

Abstract
We previously showed that silencing profilin-1 (Pfn1) expression increases breast cancer cell motility, but the underlying mechanisms have not been explored. Herein, we demonstrate that loss of Pfn1 expression leads to slower but more stable lamellipodial protrusion thereby enhancing the net protrusion rate and the overall motility of MDA-MB-231 breast cancer cells. Interestingly, MDA-MB-231 cells showed dramatic enrichment of VASP at their leading edge when Pfn1 expression was downregulated and this observation was also reproducible in other cell types including human mammary epithelial cells and vascular endothelial cells. We further demonstrate that Pfn1 downregulation results in a hyper-motile phenotype of MDA-MB-231 cells in an Ena/VASP-dependent mechanism. Pfn1-depleted cells display a strong colocalization of VASP with lamellipodin (Lpd—a PI(3,4)P2-binding protein that has been previously implicated in lamellipodial targeting of Ena/VASP) at the leading edge. Finally, inhibition of PI3-kinase (important for generation of PI(3,4)P2) delocalizes VASP from the leading edge. This observation is consistent with a possible involvement of Lpd in enhanced membrane recruitment of VASP that results from loss of Pfn1 expression. Our findings for the first time highlight a possible mechanism of how reduced expression of a pro-migratory molecule like Pfn1 could actually promote motility of breast cancer cells.
Disruption of the actin cytoskeleton is a feature of malignant cells that correlates with dysregulated expression of various actin-binding proteins (ABPs) (Wang et al., 1996; Clark et al., 2000). The malignant phenotype of tumor cells often can be reversed by experimental restoration of ABP expression, suggesting misregulation of ABPs could contribute directly to malignancy (Tanaka et al., 1995; Nikolopoulos et al., 2000). Profilin-1 (Pfn1), a ubiquitously expressed G-actin binding protein, is significantly downregulated in various types of adenocarcinoma (breast, pancreatic, hepatic) (Janke et al., 2000; Gronborg et al., 2006; Wu et al., 2006). This raises a fundamental question as to whether loss of Pfn1 expression contributes to malignant progression of tumor cells. We found that upon downregulation of Pfn1, normal human mammary epithelial cells (HMEC) exhibit disruption of cell–cell adhesion (a hallmark change that facilitates epithelial cell dissemination and migration during cancer progression) (Zou et al., 2007). Furthermore, silencing Pfn1 expression leads to increased motility and invasiveness of breast cancer cell lines and conversely, overexpression of Pfn1 dramatically suppresses the aggressive phenotype of breast cancer cells (Roy and Jacobson, 2004; Zou et al., 2007). These findings collectively suggest that Pfn1 downregulation may contribute to breast cancer invasion and metastasis.
Although Pfn1 was initially identified as a G-actin sequestering protein (Karlsson et al., 1977), it can promote actin polymerization by acting as an efficient ADP-to-ATP exchanger on G-actin and adding ATP-G-actin to the barbed but not pointed ends of actin filaments (Witke, 2004). Consistent with this model, Pfn1 depletion induces a significant reduction in F-actin content in various cell types (Ding et al., 2006; Zou et al., 2007). Gene deletion of both Pfn1 and Pfn2 [another variant of Pfn1 that is mainly expressed in the nervous system in vertebrates (Lambrechts et al., 2000)] caused impaired motility of Dictyostelium amebae, providing direct evidence for Pfn’s involvement in migration of eukaryotic cells (Haugwitz et al., 1994). Further support for Pfn1’s requirement in cell migration came from studies that showed cell motility defects in a chickadee (Pfn1 homolog)-null Drososphila mutant (Verheyen and Cooley, 1994) and in human vascular endothelial cells in which Pfn1 expression was silenced (Ding et al., 2006). Several lines of experimental evidence including slower actin-driven intracellular propulsion of bacterial pathogens (Loisel et al., 1999; Mimuro et al., 2000), impaired membrane protrusion of vascular endothelial cells as a consequence of Pfn1 depletion (Ding et al., 2006), and very recently, induction of lamellipodia by Pfn1 in a growth-factor insensitive mechanism (Syriani et al., 2008) all point to a key role of Pfn1 in driving lamellipodial protrusion during cell migration. Several important classes of ABPs that either promote nucleation and/ or elongation of actin filaments at the leading edge including those belonging to Ena/VASP [Enabled/vasodilator stimulated phosphoprotein (Ferron et al., 2007)], Wiskott–Aldrich syndrome protein [WASP; example: Neuronal or N-WASP (Suetsugu et al., 1998), WAVE or WASP-associated verprolin homology (Miki et al., 1998)] family are physiological ligands that bind Pfn1 via polyproline-rich motifs. It has thus been proposed that these proline-rich proteins, when activated by signals, act as scaffolds to spatially recruit Pfn1-actin complex to the sites of actin assembly during cell migration (Holt and Koffer, 2001).
