

Abstract
A novel class of PEGylated polyacridine peptides was developed that mediate potent stimulated gene transfer in the liver of mice. Polyacridine peptides, (Acr-X)n-Cys-PEG, possessing 2–6 repeats of Lys-acridine (Acr) spaced by either Lys, Arg, Leu or Glu, were Cys derivatized with polyethylene glycol (PEG 5000 Da) and evaluated as in vivo gene transfer agents. An optimal peptide of (Acr-Lys)6-Cys-PEG was able to bind to plasmid DNA (pGL3) with high affinity by polyintercalation, stabilize DNA from metabolism by DNAse and extend the pharmacokinetic half-life of DNA in the circulation for up to 2 hrs. A tail vein dose of PEGylated polyacridine peptide pGL3 polyplexes (1 μg in 50 μl), followed by a stimulatory hydrodynamic dose of normal saline at times ranging from 5–60 min post-DNA administration, led to a high level of luciferase expression in the liver, equivalent to levels mediated by direct hydrodynamic dosing of 1 μg of pGL3. The results establish the unique properties of PEGylated polyacridine peptides as a new and promising class of gene delivery peptides that facilitate reversible binding to plasmid DNA, protecting it from DNase in vivo resulting in an extended circulatory half-life, and release of transfection-competent DNA into the liver to mediate a high-level of gene expression upon hydrodynamic boost.
Keywords: Gene Delivery, DNA pharmacokinetics, peptide, gene expression, biodistribution
Introduction
New gene delivery agents are needed that function by efficiently mediating targeted nonviral gene delivery in vivo. Some of the most successful delivery agents developed to date, such as PEI and cationic lipids, produce robust gene transfer in vitro, but fail to mediate significant gene expression in vivo.1–8 In contrast, hydrodynamic (HD) dosing and electroporation are proven physical methods that are highly efficient in delivering plasmid DNA to animals and may find utility in humans.9–15 However, due to the invasiveness of HD dosing, there is a need for new gene delivery agents that produce gene expression at levels comparable to HD dosing, but without the requirement of high-volume administration.
Most i.v. dosed nonviral gene delivery systems are cationic polyplexes or lipoplexes, the primary exception being anionic liposomes with encapsulated DNA.16 To increase their compatibility with blood, cationic polyplexes are modified with polyethylene glycol (PEG) to block albumin binding and to “stealth” their rapid endocytosis by cells of the reticuloendothelial system (RES).1, 17–20 Despite these attempts, most PEGylated cationic polyplexes still quickly biodistribute to the liver, and are taken-up primarily by Kupffer cells.21, 22 Few, if any, of these experimental i.v. dosed gene delivery systems have produced appreciable gene expression in the liver,23, 24 especially when compared to the level of luciferase expression from an equivalent dose of DNA delivered hydrodynamically.10, 25
To develop nonviral gene delivery agents that function with greater efficiency in vivo following i.v. dosing, it is necessary to control the size, charge and metabolic stability of polyplexes.26 The cationic surface charge of most polyplexes results from residual unpaired amines on the cationic polymer used to condense DNA. Anionic polyplexes may be more blood compatible, but would require a new mode of binding polymers to DNA, not based on electrostatic interaction.
The use of polyintercalation has been shown to increase the binding affinity of small polymers and peptides to DNA.27–33 Previous studies from our group have established that polyacridine peptides possessing four Lys(Acr) residues (ε amine of Lys is modified with acridine), spaced by either a Lys or Arg, produce high affinity binding for plasmid DNA.34 Modification of polyacridine peptides with either a fusogenic peptide (melittin) or an N-glycan (high-mannose) produced gene transfer agents that formed cationic polyplexes resulting in high level selective gene transfer in vitro.34, 35 PEGylated polyacridine peptides have been shown to bind to plasmid DNA and produce unique, metabolically stable, anionic open-polyplexes that are capable of mediating gene transfer in vivo when delivered by i.m.-electroporation.36
In the present study we prepared a panel of PEGylated polyacridine peptides to establish their utility in mediating gene expression following i.v. dosing. The results establish a clear structure-activity relationship by which protection of the DNA in blood and liver by a PEGylated polyacridine peptide results in the ability to stimulate high level gene expression in liver with a delayed hydrodynamic dose of saline. The results also establish the ability to extend the circulatory half-life of a significant percentage of i.v. dosed DNA, which is an important prerequisite of achieving targeted gene delivery to organs and tissues. The synthetic adaptability of polyacridine peptides, along with the ability to prepare either anionic-open or cationic-closed-polyplexes that mediate gene transfer in vivo, demonstrate the unique importance of poly-intercalative binding to achieve gene delivery.
Materials and Methods
Unsubstituted Wang resin, 9-hydroxybenzotriazole, Fmoc-protected amino acids, O-(7-Azabenzo-triazol-1-yl)-N,N,N′,N′-tetramethylurionium hexafluorophosphate (HATU), Fmoc-Lysine-OH, and N-Methyl-2-pyrrolidinone (NMP) were obtained from Advanced ChemTech (Lexington, KY). N,N-Dimethylformamide (DMF), trifluoroacetic acid (TFA), and acetonitrile were purchased from Fisher Scientific (Pittsburgh, PA). Diisopropylethylamine, piperidine, acetic anhydride, Tris(2-carboxyethyl)-phosphine hydrochloride (TCEP), 9-chloroacridine and thiazole orange were obtained from Sigma Chemical Co. (St. Louis, MO). Agarose was obtained from Gibco-BRL. mPEG-maleimide and mPEG-OPSS (5,000 Da) were purchased from Laysen Bio (Avab, AL). D-Luciferin and luciferase from Photinus pyralis were obtained from Roche Applied Science (Indianapolis, IN). pGL3 control vector, a 5.3 kb luciferase plasmid containing a SV40 promoter and enhancer, was obtained from Promega (Madison, WI). pGL3 was amplified in a DH5α strain of Escherichia coli and purified according to manufacturer’s instructions.
Synthesis and Characterization of Polyacridine Peptides
9-Phenoxyacridine and Fmoc-Lysine(Acridine)-OH were prepared as recently reported.34, 35 The polyacridine peptides reported in Table 1 were prepared by solid phase peptide synthesis on a 30 μmol scale on an APEX 396 Synthesizer using standard Fmoc procedures including 9-hydroxybenzotriazole and HATU activation while employing double coupling of Fmoc-Lys(Acr)-OH and triple coupling for the spacing amino acid while using a 5-fold excess of amino acid over resin and omitting N-capping of truncated peptide species. Peptides were removed from resin and side chain deprotected using a cleavage cocktail of TFA/ethanedithiol/water (93:4:3 v/v/v) for 3 hrs followed by precipitation in cold ether. Precipitates were centrifuged for 10 min at 5000 × g at 4°C and the supernatant decanted. Peptides were then reconstituted with 0.1 v/v % TFA and purified to homogeneity on RP-HPLC by injecting 0.5–2 μmol onto a Vydac C18 semi-preparative column (2 × 25 cm) eluted at 5 ml per min with 0.1 v/v % TFA with an acetonitrile gradient of 20–30 v/v % over 30 min while monitoring acridine at 409 nm. The major peak was collected and pooled from multiple runs, concentrated by rotary evaporation, lyophilized, and stored at −20°C. Purified peptides were reconstituted in 0.1 v/v % TFA and quantified by absorbance (acridine ε 409 nm = 9266 M−1 cm−1 assuming additivity of ε for multiple acridines) to determine isolated yield (Table 1). Purified peptides were characterized by LC-MS by injecting 2 nmol onto a Vydac C18 analytical column (0.47 × 25 cm) eluted at 0.7 ml per min with 0.1 v/v % TFA and an acetonitrile gradient of 10–55 v/v % over 30 min while acquiring ESI-MS in the positive mode.
Table 1.
PEGylated Polyacridine Peptides
| Polyacridine Peptides | Mass (calc/obs)a | %Yield |
|---|---|---|
| (Acr-Lys)2-Cys | 988.5/988.3 | 37 |
| (Acr-Lys)4-Cys | 1855.3/1855.1 | 26 |
| (Acr-Lys)6-Cys | 2722.4/2722.0 | 20 |
| (Acr-Arg)4-Cys | 1967.4/1967.2 | 30 |
| (Acr-Leu)4-Cys | 1795.3/1795.1 | 31 |
| (Acr-Glu)4-Cys | 1859.1/1859.0 | 22 |
| PEGylated Polyacridine Peptides | Mass (calc/obs)b | %Yield |
|---|---|---|
| PEGylated-Mal-(Acr-Lys)2 | 6488/6531 | 64 |
| PEGylated-Mal-(Acr-Lys)4 | 7355/7218 | 55 |
| PEGyalted-Mal-(Acr-Lys)6 | 8222/8116 | 66 |
| PEGylated-Mal-(Acr-Arg)4 | 7467/7218 | 53 |
| PEGyalted-SS-(Acr-Arg)4 | 7467/7450 | 44 |
| PEGyalted-Mal-(Acr-Leu)4 | 7295/7110 | 46 |
| PEGyalted-Mal-(Acr-Glu)4 | 7359/7262 | 35 |
Determined by ESI-MS.
Determined by MALDI-TOF MS.
Synthesis and Characterization of PEGylated Polyacridine Peptides
PEGylation of the Cys residue on (Acr-X)n-Cys was achieved by reacting with 1 μmol of peptide with 1.1 μmol of PEG5000 Da–maleimide or PEG5000 Da-OPSS in 4 ml of 10 mM ammonium acetate buffer pH 7 for 12 hrs at RT. PEGylated peptides were purified by semipreparative HPLC as previously described and eluted with 0.1 v/v % TFA with an acetonitrile gradient of 25–65 v/v % acetonitrile while monitoring acridine at 409 nm. The major peak was collected and pooled from multiple runs, concentrated by rotary evaporation, lyophilized, and stored at −20°C. Counter-ion exchange was accomplished by chromatography on a G-25 column (2.5 × 50 cm) equilibrated with 0.1 v/v % acetic acid to obtain the peptide in an acetate salt form. The major peak corresponding to the PEG-peptide eluted in the void volume (100 ml) was pooled, concentrated by rotary evaporation, and freeze-dried. PEG-peptides were reconstituted in water and quantified by Abs409nm to determine isolated yield (Table 1). PEG-peptides were characterized by MALDI-TOF MS by combining 1 nmol with 10 μl of 2 mg per ml α-cyano-4-hydroxycinnamic acid (CHCA) in 50 v/v % acetonitrile and 0.1 v/v % TFA. Samples were spotted onto the target and ionized on a Bruker Biflex III Mass Spectrometer operated in the positive ion mode.
