710–G717. doi: 10.1152/ajpgi.2000.278.5.G710. [DOI] [PubMed] [Google Scholar]
- 38.Fichtner-Feigl S, Young CA, Kitani A, Geissler EK, Schlitt H-J, Strober W. IL-13 Signaling via IL-13R[alpha]2 Induces Major Downstream Fibrogenic Factors Mediating Fibrosis in Chronic TNBS Colitis. Gastroenterology. 2008;135:2003–2013. doi: 10.1053/j.gastro.2008.08.055. e7. [DOI] [PubMed] [Google Scholar]
- 39.Simmons JG, Pucilowska JB, Keku TO, Lund PK. IGF-I and TGF-beta 1 have distinct effects on phenotype and proliferation of intestinal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;283:G809–G818. doi: 10.1152/ajpgi.00057.2002. [DOI] [PubMed] [Google Scholar]