In addition to actin and polyproline ligands, Pfn1 also binds to membrane phosphoinositides (phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2]), phosphatidylinositol-3,4-bisphosphate [PI(3,4)P2] and phosphatidylinositol-3,4,5-triphosphate [PI(3,4,5)P3] (Lu et al., 1996). Although the physiological implications of Pfn1’s binding to membrane lipids have not been explored in detail, based on Pfn1’s ability to inhibit PI(4,5)P2 hydrolysis it has been proposed that Pfn1 might also regulate phosphoinositide metabolism in cells (Goldschmidt-Clermont et al., 1990). In fact, a recent study by our group has demonstrated that overexpression of Pfn1 in breast cancer cells dramatically suppresses growth-factor induced PI(3,4,5)P3 generation (Das et al., 2009), Since membrane phosphoinositides are critical regulators of actin cytoskeleton, Pfn1 could be an important nexus between phosphoinositide-derived signaling and cell motility.
Given the plethora of evidence demonstrating importance of Pfn1 in driving actin polymerization at the leading edge and hence playing a critical role in cell migration, it is not clear how reduced Pfn1 expression might contribute to faster motility of breast cancer cells. This was the overall goal of the present study where we have now linked loss of Pfn1 expression and hyper-motile phenotype of breast cancer cells through a Ena/VASP-dependent mechanism.
Materials and Methods
Antibodies
Polyclonal Pfn1 antibody was purchased from Santa Cruz (Santa Cruz, CA—in some experiments, a different polyclonal Pfn1 antibody that was kindly provided by Dr. Sally Zigmond of the University of Pennsylvania was used). Monoclonal GAPDH antibody is a product of Abd Serotec (Raleigh, NC). Polyclonal GFP and monoclonal VASP and FAK antibodies were obtained from Pharmingen (San Diego, CA). Polyclonal Lpd antibody was generated in Dr. Gertler’s laboratory (in some experiments, a commercially available Lpd antibody (source: Santa Cruz) was used). Polyclonal cofilin antibody was purchased from Abcam (Cambridge, MA). All cell culture reagents are products of Invitrogen (Carlsbad, CA). PI3-kinase inhibitor LY294002 was purchased from Calbiochem (Carlsbad, CA).
Plasmids and siRNA
EGFP-AP4-mito and EGFP-FP4-mito expression plasmids were generated in Dr. Gertler’s laboratory as previously described (Bear et al., 2000). The sense-strand sequence of a single-target specific Pfn1-siRNA is 5′-UAGCGACUAAACACAUCAAUU-3′. Smart-pool of non-targeting control siRNA was purchased from Dharmacon (Chicago, IL). The sense-strand sequences of smart-pool Pfn1 siRNA (also available from Dharmacon) are 5′-GGCCAGAAAUGUUCGGUGAUU-3′, 5′-GUGGUUUGAUCAACAAGAAUU-3′, 5′-CAAUGUCACUGUCACCAAGUU-3′ and 5′-GGUGGAACGCCUACAUCGAUU-3′.
Cell culture and transfection
MDA-231 breast cancer cells were cultured in EMEM media supplemented with 10% FBS, sodium pyruvate and antibiotics (Invitrogen). HMEC and HUVEC (source: Cambrex, Walkersville, MD) were cultured in a complete growth media supplied by the manufacturer. Plasmid transfection was performed using lipofectamine 2000 reagent (Invitrogen). All siRNA transfection was performed at either 50 nM (MDA-231) or 100 nM (HMEC, HUVEC) working concentration using reagents commercially available through Dharmacon according to the manufacturer’s instruction. Cytochalasin-D was purchased from Cytoskeleton, Inc. (Denver, CO).