Formulation and Characterization of PEGylated Polyacridine Peptide Polyplexes
The relative binding affinity of PEGylated polyacridine peptides for DNA was determined by a fluorophore exclusion assay.37 pGL3 (200 μl of 5 μg/ml in 5 mM Hepes pH 7.5 containing 0.1 μM thiazole orange) was combined with 0, 0.05, 0.1, 0.25, 0.35, 0.5, or 1 nmol of PEGylated polyacridine peptide in 300 μl of Hepes and allowed to bind at RT for 30 min. Thiazole orange fluorescence was measured using an LS50B fluorometer (Perkin-Elmer, U.K.) by exciting at 498 nm while monitoring emission at 546 nm with the slit widths set at 10 nm. A fluorescence blank of thiazole orange in the absence of DNA was subtracted from all values before data analysis. The data is presented as nmol of PEGylated polyacridine peptide per μg of DNA versus the percent fluorescence intensity ± the standard deviation determined by three independent measurements.
The particle size and zeta potential were determined by preparing 2 ml of polyplex in 5 mM Hepes pH 7.5 at a DNA concentration of 30μg per ml and a PEGylated polyacridine peptide stoichiometry of 0, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8 or 1 nmol per μg of DNA. The particle size was measured by quasi-elastic light scattering (QELS) at a scatter angle of 90° on a Brookhaven ZetaPlus particle sizer (Brookhaven Instruments Corporation, NY). The zeta potential was determined as the mean of ten measurements immediately following acquisition of the particle size.
The shape of PEGylated polyacridine polyplexes were determined using atomic force microscopy (AFM). pGL3 alone, or anionic PEGylated polyacridine polyplexes at 0.2 nmol of peptide per μg of DNA, were prepared at a concentration of 100 μg per ml of DNA in 10 mM Tris, 1 mM EDTA pH 8. Polyplexes were diluted to 1 μg per ml in 40 mM Hepes 5 mM nickel chloride pH 6.7 and deposited on a fresh cleaved mica surface (cationic mica) for 10 min followed by washing with deionized water. Cationic PEGylated polyacridine polyplexes prepared at 0.8 nmol of PEGylated (Acr-Lys)6 per μg of DNA, at 100 μg per ml of DNA in 10 mM Tris, 1 mM EDTA pH 8 were deposited directly on a freshly cleaved mica surface (anionic mica) and allowed to bind for 10 min prior to washing with deionized water. Images were captured using an Asylum AFM MFP3D (Santa Barbara, CA) operated in the AC-mode using a silicon cantilever (Ultrasharp NSC15/AIBS, Mikro Masch).
Gel Band Shift and DNase Protection Assay
pGL3 (1 μg), or pGL3 polyplexes (1 μg) were prepared at either 0.2 or 0.8 nmol of PEGylated (Acr-Lys)2, 4, or 6, in 20 μl of 5 mM Hepes buffer pH 7.4. The ability of polyplexes to resist digestion with DNase was determined by incubation with 0.06 U of DNase I for 10 min. DNase was inactivated by the addition of 500 μl of 0.5 mg per ml proteinase K (in 100 mM sodium chloride, 1% SDS and 50 mM Tris pH 8.0) followed by incubation at 37°C for 30 min. The polyplexes were extracted with 500 μl of phenol/chloroform/isoamyl alcohol (24:25:1) to remove PEGylated peptides, followed by precipitation of DNA with the addition of 1 ml of ethanol. The precipitate was collected by centrifugation at 13,000 g for 10 min, and the DNA pellet was dried and dissolved in 5 mM Hepes buffer pH 7.4. DNA samples were combined with 2 μl of loading buffer and applied to a 1% agarose gel (50 ml) and electrophoresed in TBE buffer at 70 V for 60 min.38 The gel was post-stained with 0.1 μg per ml ethidium bromide at 4°C overnight then imaged on a UVP Biospectrum Imaging System (Upland, California).
Pharmacokinetic Analysis of PEGylated Polyacridine Polyplexes
Radioiodinated pGL3 was prepared as previously described.39 Triplicate mice were anesthetized by i.p. injection of ketamine hydrochloride (100 mg per kg) and xylazine hydrochloride (10 mg per kg) then underwent a dual cannulation of the right and left jugular veins. An i.v. dose of 125I-DNA (3μg in 50 μl of HBM, 1.2 μCi) or 125I-DNA polyplex (3μg) was administered via the left catheter, and blood samples (10 μl) were drawn from the right catheter at 1, 3, 6, 10, 20, 30, 60, 90 and 120 min and immediately frozen, then replaced with 10 μl of normal saline. The amount of radioactivity in each blood time point was quantified by direct γ-counting followed by extraction of the DNA and separation by gel electrophoresis as described above. The gel was dried on a zeta probe membrane and autoradiographed on a Phosphor Imager (Molecular Devices, Sunnyvale CA) following a 15 hr exposure.
Biodistribution Analysis of PEGylated Polyacridine Polyplexes
Triplicate mice were anesthetized and a single catheter was placed in the left jugular vein. 125I-DNA (1.5 μg in 50 μL of HBM, 0.6 μCi) or 125I-DNA polyplexes (1.5 μg) were dosed i.v. followed by vein ligation. After 5, 15, 30, 60, or 120 min, mice were sacrificed by cervical dislocation and the major organs (liver, lung, spleen, stomach, kidney, heart, large intestine, and small intestine) were harvested, and rinsed with saline. The radioactivity in each organ was determined by direct γ-counting and expressed as the percent of the dose in the organ.
Hydrodynamic Stimulation and Bioluminescence Imaging
pGL3 (1 μg) was prepared in a volume of normal saline corresponding to 9 wt/vol % of the mouse’s body weight (1.6 – 2.3 ml based on 15–23 g mice). The DNA dose was administered by hydrodynamic dosing to tail vein to 4–5 mice in 5 sec according to a published procedure.10, 25 Mice (4–5) were also dosed tail vein with 1 μg of PEGylated polyacridine polyplex in 50 μl of HBM (5 mM Hepes, 0.27 M mannitol, pH 7.4). At times ranging from 5–120 min, a Hydrodynamic Stimulatory Dose of normal saline (9 wt/vol% of the body weight) was administered over 5 sec. At 24 hrs post-DNA dose, mice were anesthetized by 3% isofluorane, then administered an i.p. dose of 80 μl (2.4 mg) of D-luciferin (30 μg/μl in phosphate-buffered saline). At 5 min following the D-luciferin dose, mice were imaged for bioluminescence (BLI) on an IVIS Imaging 200 Series (Xenogen). BLI was performed in a light-tight chamber on a temperature-controlled, adjustable stage while isofluorane was administered by a gas manifold at a flow rate of 3%. Images were acquired at a ‘medium’ binning level and a 20 cm field of view. Acquisition times were varied (1 sec – 1 min) depending on the intensity of the luminescence. The Xenogen system reported bioluminescence as photons/sec/cm2/seradian in a 2.86 cm diameter region of interest covering the liver. The integration area was transformed to pmols of luciferase in the liver using a previously reported standard curve.40 The results were determined to be statistically significant (p ≤ 0.05) based on a two-tailed unpaired t-test.
Results
A panel of PEGylated polyacridine peptides were prepared and tested for their ability to bind and transport DNA in vivo. The peptides were designed to test the influence of DNA binding affinity on gene transfer efficiency. To examine the influence of spacing amino acid, four peptides of the general structure (Acr-X)4 were prepared, where X is either Lys, Arg, Leu or Glu (Table 1). As discussed in more detail below, a Lys spacing amino acid proved most efficient in gene transfer, which warranted an expansion of (Acr-Lys)n to include repeats of 2 and 6 (Table 1). Each polyacridine peptide also possessed a C-terminal Cys residue that was modified with PEG. During the course of synthesis, the synthetic yields of polyacridine peptides were improved by using HATU to activate amino acids during coupling. Improved yields were also the result of applying triple coupling of the spacing amino acids, omitting N-capping to avoid a side reaction with acridine, and using ethanedithiol as a scavenger during workup. Under these conditions, (Acr-Lys)n with repeats ranging from 2 to 6 were prepared in 20–37% isolated yield (Table 1). Each polyacridine peptide produced an ESI-MS consistent with the calculated mass (Table 1).
Conjugation of each polyacridine peptide with PEG was accomplished by reaction of the Cys residues with either PEG-maleimide or PEG-OPSS, resulting in PEGylated polyacridine peptides with a reducible disulfide or a non-reducible maleimide linkage (Fig. 1). The reaction was monitored by RP-HPLC in which the polyacridine peptide (Fig. 2A) was mostly consumed, resulting in formation of a later eluting PEG-peptide (Fig. 2B). Preparative RP-HPLC purification produced PEG peptides free of unreacted polyacridine peptide and PEG as established by analytical RP-HPLC and MALDI-TOF analysis (Fig. 2C). Each PEGylated polyacridine peptide produced a MALDI-TOF MS with multiple peaks due to the polydispersity of PEG5000 Da, with average mass closely correlated to the calculated mass for the PEGylated peptide (Table 1).
Figure 1. Synthetic Strategy for PEGyalted Polyacridine Peptides.
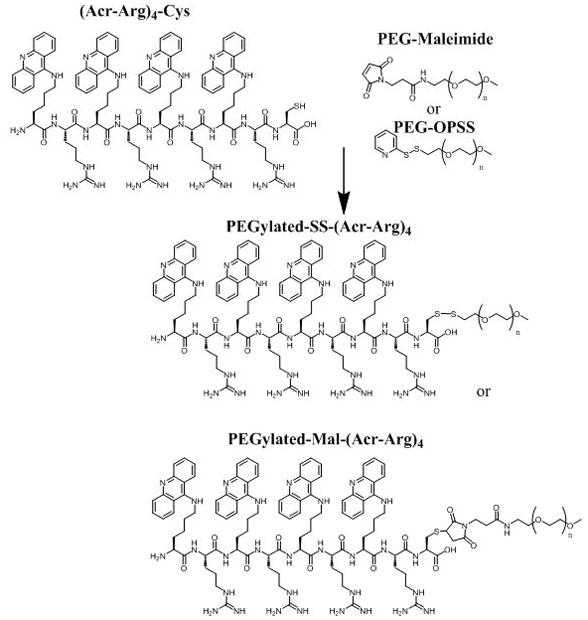
The approach used to prepare PEGylated-Mal-(Acr-Arg)4 and PEGylated-SS-(Acr-Arg)4 is demonstrated as an example of how all other polyacridine PEG-peptides described in Table 1 were prepared. (Acr-Arg)4-Cys (where Acr is Lys modified on the ε-amine with an acridine) was prepared by solid phase peptide synthesis. The Cys thiol was then reacted with either 5 kDa PEG-maleimide or PEG-OPSS (n = 109), resulting in PEGylated-Mal-(Acr-Arg)4 or PEGylated-SS-(Acr-Arg)4.