Immunostaining
For immunostaining, cells were washed with PBS, fixed with 3.7% formaldehyde for 15 min, permeabliized with 0.5% Triton X-100 for 5 min and then blocked with 10% goat-serum for 1 h at room temperature. After incubating with the primary antibodies (VASP and/or Lpd at 1:100 dilution; Pfn1 antibody at 1:50 dilution) for 1 h at room temperature, cells were washed two times with PBS containing 0.02% tween and 2 times with PBS and then incubated with secondary antibodies (source: Jackson Immunoresearch, West Grove, PA) with or without alexa-488 phalloidin (Invitrogen) for an additional hour at room temperature. Stained cells were washed two times with PBS containing 0.02% tween, two times with PBS, and then once with distilled water before mounting on slides for imaging using a 60× oil-immersion objective on an Olympus IX71 inverted microscope. Quantitative fluorescence analyses of images were performed by Metamorph software. Specifically, for VASP and Lpd quantification at the leading edge, images were first background subtracted (the average intensity of cell-free area on the coverslips was used for background correction), and the average fluorescence intensity at the very leading edge (identified by phalloidin counterstaining) was then calculated based on 15–20 line scan measurements across the lamellipodia.
Immunoblotting
Total cell lysate was prepared by extracting cells with warm 1× sample buffer containing protease inhibitor. For immunoblotting, the antibodies were used at the following concentrations: GAPDH (1:2,000), Pfn1 (1:200), VASP (1:1,000), and cofilin (1:3,000).
Time-lapse cell motility assay
Cells, sparsely plated overnight on collagen-coated 35-mm culture dish, were imaged for 2–3 h at a 1-min time-interval between the successive image frames. For all time-lapse recordings, proper environmental conditions (37°C/pH 7.4) were maintained by placing the culture dish in a microincubator. Cell trajectory was constructed by frame-by-frame analyses of the centroid positions (x, y) of cell-nuclei (assumed to be the representations of cell-bodies). The change in the direction of centroid movement between successive image frames (i − 1, i, i + 1) was calculated as Δθi = θi, i+1 − θi, i−1 (θi, i+1 = cos−1(xi+1 − xi)/sqrt ((xi+1 − xi)2) + ((yi+1 − yi)2)) and from these values, the standard deviation of Δθ was computed. For kymography, additional time-lapse movies were recorded for 10–20 min at a 5-sec time-interval. Kymographs marking the beginning to the end of protrusion were constructed based on 1-pixel wide (0.3 μm) lines drawn at multiple locations (3–4) across the protruding membrane. Membrane fluctuation < 4 pixels (1.2 μm) was disregraded for quantitative analyses. All images were acquired and analyzed using Metamorph and NIH ImageJ softwares, respectively.
Statistics and data representation
All statistical tests were performed with ANOVA followed by Newman–Keuls post-hoc test for multiple comparison whenever applicable, and a “P” value less than 0.05 was considered to be statistically significant. In most cases, experimental data were represented as box and whisker plots where dot represents the mean, middle line of box indicates median, top of the box indicates 75th percentile, bottom of the box measures 25th percentile and the two whiskers indicate the 10th and 90th percentiles, respectively.