Figure 2. RP-HPLC Analysis of Polyacridine PEG-Peptide Synthesis.

Reaction of (Acr-Arg)4-Cys with 1.1 mol equivalents of PEG-Mal (panel A), results in the formation of PEGylated-Mal-(Acr-Arg)4 detected at 280 nm with simultaneous consumption of (Acr-Arg)4 and formation of dimeric peptide ((Acr-Arg)4-Cys)2 (panel B). The HPLC purified product PEGylated-Mal-(Acr-Arg)4 rechromatographed on RP-HPLC as a single peak (panel C) and is characterized by MALDI-TOF MS (panel C, inset), resulting in an observed m/z corresponding to the calculated mass (Table 1). The preparation of PEGylated-Mal-(Acr-X)n and PEGylated-SS-(Acr-X)n peptides described in Table 1 produced equivalent chromatographic evidence.
The relative binding affinity of PEGylated polyacridine peptides for pGL3 was compared by determining the concentration of peptide that displaces a thiazole orange intercalator dye, resulting in decreased fluorescence. Comparison of PEGylated (Acr-X)4 (X is either Arg, Lys, Leu or Glu) established the importance of cationic amino acids to increase binding affinity. Both the Lys and Arg analogue demonstrated high affinity by completely displacing thiazole orange at 0.4 nmol of peptide per μg of DNA. Conversely, the Leu analogue demonstrated weaker binding, resulting in full displacement at 1 nmol, and the Glu analogue was determined to have the lowest affinity by only achieving 40% displacement at 1 nmol (Fig. 3A).
Figure 3. DNA Binding Affinity of PEGylated Polyacridine Peptides.

A thiazole orange displacement assay was used to determine the relative binding affinity of polyacridine PEG peptides for DNA. Polyacridine PEG peptides when titrating 0.2 to 1 nmol of PEGylated (Acr-Arg)4 (●), PEGylated (Acr-Lys)4 (○), PEGylated (Acr-Leu)4 (▼), or PEGylated (Acr-Glu)4 (△), with 1 μg of pGL3 and 0.1 μM thiazole orange in 0.5 ml of 5 mM Hepes pH 7.0 prior to measuring thiazole fluorescence intensity. The results in Panel A established that PEGylated (Acr-Arg)4 and PEGylated (Acr-Lys)4 possessed higher affinity for DNA compared to PEGylated (Acr-Glu)4 and PEGylated (Acr-Leu)4. In Panel B the relative affinity of PEGylated (Acr-Lys)2, PEGylated (Acr-Lys)4, and PEGylated (Acr-Lys)6 are compared. The results establish that both the number of Acr and the spacing amino acid contribute to the DNA binding affinity.
A similar comparison of the binding affinities for PEGylated (Acr-Lys)2, 4 and 6 established a relationship of increasing affinity with increasing the number of Acr, such that PEGylated (Acr-Lys)6 completely displaced thiazole orange at 0.2 nmols of peptide per μg of DNA (Fig. 3B). The importance of the spacing amino acid was further established by PEGylated (Acr-Lys)2 which possessed greater DNA binding affinity than PEGylated (Acr-Glu)4 (Fig. 3A and B).
The size and charge of DNA polyplexes are often a function of the stoichiometry of peptide bound to DNA. To examine this relationship for PEGylated polyacridine polyplexes, QELS particle size and zeta potential were measured as a function of peptide to DNA ratio. One unusual property of PEGylated polyacridine polyplexes was their QELS mean diameter remained constant throughout the titration (Fig. 4A–F). Each of the polyplexes had an apparent mean diameter of approximately 100–200 nm, with the exception of PEGylated (Acr-Leu)4 and PEGylated (Acr-Glu)4 which produced polyplexes of an 200–400 nm mean diameter (Fig. 4B and C).
Figure 4. Size and Charge of PEGylated Polyacridine Polyplexes.

The QELS particle size (-▲-) and zeta potential (-●-) of polyplexes, prepared at concentrations ranging from 0.2–1 nmol of peptide per μg of DNA, are illustrated for PEGylated (Acr-Arg)4 (A), PEGylated (Acr-Leu)4 (B), PEGylated (Acr-Glu)4 (C), PEGylated (Acr-Lys)2 (D), PEGylated (Acr-Lys)4 (E) or PEGylated (Acr-Lys)6 (F). The results establish no significant change in particle size throughout the titration, whereas the zeta potential increases from −20 to 0 mV when titrating with peptides containing spacing amino acids Arg, Lys or Leu (panels A, B, D). Comparison of PEGylated (Arc-Lys)n repeats of n = 2, 4 and 6 (panel D, E and F) results in polyplexes that titrate to final zeta potential of −10, −2 and 5 mV, respectively.
A second unusual property of PEGylated polyacridine polyplexes was the observed zeta potential at each peptide stoichiometry (Fig. 4). Titration with PEGylated (Acr-Arg)4 resulted in an increase in zeta potential from −15 mV to a maximum of −2 mV at 0.4 nmols of peptide per μg of DNA or higher (Fig 4A). Similar zeta potential titration curves were determined for PEGylated (Acr-Leu)4, (Acr-Lys)2 and (Acr-Lys)4, each of which resulted in polyplexes with a negative zeta potential when fully titrated (Fig. 4B, D and E). In contrast, PEGylated (Acr-Glu)4 produced no change in zeta potential during the titration (Fig. 4C), and titration with PEGylated (Acr-Lys)6 produced zeta potentials that increased from −5 mV to a maximum of +9 mV during the titration (Fig. 4F). These data suggested that, unlike polylysine or PEI polyplexes that condense DNA into smaller polyplexes of 50–100 nm with charge of +25 mV,41, 42 polyacridine peptides bind without dramatically changing DNA size and can be, in certain cases, titrated to completion while maintaining a negative zeta potential.
The shape and relative charge of PEGylated polyacridine polyplexes was further examined by atomic force microscopy (AFM) (Fig. 5). Polyplexes prepared at 0.8 nmol per μg of DNA using PEGylated (Acr-Arg)4, (Acr-Lys)4, (Acr-Leu)4 and (Acr-Glu)4, each produced anionic open-polyplexes that bound to cationically charged mica (Fig. 5A-D). The relative size and shape appeared as open-polyplexes of approximately 0.2–0.3μm, relative to naked plasmid DNA which also bound to cationic mica to produce a more open coiled structure of approximately 0.5 μm diameter (Fig. 5E). Examination of AFM images of PEGylated (Acr-Lys)6 polyplexes revealed anionic open-polyplexes that bound to cationic mica at 0.2 nmol per μg of DNA (Fig. 5F). Alternatively, 0.8 nmol per μg of DNA produced cationic closed-polyplexes of slightly smaller size that bound to anionic mica (Fig. 5G). Cationic closed-polyplexes were only observed on anionic mica, whereas an attempt to achieve binding to cationic mica resulted in images devoid of visible polyplexes (Fig. 5H). These results illustrate the unique shape of anionic open-polyplexes as well as the ability to manipulate the charge of PEGylated (Acr-Lys)6 polyplexes to be either cationic or anionic and change shape from open to closed-polyplexes depending on peptide stoichiometry. Anionic PEGylated open-polyplexes should minimize binding to serum proteins and extend the pharmacokinetic half-life of plasmid DNA, provided that the DNA remains protected from premature metabolism.
Figure 5. Shape of PEGylated Polyacridine Polyplexes.
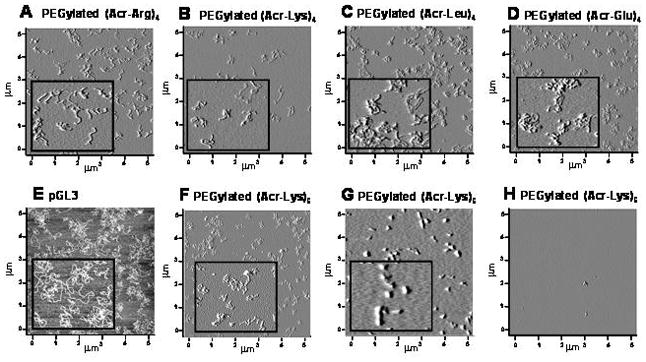
Atomic force microscopy (AFM) was used to analyze the shape of DNA polyplexes prepared at 0.8 nmol per μg of DNA with (A) PEGylated (Acr-Arg)4 (+) mica, (B) PEGylated (Acr-Lys)4 (+) mica, (C) PEGylated (Acr-Leu)4 (+) mica, (D) PEGylated (Acr-Glu)4 (+) mica, or (E) pGL3 (+) mica, (F) 0.2 nmol of PEGylated (Acr-Lys)6 (+) mica, (G) 0.8 nmol of PEGylated (Acr-Lys)6 (−) mica, and (H) 0.8 nmol of PEGylated (Acr-Lys)6 (+) mica. Anionic PEGylated polyacridine polyplexes produced open polyplex structures (A-D, F) that appeared slightly more coiled than plasmid DNA (E), where cationic PEGylated polyacridine polyplexes produced closed polyplex structures (G). Panel H demonstrates that cationic polyplexes do not bind to cationic mica. Each inset represents a 1 × 1 μm enlargement.
To investigate the metabolic stability of DNA polyplexes, PEGylated (Acr-Lys)2, 4, and 6 polyplexes were digested with DNase, followed by phenol-chloroform extraction to remove peptide and gel electrophoresis to determine the status of the plasmid DNA (Fig. 6). Unique band shifts occurred for PEGylated (Acr-Lys)2, 4, and 6 polyplexes prepared at either 0.2 or 0.8 nmols of peptide per μg of plasmid DNA (Fig. 6, lane 2 and 3 of panels A, B, C). However, phenol-chloroform extraction removed the PEG-peptide allowing recovery of the DNA which migrated coincident with control (Fig. 6, lane 4 of panels A, B and C). When challenged with a DNase digestion, PEGylated (Acr-Lys)2 or 4 polyplexes prepared at 0.2 nmol per μg of DNA failed to protect DNA from metabolism (Fig. 6, panel A and B, lane 5 and 6). In contrast, polyplexes formed at 0.2 nmols PEGylated (Acr-Lys)6, protected the DNA from DNase resulting in the recovery of bands following extraction (Fig. 6, panel C, lanes 5 and 6). At a higher stoichiometry of 0.8 nmols, PEGylated (Acr-Lys)2, 4 and 6 each protected DNA from metabolism (Fig. 6, panel A–C, lanes 7 and 8). These results establish a clear correlation between polyacridine DNA binding affinity (Fig. 3) and metabolic stability of polyplexes. Most importantly, PEGylated (Acr-Lys)6 is able to form DNase stable anionic open-polyplexes when prepared at 0.2, as well as cationic closed-polyplexes prepared at 0.8 nmol per μg of DNA.