Results
Loss of Pfn1 expression leads to faster random motility of MDA-231 breast cancer cells
In our previous study, MDA-MB-231 (MDA-231), a highly invasive and metastatic breast cancer cell line, was found to be more motile in both wound-healing and transwell-based migration assays when Pfn1 expression was silenced (Zou et al., 2007). However, neither of these two migration assays allows for detailed investigation of cell motility and particularly in wound-healing assay, the speed of cell migration can also be influenced by cell-cell contact. Therefore, we examined the effect of Pfn1 knockdown on the motility of MDA-231 cells at the single-cell level to identify possible mechanisms underlying of the resulting increased motility of breast cancer cells. Specifically, we conducted time-lapse imaging of individual MDA-231 cells which were transiently transfected with either a pool of non-targeting control siRNAs or our previously validated siRNA targeting a single region of Pfn1-mRNA (Ding et al., 2006; Zou et al., 2007). As an additional control, cells were also subjected to mock transfection. Immunoblotting of total cell lysate (TCL) extracted 72 h after siRNA transfection showed strong Pfn1 bands for either of the two control groups of cells, but virtually undetectable signal for Pfn1-siRNA treated cells (Fig. 1A). There was also no detectable change in Pfn1 expression between mock and control-siRNA transfection groups. These results demonstrate that we are able to achieve very close to 100% transfection efficiency and near complete suppression of Pfn1 expression in MDA-231cells by siRNA treatment (this was also independently confirmed by immunostaining data as shown in Supplemental Fig. S1). Figure 1B shows the actual trajectories of different groups of cells obtained from 2-h time-lapse recordings (data pooled from 3 experiments). From cell trajectory analyses, we found that the average speed of migration of Pfn1-deficient MDA-231 cells (~1.0 μm/min) was 1.9-fold higher than that of mock-transfected (~0.52 μm/min) and control-siRNA treated cells (~0.53 μm/min); there was no significant difference in the average speed of cell migration between our mock and control-siRNA treatment groups thus confirming the specificity of our findings (Fig. 1C). As an additional validation of our results, we repeated these time-lapse motility experiments with a set of pooled siRNAs that targets different regions of Pfn1-mRNA and we found close to 1.5-fold increase in the average speed of MDA-231 cell migration as a result of Pfn1 downregulation (data not shown). A 20% difference in the average speed of migration between single (~1.0 μm/min) and pooled (~0.8 μm/min) Pfn1 siRNA-treated cells could be due to marginally better silencing of Pfn1 achieved with a single siRNA treatment (unpublished observation).
Fig. 1.

Silencing Pfn1 expression leads to faster motility of MDA-231 breast cancer cells and HMEC in time-lapse assay. A: Pfn1 immunoblot of MDA-231 cell lysates 72 h after siRNA transfection (the GAPDH blot serves as the loading control). B: Trajectories of individual MDA-231 cells of different experimental groups in time-lapse motility assay (data pooled from three independent experiments). C: A box and whisker plot representing the relative comparison of the average speed of migration between different treatment groups for MDA-231 cells and HMEC (the insets shows Pfn1 downregulation in HMEC 96 h after siRNA treatment where GAPDH immunoblot serves as the loading control). All results arenormalized to the average value calculated for the control-siRNA treated cells and “n” indicates then number of cells analyzed in each treatment group. The asterisk marks represent P < 0.05.
Since breast cancer cells are of epithelial origin, we next analyzed the effect of Pfn1 depletion on the random motility of normal HMEC which express Pfn1 at higher levels than breast cancer cell lines including MDA-231 cells (Janke et al., 2000; Zou et al., 2007) and also show better chemotactic migration in transwell assay in the absence of Pfn1 expression as demonstrated in our previous study (Zou et al., 2007). Similar to our observation in MDA-231 cells, depletion of Pfn1 in HMEC led to a 2.6-fold increase in the average speed of cell migration in time-lapse assay (Fig. 1C–the immunoblot in the inset confirm siRNA-mediated suppression of Pfn1 expression in HMEC). Overall, these time-lapse motility observations for HMEC and MDA-231 cells are consistent with our previously reported wound-healing and transwell migration data (Zou et al., 2007).
We also asked whether loss of Pfn1 expression affects the directionality of MDA-231 cell movement. From the sequence of images of individual cells for different experimental groups (shown in Fig. 2A), it became apparent that Pfn1-depleted cells change direction of migration less frequently than either of the two control groups of cells. To quantitatively represent this feature, we first scored the change in direction of centroid movement (Δθ) as a function of time from which we computed the standard deviation of Δθ (a higher value indicates larger fluctuation in the directionality of movement). As summarized in the form of a box and whisker plot in Figure 2B, a somewhat smaller standard deviation of Δθ of Pfn1-sRNA treated cells compared to either of the two control groups of cells suggests that Pfn1-depletion moderately enhances the directional persistence of motility of MDA-231 cells.
Fig. 2.

SilencingPfn1expressionenhancesdirectionalpersistenceofMDA-231cellmigration.A:Sequencesofimagesfromasetofrepresentative time-lapse experiments of MDA-231 cells conducted for 2 h (arrow—direction of protrusion; scale—10 μm). B: Relative comparison of the standard deviation of Δθ (a measure of the change in the direction of centroid movement) during motility between different treatment groups of MDA-231 cells. The asterisk mark represents P < 0.05.