Figure 6. Metabolic Stability of PEGylated Polyacridine Polyplexes.
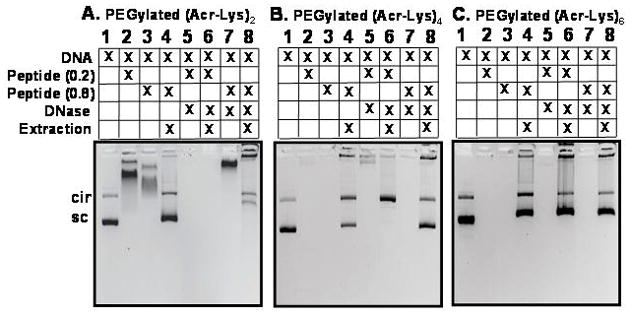
Agarose gel electrophoresis of (1) plasmid DNA, (2) PEGylated (Acr-Lys)n polyplex (n =2, 4 or 6) at 0.2 nmol of peptide per μg of DNA, (3) PEGylated (Acr-Lys)n polyplex at 0.8 nmol of peptide per μg of DNA, (4) release of DNA from PEGylated (Acr-Lys)n polyplex at 0.8 nmol per μg of DNA, (5) PEGylated (Acr-Lys)n polyplex at 0.2 nmol per μg of DNA following DNase digest, (6) released PEGylated (Acr-Lys)n polyplex at 0.2 nmol per μg of DNA following DNase digest, (7) PEGylated (Acr-Lys)n polyplex at 0.8 nmol per μg of DNA following DNase digest, (8) released PEGylated (Acr-Lys)n polyplex at 0.8 nmol per μg of DNA following DNase digest. The results establish the partial or complete protection of DNA from DNase at 0.8 nmol of (Acr-Lys)2-PEG (panel A lane 8) and (Acr-Lys)4-PEG (panel B lane 8), and the complete protection of DNA from DNase at 0.2 and 0.8 nmol of (Acr-Lys)6-PEG (panel C lane 6 and 8).
PEGylated polyacridine peptides were also subjected to trypsin digestion to determine if they could be digested by a common serine protease, suggesting perhaps they could also be more easily cleared from cells. The results established that even as little as 100 mU of trypsin catalyzed the formation of a dipeptide of Lys-Lys(Acr) with mass of 451 g/mol, to a reaction completion within 1 hr. Thereby, the high-affinity DNA binding of PEGylated polyacridine peptides is greatly reduced upon proteolytic digestion.
To determine if polyacridine peptides would mediate gene transfer, we chose to examine the ability of PEGylated DNA polyplexes to produce luciferase expression in the liver of mice, 24 hrs following a 1 μg dose of pGL3. Hydrodynamic dosing of 1 μg of pGL3 produced 108 photon/sec/cm2/sr determined using a calibrated bioluminescence assay (Fig. 7A, HD DNA). When pGL3 (1 μg in 50 μl) was dosed via the tail vein without hydrodynamic delivery, there was no detectable luciferase expression (not shown). In an attempt to stimulate gene expression, PEGylated polyacridine peptide polyplexes were administered via the tail vein (1 μg of pGL3 in 50 μl) and after 30 min a hydrodynamic dose (9 wt/v %, or approximately 2 ml) of saline was used to stimulate liver uptake and gene expression (Fig. 7A). In the subsequent studies described below, stimulated expression produced 107–109 photon/sec/cm2/sr, the magnitude being dependent on the structure of the PEGylated polyacridine peptide, the time delay between primary dose and stimulation dose, the charge ratio at which the polyplex was prepared, and the amount of DNA dosed.
Figure 7. Stimulated In Vivo Gene Expression Using PEGylated Polyacridine Polyplexes.
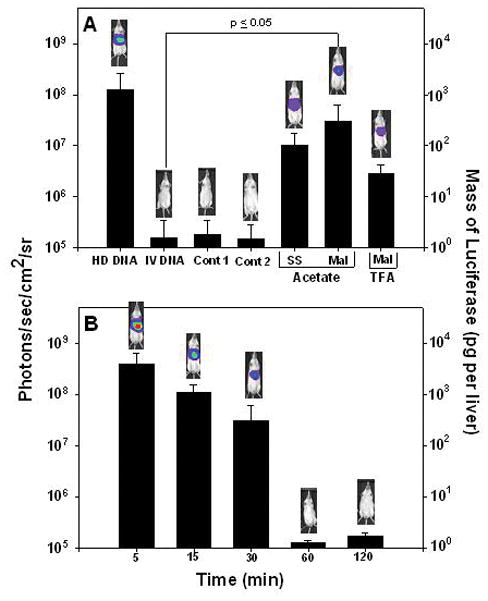
Direct HD dosing of 1 μg of pGL3 in multiple mice results in a mean BLI response of 108 photons/sec/cm2/sr at 24 hrs following dosing (panel A, HD DNA). Alternatively, a 24 hr BLI analysis of mice tail vein dosed with pGL3 (1 μg in 50 μl) in complex with 0.5 nmol of either PEGylated-Mal-(Acr-Arg)4 (panel A, Mal) or PEGylated-SS-(Acr-Arg)4 (panel A, SS) followed by hydrodynamic stimulation with 2 ml of saline delivered 30 min after DNA delivery, results in approximately 107 photons/sec/cm2/sr (panel A). Omission of HD stimulation (not shown) or PEGylated polyacridine peptide (panel A, i.v. DNA) results in no expression (105 photons/sec/cm2/sr). Likewise, HD stimulation after 30 min failed to produce measurable expression from a 1 μg pGL3 dose in complex with PEG-Cys-Trp-Lys18 (panel A, Cont 1) or a PEGylated glycoprotein described previously 44 (panel A, Cont 2). An acetic acid counter ion on PEGylated-Mal-(Acr-Arg)4 resulted in nearly 10-fold increase in expression relative to a TFA counter ion (panel A). Varying the dwell time of the HD stimulation from 5–120 min following a 1 μg dose of pGL3 in complex with 0.5 nmol of PEGylated-Mal-(Acr-Arg)4 established a maximum of 30 min to retain expression at 107 photons/sec/cm2/sr or higher measured at 24 hrs post administration (panel B). Statistical analysis was performed using a two-tailed unpaired t-test (*p ≤0.05).
At a fixed dose of 1 μg of pGL3 and a time delay of 30 min between primary and stimulatory dose, polyplexes prepared at 0.5 nmol of PEGylated (Acr-Arg)4, in which the PEG was attached by a maleimide (Mal) or a reducible disulfide (SS) linkage, were administered to mice and analyzed by BLI after 24 hrs. Approximately 5-fold higher luciferase expression was observed for Mal versus SS, indicating that a reducible linkage offered no advantage for stimulated expression (Fig. 7A). Comparison of the Mal peptide prepared as either an acetate or TFA salt form (ion paired with Arg) resulted in an approximately 10-fold loss of gene transfer for the TFA form (Fig. 7A). These results justified the use of Mal peptides in the acetate salt form for further comparisons.
Several controls were applied to further establish that PEGylated polyacridine peptides were necessary to achieve stimulated expression. The administration of a 1 μg dose of pGL3 followed by a stimulatory dose of saline after 30 min resulted in no detectable luciferase expression at 24 hrs (Fig. 7A, IV DNA). Likewise, there was no detectable luciferase expression with a 1 μg dose of pGL3 prepared as a PEGylated-Cys-Trp-Lys18 polyplex (Fig. 7A, cont 1),43 or PEGylated glycoprotein polyplex (Fig. 7A, cont 2).44 These controls confirmed that naked DNA was not responsible for the observed expression and also established that not all gene formulations were capable of mediating stimulated expression.
The stimulated expression mediated by PEGylated (Acr-Arg)4 polyplexes (1 μg) was examined to determine the influence of the delay time between primary and stimulatory dose. Varying the delay from 5–120 min established a 10-fold loss in luciferase expression between 5 and 30 min, followed by a complete loss of stimulated expression at 60 min and longer (Fig. 7B). Interestingly, the magnitude of stimulated gene expression at a delay time of 5 min was approximately 5-fold greater than direct hydrodynamic dosing of 1 μg of pGL3 (Fig. 7A and B). Controls that either decreased the volume of the hydrodynamic stimulatory dose or the speed at which it was administered both led to no gene expression.
To determine how the spacing amino acid influence stimulated gene expression, PEGylated peptides of general structure (Acr-X)4, possessing X as either Lys, Arg, Leu or Glu were used to prepare polyplexes that were dosed in mice and stimulated to express luciferase. While the DNA dose was fixed at 1 μg, and the time delay was 30 min, the stoichiometry of PEGylated peptide to DNA was adjusted based on the results of zeta potential titration (Fig. 4) to favor complete polyplex formation. The results established that only PEGylated peptides possessing an Arg or Lys spacing amino acid produced appreciable levels of luciferase expression (Fig. 8A), whereas peptides with Glu and Leu spacers were essentially inactive.
Figure 8. Structure-Activity Relationships for Stimulated Gene Expression.
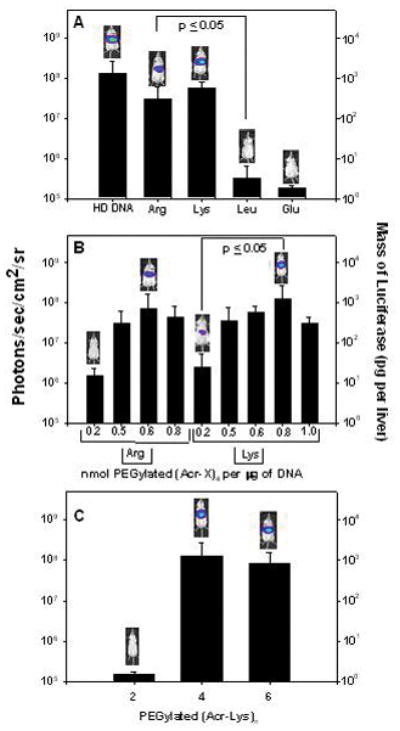
The BLI analysis at 24 hrs following tail vein dosed and HD stimulated (30 min post-DNA administration) pGL3 (1 μg in 50 μl) in complex with 0.5 nmol of either (Acr-Arg)4-Cys-Mal-PEG (panel A, Arg), 0.6 nmol of (Acr-Lys)4-Cys-Mal-PEG (panel A, Lys), 1 nmol of (Acr-Leu)4-Cys-Mal-PEG (panel A, Leu), or 0.8 nmol of (Acr-Glu)4-Cys-Mal-PEG (panel A, Glu) are compared with direct HD delivery of 1 μg of pGL3. The results establish polyacridine PEG-peptides with Arg and Lys spacing amino acids mediate 107–108 photons/sec/cm2/sr whereas substitution with Leu and Glu results in negligible expression. Varying only the stoichiometry of PEGylated polyacridine peptide to DNA for (Acr-Arg)4-Cys-Mal-PEG (panel B, Arg) and (Acr-Lys)4-Cys-Mal-PEG (panel B, Lys), established a maximal expression at 0.6 for Arg and 0.8 for Lys (panel B). Direct comparison of HD stimulated gene expression using (Acr-Lys)n-Cys-Mal-PEG (where n = 2, 4, or 6) in complex with 1 μg of pGL3 established the equivalency of 0.8 of (Acr-Lys)4-Cys-Mal-PEG with 0.2 nmol of (Acr-Lys)6-Cys-Mal-PEG, respectively (panel C), relative to (Acr-Lys)2-Cys-Mal-PEG which mediated negligible expression. Statistical analysis was performed using a two-tailed unpaired t-test (*p ≤0.05).
The stoichiometry of both Lys and Arg spaced PEGylated polyacridine peptide were varied from 0.2–1 nmol of peptide per μg of DNA, then dosed via tail vein and stimulated after 30 min. A 1 μg open polyplex prepared with 0.6 nmol of PEGylated (Acr-Arg)4 or 0.8 nmol of PEGylated (Acr-Lys)4 mediated maximal luciferase expression when assayed by BLI at 24 hrs (Fig. 8B). Both the Lys and Arg spaced PEGylated peptide mediated approximately 100-fold lower gene expression at 0.2 nmols of peptide compared to the stoichiometry of maximal expression (Fig. 8B). Likewise, at stoichiometries greater than maximal expression, a decline in expression was observed. These results suggested that a Lys spacer may have a slight advantage over Arg, which then prompted an investigation into varying the number of (Acr-Lys) repeats.
Comparison of the stimulated gene expression (30 min) mediated by PEGylated (Acr-Lys)2, 4 and 6 polyplexes (1 μg pGL3) prepared at an optimized stoichiometry of 1, 0.8 and 0.2 nmols of peptide respectfully (based on zeta potential, Fig. 4), established that PEGylated (Acr-Lys)2 was completely inactive. In contrast, PEGylated (Acr-Lys)6 at 0.2 nmols matched the activity of PEGylated (Acr-Lys)4 at 0.8 nmols (Fig. 8C). These results correlate with PEGylated (Acr-Lys)6 having a higher binding affinity for DNA (Fig. 3), with a greater ability to protect DNA from metabolism (Fig. 4), and with the ability to mediate stimulated gene expression even at a low stoichiometry of 0.2 nmols per μg of DNA (Fig. 8).
When PEGylated (Acr-Lys)6 polyplexes were administered to mice to determine how changing stoichiometry influenced gene expression, the results established that a 1 μg dose of pGL3 polyplex, stimulated at 30 min, produced equivalent gene expression across the range of 0.2–0.8 nmols of peptide (Fig. 9A). These results are in sharp contrast to those determined for PEGylated (Acr-Lys)4 (Fig. 8B), in which the gene expression was highly dependent on the stoichiometry of peptide to DNA. Furthermore, since PEGylated (Acr-Lys)6 was the only peptide to produce a zeta potential titration that started anionic (−5 mV) at 0.2 nmol and titrated to cationic (+9 mV) at 0.8 nmols (Fig. 4F), it appeared that the stimulated gene expression mediated by PEGylated (Acr-Lys)6 is fully functional as both an anionic open polyplex (Fig. 5F), and as an cationic closed polyplex (Fig 5G).
Figure 9. Optimal Parameters for Stimulated Gene Expression of Polyacridine PEG-peptide DNA Polyplexes.
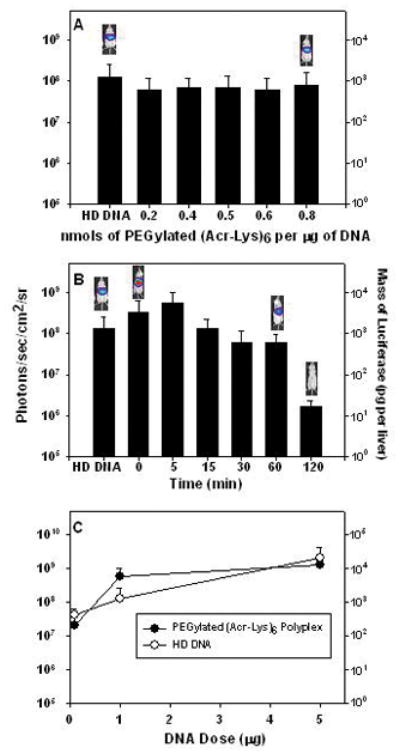
In panel A, the level of expression measured at 24 hrs, following HD stimulation 30 min after DNA dosing, remains nearly constant when delivering (Acr-Lys)6-Cys-Mal-PEG pGL3 polyplexes prepared at stoichiometries ranging from 0.2–0.8 nmols of peptide per μg of DNA (panel A). The results in panel B illustrate that varying the HD stimulation delay-time following delivery of (Acr-Lys)6-Cys-Mal-PEG pGL3 polyplexes results in expression of approximately 108 photons/sec/cm2/sr up to 60 min, whereas the expression decreased nearly 100-fold when delaying HD stimulation to 120 min (panel B). The dose-response curve for in vivo gene expression mediated delivery of (Acr-Lys)6-Cys-Mal-PEG pGL3 polyplexes with 5 min delay in stimulation (●) is compared with direct HD of pGL3 (○). The luciferase expression at 24 hrs determined by BLI established that HD delivery of 1 μg of (Acr-Lys)6-Cys-Mal-PEG polyplex is approximately 5-fold more efficient than the HD delivery of pGL3 (panel C).
To determine if the unique DNA binding properties of PEGylated (Acr-Lys)6 would also extend the delay time allowed between primary and stimulatory dose, polyplexes (1 μg) were prepared with 0.2 nmols of peptide and administered with a time delay varying from 0 to 120 min. Delaying the stimulation from 5 to 60 min resulted in only a slight 2–3 fold decrease in expression, whereas with a 120 min delay the expression decreased 100-fold (Fig. 9B). Using a zero delay time resulted in a nearly 5-fold increase in gene expression relative to dose equivalent direct HD administration of pGL3 (Fig. 9B). These results demonstrate that PEGylated (Acr-Lys)6 mediates significant high-level gene expression, even with a 60 min delay time, compared to PEGylated (Acr-Arg)4 which failed to mediate detectable expression following a 60 min delay in stimulation (Fig. 7B).
To establish the dose-equivalency of direct HD administration of DNA, relative to the stimulated expression of PEGylated polyacridine polyplexes, a dose-response experiment was performed (Fig. 9C). The BLI detected expression at 24 hrs post-DNA delivery was determined for direct HD delivered pGL3, dosed at 0.1, 1 and 5 μg of DNA (Fig. 9C). This was compared with the 24 hr luciferase expression from identical DNA doses of PEGylated (Acr-Lys)6 polyplexes prepared with 0.2 nmol of peptide and administered with a 5 min delay in stimulation. A nearly linear dose-response curve was identified for direct HD dosing of pGL3, whereas PEGylated (Acr-Lys)6 polyplexes showed an increase from 0.1–1 μg, followed by a plateau at 5 μg dose (Fig. 9C). At a dose of 1 μg of DNA, PEGylated (Acr-Lys)6 mediated approximately 5-fold greater gene expression (Fig. 9C).
To gain further mechanistic insight into the reason that PEGylated polyacridine polyplexes mediate stimulated gene expression, pharmacokinetic and biodistribution studies were performed. Following i.v. dosing of 125I-DNA or PEGylated (Acr-Lys)2 polyplexes resulted in a rapid 10-fold loss of radioactivity from the blood within 20–30 min, followed by an apparent long half-life (Fig. 10A). Isolation of 125I-DNA from blood time points, followed by gel electrophoresis and autoradiography demonstrated that plasmid DNA was rapidly degraded to fragments within 1 min (Fig. 10C). Likewise, electrophoretic analysis of blood time points following dosing of PEGylated (Acr-Lys)2 125I-DNA polyplexes demonstrated complete loss of plasmid DNA within 20 min (Fig. 10D), suggesting that the modest binding from PEGylated (Acr-Lys)2 leads to slightly delayed metabolism.
Figure 10. Pharmacokinetic and Biodistribution Analysis of PEGylated Polyacridine Polyplexes.
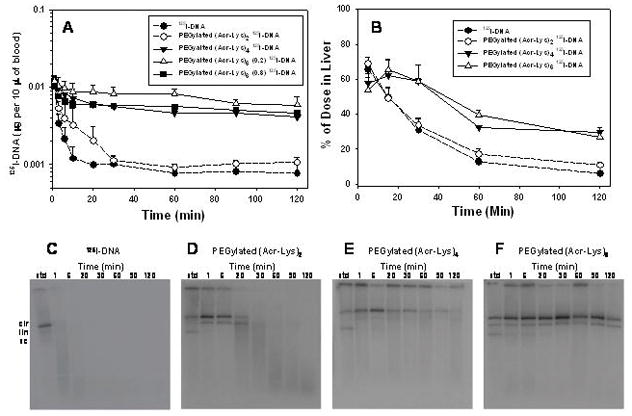
The pharmacokinetic profile for PEGylated (Acr-Lys)2, PEGylated (Acr-Lys)4, and PEGylated (Acr-Lys)6 125I-DNA polyplexes is compared with 125I-DNA (panel A). Extraction of the 125I-DNA from blood time points followed by agarose electrophoresis and autoradiography produced the images in C–F. Biodistribution analysis of PEGylated polyplexes resulted in comparison of the liver accumulation and elimination over time (panel B). The results establish that PEGylated (Acr-Lys)6 stabilizes DNA in the blood for up to two hours.