Effect of Pfn1 depletion on lamellipodial protrusion
Lamellipodial protrusion initiates and defines the direction of cell movement. Among its different functions, Pfn1’s role has been best described in the context of membrane protrusion during cell migration. We therefore analyzed the leading edge movement of MDA-231 cells for different transfection conditions from kymographs of 1-pixel (0.3 μm) wide lines that were drawn normal to the leading edge and in the direction of protrusion. Figure 3A depicts a set of representative kymographs of the protrusion events of MDA-231 cells for different siRNA transfection conditions where leading edge traces (marked by the arrows) show cycles of typical lamellipodial protrusion and withdrawal (resemble saw-tooth waveforms as outlined by the boxes). Quantitative analyses of kymographs showed that the actual protrusion velocity (this is equal to the slope of the ascending portion of a saw-tooth waveform) of Pfn1-depleted MDA-231 cells (=3.5 μm/min) is almost twofold less than that of control siRNA-treated cells (=7.2 μm/min; Fig. 3B). However, since MDA-231 cells actually exhibit a gain in overall migration speed after Pfn1 depletion, further explanation was needed. During cell motility, a protruding lamellipodium often undergoes partial or complete retraction (also true for MDA-231 cells as shown in Fig. 3A) and therefore it is the net membrane protrusion that correlates with the efficacy of whole cell movement. When we analyzed the rate of net membrane protrusion of MDA-231 cells, we found that Pfn1-deficient cells have a significantly greater net protrusion velocity (=1.0 μm/min) compared to control siRNA treated cells (=0.6 μm/min; Fig. 3C). To explain this observation, we further examined the lamellipodial withdrawal events which showed that on an average, withdrawal of protrusion was more frequent in control (1/min) than in Pfn1-depleted MDA-231 cells (0.5/min; Fig. 3D). Also, the magnitude of lamellipodial withdrawal significantly decreases when Pfn1 expression is suppressed (Fig. 3E).
Fig. 3.
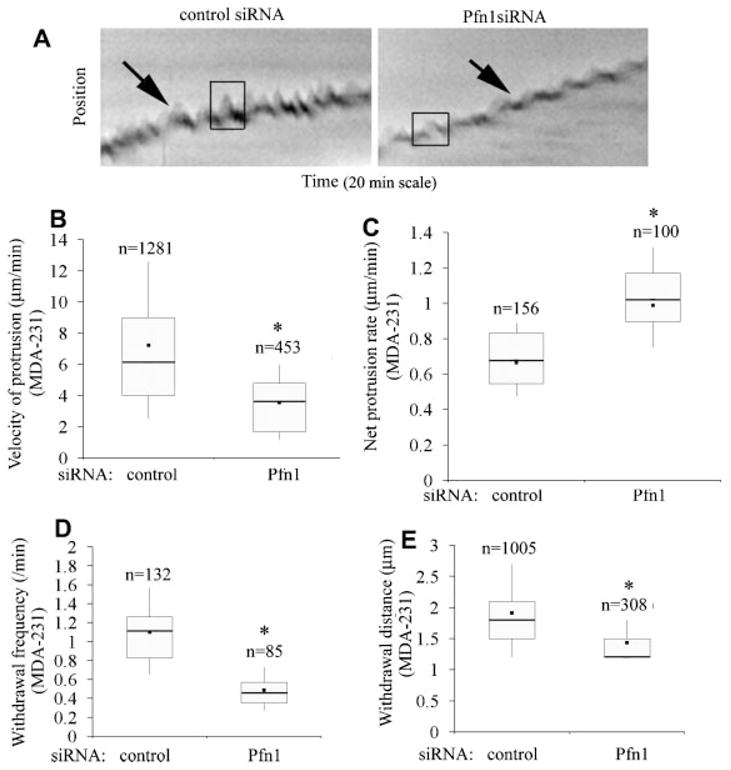
Loss of Pfn1 expression leads to slower but more stable lamellipodial protrusion in MDA-231 cells. A: Kymographs representing lamlellipodial dynamics of MDA-231 cells with or without Pfn1 depletion (leading edge traces are marked by the black arrows and the boxes outlining the saw-tooth waveforms represent typical lamellipodial protrusion and withdrawal events). B: A box and whisker plots comparing the actual protrusion velocities between control and Pfn1-siRNA treated MDA-231 cells (“n” represents the total number of protrusion events characterized by the saw-tooth waveforms).C:AboxandwhiskerplotcomparingthenetprotrusionvelocityofMDA-231cellswithorwithoutPfn1 depletion (“n” indicates the number of kymographs analyzed). D, E: Box and whisker plots comparing the frequency (part D) and average distance (part E) of lamellipodial withdrawal (i.e., the descending part of the saw-tooth wave forms) between control and Pfn1-si RNA treated MDA-231cells (“n” in parts D and E represent the total number of kymographs and with drawal events, respectively).These data are based on analyses of 30 cells for each transfection condition pooled from three independent experiments. The asterisk marks represent P < 0.05.