The pharmacokinetic profile for PEGylated (Acr-Lys)4 and 6 125I-DNA polyplexes were both very similar and distinct from those of 125I-DNA and PEGylated (Acr-Lys)2 125I-DNA polyplexes (Fig. 10A). The radioactivity in blood only decreased 3–4 fold in the first 20 min and proceeded with an apparent long half-life (Fig. 10A). Recovery of 125I-DNA from PEGylated (Acr-Lys)4 and 6 polyplexes resulted in an agarose gel autoradiographic analysis that established the blood stability of plasmid DNA up to 30 min for PEGylated (Acr-Lys)4 (Fig. 10E), and up to 182 min for PEGylated (Acr-Lys)6 (Fig. 10F). Non-linear least squares analysis of the radioactivity in blood over time was used to calculate an apparent α-half-life of 2–3 min and a β-half-life of 65 min for PEGylated (Acr-Lys)4 and 120 min for PEGylated (Acr-Lys)6 (Table 2). For the purpose of comparison, complete metabolic stability of plasmid DNA was assumed in the pharmacokinetic calculation, even though the gel electrophoretic and autoradiographic analysis for PEGylated (Acr-Lys)2 and (Acr-Lys)4 establish the differential loss of intact DNA over time. The apparent clearance rate decrease and the mean residence time increased as the β-half-live increased, whereas the apparent volume of distribution remained nearly constant. This resulted in an increase in the AUC as the stability of the polyplex increased (Table 2).
Table 2.
Pharmacokinetic Parameters for PEGylated Polyacridine Polyplexes
| Polyacridine Peptide Polyplex | t1/2αa (min) | t1/2βb (min) | Vol Disc (ml) | CLd (ml/min) | MRTe (min) | AUCf (μg*min)/ml |
|---|---|---|---|---|---|---|
| PEGylated (Acr-Lys)2 125I-DNAg | 0.7+/−0.0 | 15.2+/−0.8 | 42.8+/−0.1 | 2.3+/−0.0 | 18.9+/−0.1 | 0.1+/−0.0 |
| PEGylated (Acr-Lys)4 125I-DNAh | 2.3+/−0.3 | 65.6+/−13.5 | 37.4+/−1.9 | 0.4+/−0.1 | 92.2+/−18.9 | 0.7+/−0.1 |
| PEGylated (Acr-Lys)6 125I-DNAi | 1.8+/−0.9 | 181.5+/−33.4 | 31.6+/−1.3 | 0.1+/−0.0 | 260.8+/−47.8 | 2.5+/−0.4 |
Calculated α-half-life.
Calculated β-half-life.
Volume of distribution.
Total body clearance rate.
Mean residence time.
Area under the curve.
Calculated using blood cpm values over 20 min, assuming complete DNA stability.
Calculated using blood cpm values over 60 min, assuming complete DNA stability.
Calculated using blood cpm values over 120 min, assuming complete DNA stability.
The unique long pharmacokinetic β-half-life for PEGylated (Acr-Lys)4 and 6 polyplexes and the nearly coincident volume of distribution suggested there would be similar biodistribution to the organs. The experimental result established that the liver was the major site of biodistribution for PEGylated (Acr-Lys)2, 4 and 6 125I-DNA polyplexes (Table 3). In each of these experiments, it was not possible to extract 125I-DNA from liver or other tissues to determine its metabolic status, as was performed for DNA in blood.
Table 3.
Biodistribution Parameters for PEGylated Polyacridine Polyplexes
| Polyacridine Peptide Polyplex | Time (min) | Blooda | Liverb | Lungb | Spleenb | Stomachb | Kidneyb | Heartb | LIb | SIb | Totalc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 125I-DNA | 5 | 14.4± 5.8 | 65.7±2.5 | 6.1±2.2 | 2.8±0.3 | 0.2±0.0 | 0.9±0.2 | 0.1±0.0 | 0.2±0.1 | 0.4±0.1 | 84.7±11.2 |
| 30 | 6.8±1.7 | 31.1±6.2 | 1.4±0.1 | 2.1±0.6 | 1.6±1.3 | 3.5±1.0 | 0.2±0.0 | 1.6±0.7 | 2.8±0.3 | 51.1±11.9 | |
| 60 | 5.2±1.6 | 12.7±2.0 | 0.9±0.2 | 1.3±0.2 | 4.5±1.0 | 3.5±0.6 | 0.2±0.0 | 1.1±0.2 | 2.1±0.3 | 31.5± 6.2 | |
| 120 | 5.2±1.5 | 6.1±1.5 | 0.5±0.2 | 0.8±0.2 | 9.4±3.2 | 1.6±0.9 | 0.1±0.1 | 1.9±0.8 | 2.6±0.5 | 28.2±8.9 | |
| PEGylated (Acr-Lys)2125I-DNA | 5 | 26.4±14.0 | 68.9±3.6 | 1.9±0.1 | 5.3±1.8 | 0.2±0.0 | 0.8±0.5 | 0.1±0.1 | 0.3±0.2 | 0.4±0.1 | 104.2±20.5 |
| 30 | 7.6±1.3 | 33.6±4.0 | 1.0±0.3 | 1.7±0.4 | 1.5±0.3 | 2.8± 0.6 | 0.2±0.1 | 1.9 ±0.5 | 2.5±0.5 | 52.8±8.1 | |
| 60 | 6.2±0.6 | 17.1±3.1 | 0.7±0.2 | 0.7±0.5 | 6.1±2.0 | 2.6± 1.1 | 0.2±0.1 | 2.3±0.5 | 3.1±1.1 | 39.1±9.3 | |
| 120 | 7.2±1.0 | 10.9±1.5 | 0.8±0.2 | 1.0 ±0.4 | 8.3±1.7 | 2.3± 0.4 | 0.2±0.0 | 1.9±0.4 | 3.6±0.7 | 36.2±6.4 | |
| PEGylated (Acr-Lys)4125I-DNA | 5 | 54.1±2.7 | 57.5±5.8 | 2.2±0.9 | 2.1±0.5 | 0.3±0.2 | 0.9±0.5 | 0.2±0.1 | 0.3±0.2 | 0.5±0.2 | 118.0±11.1 |
| 30 | 37.3±4.4 | 58.6±9.7 | 1.0±0.1 | 4.5±2.3 | 0.5±0.1 | 1.4± 0.9 | 0.1±0.1 | 0.5±0.1 | 0.9±0.5 | 104.8±18.3 | |
| 60 | 31.2±2.2 | 32.2±1.2 | 0.8±0.3 | 4.7±0.3 | 2.6±0.4 | 2.1 ±0.8 | 0.2±0.0 | 1.1±0.2 | 2.7±0.7 | 77.6± 6.1 | |
| 120 | 27.4±3.2 | 29.9±2.7 | 1.0±0.2 | 4.1±2.4 | 5.3±2.4 | 2.7± 1.5 | 0.2±0.1 | 1.5±0.6 | 1.7±0.1 | 73.9±13.3 | |
| PEGylated (Acr-Lys)6125I-DNA | 5 | 65.8±14.3 | 53.8±7.0 | 1.3±0.5 | 6.8±3.1 | 0.4±0.2 | 0.7±0.1 | 0.2±0.0 | 0.2±0.1 | 0.5±0.0 | 129.6±25.3 |
| 30 | 54.9±12.3 | 58.7±2.0 | 0.9±0.2 | 11.7±1.1 | 0.7±0.2 | 1.1± 0.1 | 0.2±0.1 | 0.5±0.3 | 0.9±0.2 | 129.6±16.5 | |
| 60 | 55.2 ± 7.6 | 39.6±2.8 | 0.6±0.1 | 15.4±0.1 | 1.3±0.3 | 1.3 ±0.4 | 0.1±0.1 | 0.8±0.3 | 1.5±0.2 | 115.9±11.8 | |
| 120 | 39.4±10.2 | 26.8±2.1 | 0.8±0.2 | 18.0±1.4 | 3.2±2.8 | 1.5 ±0.2 | 0.1±0.0 | 1.1±0.7 | 2.2±0.3 | 93.2±17.9 | |
Percent of dose based on pharmacokinetic analysis.
Percent of dose based on gamma counting of tissue.
Total percent of dose recovered.
The biodistribution of 125I-DNA and PEGylated (Acr-Lys)2 125I-DNA polyplexes both reached a maximum of approximately 65% in the liver at 5 min, followed by a decrease to less than 10% by 120 min (Fig. 10B). In contrast, when dosing PEGylated (Acr-Lys)4 and 6 125I-DNA polyplexes the percent of radioactive dose in liver was approximately 55% at 5 min, which accumulated to over 60% at 15 min, followed by a decline to 30–40% at 120 min (Fig. 10B). By comparison, a maximum of 1–2% of the total radioactive dose distributed to lungs, heart, kidneys and intestine at all time points (Table 3). The exception was the spleen which showed a steady accumulation over time reaching 18% at 2 hrs when dosing PEGylated (Acr-Lys)6 polyplexes. In addition to the liver and spleen, the blood accounted for the majority of the radioactive dose, which ranged from 65–40% in the circulation during 2 hrs following dosing of PEGylated (Acr-Lys)6 polyplexes.
Discussion
PEGylated polyacridine peptides were designed to bind to plasmid DNA for the purpose of making it more blood compatible as a first step toward ultimately adding a targeting ligand and sub-cellular targeting peptides, needed to complete the delivery system and achieve significant in vivo gene expression following i.v. dosing, without the requirement of an additional stimulation. To accomplish this goal, an optimal polyacridine peptide would need to bind to DNA with sufficient affinity to protect from DNase metabolism in the circulation, while being able to release the DNA inside the cell to gain access to the nucleus.
It is likely there are many unique sequences of polyacridine peptides that accomplish this goal. The aim of the structure-activity study described was to determine a relationship between the number of Acr, the binding affinity for DNA, the polyplex pharmacokinetic half-life, and the magnitude of stimulated expression in mice. Initially, a nine amino acid peptide allowed the incorporation of four Acr spaced by a hydrophobic (Leu), anionic (Glu), or cationic (Lys and Arg) residue, along with a C-terminal Cys for modification. Prior studies from our group established that spacing amino acids that lacked a bulky side-chain or protecting group (Gly or Ala) resulted in low peptide yields. During the course of this study it was necessary to further optimize peptide yield to prepare (Acr-Lys)6-Cys. While it is possible to prepare even longer polyacridine peptides of this design, we found the yields began to diminish. Each of the polyacridine peptides was coupled to PEG5000Da, resulting in PEGylated polyacridine peptide that produced a single symmetrical peak on RP-HPLC and a MALDI-TOF MS that verified the structure.
The primary structural features that influence the level of gene expression mediated by PEGylated polyacridine peptides was the presence of at least four Acr residues combined with cationic (Lys or Arg) spacing amino acid. PEGylated polyacridine peptides possessing four Acr residues spaced by either Glu or Leu demonstrated weak binding to DNA (Fig. 3A), resulting in no stimulated gene expression (Fig. 8A), presumably due to the inability to protect DNA from metabolism in vivo. Conversely, PEGylated (Acr-Lys)4 or (Acr-Arg)4 possessed higher affinity for binding with DNA (Fig. 3A), which correlated with their ability to produce stimulated expression (Fig. 8A), due to their ability to stabilize DNA from premature metabolism in vivo (Fig. 10E). We previously reported that PEGylated Cys-Trp-Lys18 formed polyplexes that readily dissociate in the blood, resulting in rapid DNA metabolism.22 It was therefore anticipated that stimulation of i.v. dosed PEGylated Cys-Trp-Lys18 polyplexes would not produce gene expression (Fig. 7A, cont 1). Thus, a combination of polyintercalation and ionic binding to plasmid DNA results in improved affinity and stable polyplexes, where either functionality alone is insufficient.
One of the more unique aspects of PEGylated polyacridine polyplexes is their open polyplex structures that resemble naked DNA (Fig. 5). PEGylated (Acr-Lys)4 and (Acr-Arg)4 produced anionic open-polyplexes across the titration range of 0.2–0.8 nmols of peptide (Fig. 4A and E). When i.v. dosed and allowed to circulate for 30 min, the level of stimulated expression was dependent upon the stoichiometry of peptide to DNA, resulting in a parabolic peptide dose-response curve (Fig. 8B). These results suggested that the DNA stability afforded by PEGylated (Acr-Lys)4 and (Acr-Arg)4, was dependent on peptide stoichiometry, with higher stoichiometries resulting in charge neutral polyplexes (Fig. 4A) that stabilize DNA from metabolism (Fig. 6B) and demonstrate diminished gene transfer efficiency (Fig. 8B).
PEGylated (Acr-Lys)6 binds to DNA with higher affinity compared to all other peptides studied (Fig. 3). It also possessed an unusual zeta potential titration curve that produced anionic open-polyplexes at 0.2 nmols of peptide, which convert to cationic closed-polyplexes at 0.8 nmols and higher (Fig. 4F and Fig 5F & G). The DNase stability afforded to PEGylated (Acr-Lys)6 polyplexes at both 0.2 and 0.8 nmols of peptide (Fig. 6C) resulted in equivalent, high-level stimulated gene expression at each stoichiometry from 0.2–0.8 nmols (Fig. 9A). The greater affinity also led to greater stimulated expression for longer delay times compared to PEGylated (Acr-Arg)4 polyplexes, such that following the administration of 1 μg of DNA, a 1 hour delayed stimulation produced high-level gene expression, and at 2 hour delay measurable expression was still detected (Fig. 9B). These results established that PEGylated (Acr-Lys)6 polyplexes mediated equal gene expression as either closed or open-polyplex structures, and suggest that enhanced binding affinity translates to higher expression at longer delay times due to postponing metabolism.
Pharmacokinetic and biodistribution studies were used to gain insight into the underlying mechanism of how delayed hydrodynamic stimulation caused PEGylated (Acr-Lys)6 polyplexes to mediate gene expression in the liver. The most striking result was the apparent long pharmacokinetic half-life of PEGylated (Acr-Lys)4 and PEGylated (Acr-Lys)6 polyplexes, compared to the rapid loss of 125I-DNA and PEGylated (Acr-Lys)2 polyplexes (Fig. 10A). Electrophoretic analysis of the 125I-DNA recovered from blood clearly established that PEGylated (Acr-Lys)6 stabilized DNA in the circulation for 2 hrs (Fig. 10F), whereas PEGylated (Acr-Lys)4 stabilized open circular DNA bands for at least 30 min (Fig. 10E), and PEGylated (Acr-Lys)2 for no more than 20 min (Fig. 10D). Most importantly, approximately 40% of the radioactive dose remained in the blood after 2 hrs when dosing PEGylated (Acr-Lys)6 125I-DNA polyplexes (Table 3), and the pharmacokinetic profile appears to be the same whether dosing anionic-open or cationic-closed polyplexes (Fig. 10A).
These results cannot be directly compared with several prior studies that reported long circulating DNA or siRNA formulations by analyzing the pharmacokinetics of polyplexes by incorporating a fluorophore or radiolabel into the carrier component.16, 45–47 Three prior studies are more closely related in their direct analysis of pharmacokinetics using radiolabeled DNA48, 49 or by quantifying DNA in blood and tissues by PCR.50 These reports establish the ability to extend the half-life of DNA in the circulation, but stop short of establishing the metabolic status of recovered DNA. This is the first study to demonstrate stabilized plasmid DNA circulating in mice by gel electrophoresis and autoradiography. The metabolic stability of DNA is directly related to the mode and affinity of binding achieved by PEGylated (Acr-Lys)6. A similar PEGylated polylysine peptide (PEG-Cys-Trp-Lys18) also binds with high affinity to DNA through ionic interaction and forms cationic-closed polyplexes.43 However, despite being a longer peptide than PEGylated (Acr-Lys)6, PEG-Cys-Trp-Lys18 polyplexes rapidly dissociate following i.v. dosing leading to the complete metabolism of plasmid DNA in the circulation within 3 min.22 Thereby, it is the combination of polyintercalation and cationic binding that appears to be essential to achieve DNA stability during circulation, whereas PEGylated peptides that bind DNA by either mode individually are insufficient. This unique feature suggests that PEGylated (Acr-Lys)6 polyplexes may find application in targeting DNA to tissues outside the liver.
Analysis of the tissue distribution of 125I-DNA polyplexes over time established the liver as the major site of distribution accounting for approximately 54–69% of the dose within the first 5 min, with only minor (<1%) distribution to other organs. PEGylated (Acr-Lys)4 and PEGylated (Acr-Lys)6 polyplexes produced a distinct liver distribution and metabolism profile, with maximal accumulation of 60% at 20 min followed by a decrease to 30% over two hrs (Fig. 10A). The liver biodistribution profile of 125I-DNA and PEGylated (Acr-Lys)2 125I-DNA polyplexes were coincident with maximal accumulation of 65% at 5 min, followed by a decrease to 10% over 2 hrs. The stability of plasmid DNA in the blood afforded by PEGylated (Acr-Lys)6 appears to extrapolate to similar stability in the liver. Taken together, the pharmacokinetic and biodistribution data support a hypothesis in which the DNA that distributes to the liver is primarily responsible for the stimulated expression. However, liver distribution is not sufficient, the DNA must also be sufficiently metabolically stabilized to mediate significant stimulated expression with delay times up to an hr. The hypothesis that liver associated DNA, and not the DNA in blood, is responsible for stimulated expression is deduced by comparison of the level of stimulated expression from PEGylated (Acr-Lys)6 polyplexes (Fig. 9B), which decrease by nearly 100-fold between 60 and 120 min while the level of intact DNA in the blood remains constant over that time period (Fig. 10F). It is unlikely that PEG (Acr-Lys)6 spontaneously dissociates from DNA upon entering the cell or nucleus. Perhaps, PEG (Acr-Lys)6 possess affinity for chromosomal DNA, which acts as a sink, providing a mechanism for the release of plasmid DNA in the nucleus.
In conclusion, this is the first report of hydrodynamically stimulated gene expression from a nonviral delivery system that mirrors the level of expression produced by the same dose administered by direct hydrodynamic dosing. The unique attributes of PEGylated polyacridine peptides establish their ability to form open or closed polyplex structures that stabilize DNA from metabolism in mice and allow a stimulation of a high volume rapid dose of saline to complete the gene transfer, even after a 1 hour delay following the primary dose. This is still only a starting point toward the development of a nonviral delivery system that produces high level expression in animals without stimulation. However, given the modularity of polyacridine peptides, it should be possible to build multi-component gene delivery systems that drive the DNA further toward the nucleus, to ultimately achieve this aim.
Acknowledgments
The authors gratefully acknowledge support from NIH Grant DK066212.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Burke RS, Pun SH. Extracellular barriers to in Vivo PEI and PEGylated PEI polyplex-mediated gene delivery to the liver. Bioconjug Chem. 2008;19(3):693–704. doi: 10.1021/bc700388u. [DOI] [PubMed] [Google Scholar]
- 2.Boeckle S, von Gersdorff K, van der Piepen S, Culmsee C, Wagner E, Ogris M. Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. J Gene Med. 2004;6(10):1102–11. doi: 10.1002/jgm.598. [DOI] [PubMed] [Google Scholar]
- 3.Rudolph C, Schillinger U, Plank C, Gessner A, Nicklaus P, Muller R, et al. Nonviral gene delivery to the lung with copolymer-protected and transferrin-modified polyethylenimine. Biochim Biophys Acta. 2002;1573(1):75–83. doi: 10.1016/s0304-4165(02)00334-3. [DOI] [PubMed] [Google Scholar]
- 4.Guillaume C, Delepine P, Droal C, Montier T, Tymen G, Claude F. Aerosolization of cationic lipid-DNA complexes: lipoplex characterization and optimization of aerosol delivery conditions. Biochem Biophys Res Commun. 2001;286(3):464–71. doi: 10.1006/bbrc.2001.5418. [DOI] [PubMed] [Google Scholar]
- 5.Mahato RI, Anwer K, Tagliaferri F, Meaney C, Leonard P, Wadhwa MS, et al. Biodistribution and gene expression of lipid/plasmid complexes after systemic administration. Hum Gene Ther. 1998;9(14):2083–99. doi: 10.1089/hum.1998.9.14-2083. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Rizzo MA, Bhattacharya S, Huang L. Characterization of cationic lipid-protamine-DNA (LPD) complexes for intravenous gene delivery. Gene Ther. 1998;5(7):930–7. doi: 10.1038/sj.gt.3300683. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Qi H, Huang L, Liu D. Factors controlling the efficiency of cationic lipid-mediated transfection in vivo via intravenous administration. Gene Ther. 1997;4(6):517–23. doi: 10.1038/sj.gt.3300424. [DOI] [PubMed] [Google Scholar]
- 8.Felgner PL, Tsai YJ, Sukhu L, Wheeler CJ, Manthorpe M, Marshall J, et al. Improved cationic lipid formulations for in vivo gene therapy. Ann N Y Acad Sci. 1995;772:126–39. doi: 10.1111/j.1749-6632.1995.tb44738.x. [DOI] [PubMed] [Google Scholar]
- 9.Al-Dosari MS, Knapp JE, Liu D. Adv Genet. Vol. 54. Academic Press; 2005. Hydrodynamic Delivery; pp. 65–82. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6(7):1258–66. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 11.Molnar MJ, Gilbert R, Lu Y, Liu AB, Guo A, Larochelle N, et al. Factors influencing the efficacy, longevity, and safety of electroporation-assisted plasmid-based gene transfer into mouse muscles. Mol Ther. 2004;10(3):447–55. doi: 10.1016/j.ymthe.2004.06.642. [DOI] [PubMed] [Google Scholar]
- 12.Dean DA, Machado-Aranda D, Blair-Parks K, Yeldandi AV, Young JL. Electroporation as a method for high-level nonviral gene transfer to the lung. Gene Ther. 2003;10(18):1608–15. doi: 10.1038/sj.gt.3302053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fattori E, La Monica N, Ciliberto G, Toniatti C. Electro-gene-transfer: a new approach for muscle gene delivery. Somat Cell Mol Genet. 2002;27(1–6):75–83. doi: 10.1023/a:1022927822244. [DOI] [PubMed] [Google Scholar]
- 14.Li S, MacLaughlin FC, Fewell JG, Gondo M, Wang J, Nicol F, et al. Muscle-specific enhancement of gene expression by incorporation of SV40 enhancer in the expression plasmid. Gene Ther. 2001;8(6):494–7. doi: 10.1038/sj.gt.3301419. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, LH A Syringe Electrode Device for Simultaneous Injection of DNA and Electrotransfer. Mol Ther. 2002;5:323–328. doi: 10.1006/mthe.2002.0540. [DOI] [PubMed] [Google Scholar]
- 16.Heyes J, Palmer L, Chan K, Giesbrecht C, Jeffs L, MacLachlan I. Lipid encapsulation enables the effective systemic delivery of polyplex plasmid DNA. Mol Ther. 2007;15(4):713–20. doi: 10.1038/sj.mt.6300101. [DOI] [PubMed] [Google Scholar]
- 17.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999;6(4):595–605. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 18.Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different Strategies for Formation of PEGylated EGF-Conjugated PEI/DNA Complexes for Targeted Gene Delivery. Bioconj Chem. 2001;12:529–537. doi: 10.1021/bc0001488. [DOI] [PubMed] [Google Scholar]
- 19.Mannisto M, Vanderkerken S, Toncheva V, Elomaa M, Ruponen M, Schacht E, et al. Structure-activity relationships of poly(L-lysines): effects of pegylation and molecular shape on physicochemical and biological properties in gene delivery. J Control Release. 2002;83(1):169–82. doi: 10.1016/s0168-3659(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 20.Kwok KY, Park Y, Yongsheng Y, McKenzie DL, Rice KG. In Vivo Gene Transfer using Sulfhydryl Crosslinked PEG-peptide/Glycopeptide DNA Co-Condensates. J Pharm Sci. 2003;92(6):1174–1185. doi: 10.1002/jps.10384. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Park Y, Man S, Liu Y, Rice KG. Cross-linked low molecular weight glycopeptide-mediated gene delivery: relationship between DNA metabolic stability and the level of transient gene expression in vivo. J Pharm Sci. 2001;90(12):2010–22. doi: 10.1002/jps.1152. [DOI] [PubMed] [Google Scholar]
- 22.Collard WT, Yang Y, Kwok KY, Park Y, Rice KG. Biodistribution, metabolism, and in vivo gene expression of low molecular weight glycopeptide polyethylene glycol peptide DNA co-condensates. J Pharm Sci. 2000;89(4):499–512. doi: 10.1002/(SICI)1520-6017(200004)89:4<499::AID-JPS7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 23.Nishikawa M, Yamauchi M, Morimoto K, Ishida E, Takakura Y, Hashida M. Heptocyte-targeted in vivo gene expression by intraveneous injection of plasmid DNA complexed with synthetic multi-functional gene delivery system. Gene Ther. 2000;7:548–555. doi: 10.1038/sj.gt.3301140. [DOI] [PubMed] [Google Scholar]
- 24.Schuster MJ, Wu GY, Walton CM, Wu CH. Multicomponent DNA carrier with a vesicular stomatitis virus G-peptide greatly enhances liver-targeted gene expression in mice. Bioconj Chem. 1999;10:1075–1083. doi: 10.1021/bc990071r. [DOI] [PubMed] [Google Scholar]
- 25.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10(10):1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 26.Dash PR, Read ML, Barrett LB, Wolfert MA, Seymour LW. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther. 1999;6:643–650. doi: 10.1038/sj.gt.3300843. [DOI] [PubMed] [Google Scholar]
- 27.Ueyama H, Waki M, Takagi M, Takenaka S. Novel synthesis of a tetra-acridinyl peptide as a new DNA polyintercalator. Nucleic Acids Symp Ser. 2000;(44):133–4. doi: 10.1093/nass/44.1.133. [DOI] [PubMed] [Google Scholar]
- 28.Haensler J, Szoka JFC. Synthesis and characterization of a trigalactosylated bisacridine compound to target DNA to hepatocytes. Bioconjug Chem. 1993;4(1):85–93. doi: 10.1021/bc00019a012. [DOI] [PubMed] [Google Scholar]
- 29.Boulanger C, Di Giorgio C, Vierling P. Synthesis of acridine-nuclear localization signal (NLS) conjugates and evaluation of their impact on lipoplex and polyplex-based transfection. Eur J Med Chem. 2005;40(12):1295–306. doi: 10.1016/j.ejmech.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Shiraishi T, Hamzavi R, Nielsen PE. Targeted Delivery of Plasmid DNA into the Nucleus of Cells via Nuclear Localization Signal Peptide Conjugated to DNA Intercalating Bis-and Trisacridines. Bioconj Chem. 2005;16(5):1112–1116. doi: 10.1021/bc050093f. [DOI] [PubMed] [Google Scholar]
- 31.Vinogradov SV, Zhang H, Mitin A, Warren G. Intercalating Conjugates of Peg with Nuclear Localization Signal (Nls) Peptide. Polymer Prepr. 2008;49(2):434–435. [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, Mitin A, Vinogradov SV. Efficient transfection of blood-brain barrier endothelial cells by lipoplexes and polyplexes in the presence of nuclear targeting NLS-PEG-acridine conjugates. Bioconjug Chem. 2009;20(1):120–8. doi: 10.1021/bc8003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Vinogradov SV. Short biodegradable polyamines for gene delivery and transfection of brain capillary endothelial cells. J Control Release. 2010;143(3):359–66. doi: 10.1016/j.jconrel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baumhover NJ, Anderson K, Fernandez CA, Rice KG. Synthesis and In Vitro Testing of New Potent Polyacridine-Melittin Gene Delivery Peptides. Bioconj Chem. 2010;21:74–86. doi: 10.1021/bc9003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson K, Fernandez CA, Rice KG. N-Glycan Targeted Gene Delivery to the Dendritic Cell SIGN Receptor. Bioconj Chem. 2010 doi: 10.1021/bc1000824. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez CA, Baumhover NJ, Anderson K, Rice KG. Discovery of Metabolically Stablized Electronegative Polyacridine-PEG Peptide DNA Open Polyplexes. Bioconj Chem. 2010;21:723–30. doi: 10.1021/bc900514s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wadhwa MS, Collard WT, Adami RC, McKenzie DL, Rice KG. Peptide-mediated gene delivery: influence of peptide structure on gene expression. Bioconj Chem. 1997;8(1):81–8. doi: 10.1021/bc960079q. [DOI] [PubMed] [Google Scholar]
- 38.Adami RC, Rice KG. Metabolic stability of glutaraldehyde cross-linked peptide DNA condensates. J Pharm Sci. 1999;88:739–746. doi: 10.1021/js990042p. [DOI] [PubMed] [Google Scholar]
- 39.Terebesi J, Kwok KY, Rice KG. Iodinated plasmid DNA as a tool for studying gene delivery. Anal Biochem. 1998;263:120–123. doi: 10.1006/abio.1998.2834. [DOI] [PubMed] [Google Scholar]
- 40.Rettig G, McAnuff M, Kim J, Liu D, Rice KG. Qantitative Bioluminscence Imaging of Transgene Expression In Vivo. Anal Biochem. 2006;335:90–94. doi: 10.1016/j.ab.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 41.Abdallah B, Hassan A, Benoist C, Goula D, Behr JP, Demeneix BA. A powerful nonviral vector for in vivo gene transfer into the adult mammalian brain: polyethylenimine. Hum Gene Ther. 1996;7(16):1947–54. doi: 10.1089/hum.1996.7.16-1947. [DOI] [PubMed] [Google Scholar]
- 42.Blessing T, Remy JS, Behr JP. Monomolecular collapse of plasmid DNA into stable virus-like particles. Proc Natl Acad Sci U S A. 1998;95(4):1427–31. doi: 10.1073/pnas.95.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwok KY, McKenzie DL, Evers DL, Rice KG. Formulation of highly soluble poly(ethylene glycol)-peptide DNA condensates. J Pharm Sci. 1999;88(10):996–1003. doi: 10.1021/js990072s. [DOI] [PubMed] [Google Scholar]
- 44.Chen Cp, Kim Js, Liu D, Rettig GR, McAnuff MA, Martin ME, et al. Synthetic PEGylated Glycoproteins and Their Utility in Gene Delivery. Bioconj Chem. 2007;18(2):371–378. doi: 10.1021/bc060229p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunath K, von Harpe A, Petersen H, Fischer D, Voigt K, Kissel T, et al. The Structure of PEG-Modified Poly(Ethylene Imines) Influences Biodistribution and Pharmacokinetics of Their Complexes with NF-κB Decoy in Mice. Pharm Res. 2002;19(6):810–817. doi: 10.1023/a:1016152831963. [DOI] [PubMed] [Google Scholar]
- 46.Li S-D, Chen Y-C, Hackett MJ, Huang L. Tumor-targeted Delivery of siRNA by Self-assembled Nanoparticles. Mol Ther. 2007;16(1):163–169. doi: 10.1038/sj.mt.6300323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morille M, Montier T, Legras P, Carmoy N, Brodin P, Pitard B, et al. Long-circulating DNA lipid nanocapsules as new vector for passive tumor targeting. Biomaterials. 2010;31(2):321–329. doi: 10.1016/j.biomaterials.2009.09.044. [DOI] [PubMed] [Google Scholar]
- 48.Oupicky D, Ogris M, Howard KA, Dash PR, Ulbrich L, Seymour LW. Importance of Lateral an Steric Stabilization of Polyelectrolyte Gene Delivery Vectors for Extended Systemic Circulation. Mol Ther. 2002;5:463–472. doi: 10.1006/mthe.2002.0568. [DOI] [PubMed] [Google Scholar]
- 49.Ko YTKA, Hartner WC, Papahadjopoulos-Sternberg B, Torchilin VP. Self-assembling micelle-like nanoparticles based on phospholipid-polyethyleneimine conjugates for systemic gene delivery. J Control Release. 2009:132–138. doi: 10.1016/j.jconrel.2008.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Q-H, Wu C, Manickam D, Oupický D. Evaluation of Pharmacokinetics of Bioreducible Gene Delivery Vectors by Real-time PCR. Pharm Res. 2009;26(7):1581–1589. doi: 10.1007/s11095-009-9847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]