We also compared our protrusion data of MDA-231 cells with that of human umbilical vein endothelial cells (HUVEC) since we have shown previously that HUVEC exhibit reduced spreading and significantly slower motility when Pfn1 expression is silenced (Ding et al., 2006), and this phenotypic change is consistent with the traditionally conceived pro-migratory role of Pfn1. We therefore performed similar kymograph analyses of HUVEC with or without Pfn1 depletion. On a comparative scale, although the amplitude of the oscillatory movement of lamellipodia of control HUVEC is much greater than that exhibited by MDA-231 cells (Fig. 4A), the effect of Pfn1 depletion on the actual protrusion velocity was similar in these two cell types. We found a 1.8-fold reduction in the actual protrusion velocity of HUVEC when Pfn1 expression was silenced (Fig. 4B). The frequency of withdrawal of protrusions for HUVEC decreases when Pfn1 expression is downregulated (Fig. 4C). This is expected since the frequency of actual membrane protrusion is also less in Pfn1-depleted HUVEC. In summary, slower velocity of lamellipodial protrusion resulting from loss of Pfn1 expression is consistent with previously demonstrated inhibitory effect of Pfn1 depletion on intracellular propulsion of bacterial pathogens (Loisel et al., 1999; Mimuro et al., 2000) as well as on whole cell motility of HUVEC (Ding et al., 2006). As for MDA-231 cells, our data suggest that loss of Pfn1 expression cells is associated with slower but more stable (resistant to withdrawal) lamellipodial protrusion.
Fig. 4.
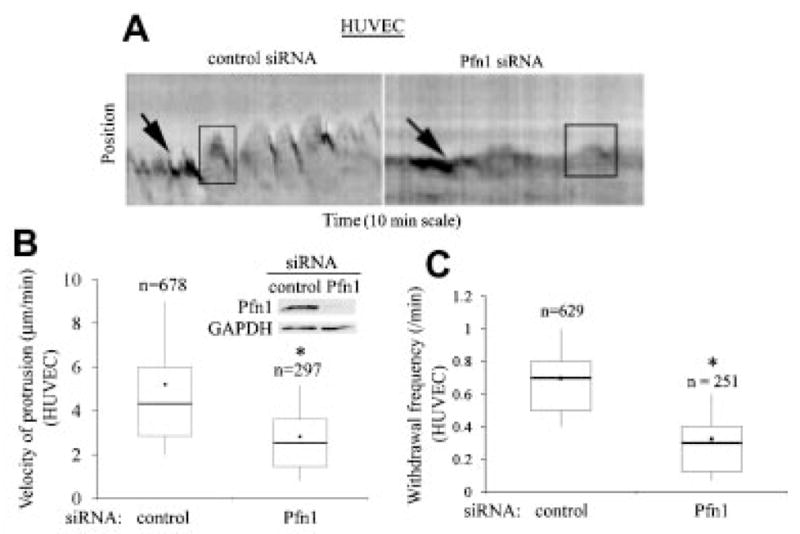
Loss of Pfn1 expression leads to slower protrusion in HUVEC cells. A: Kymographs representing lamlellipodial dynamics of HUVEC with or without Pfn1 depletion (leading edge traces are marked by the black arrows and the boxes outlining the saw-tooth waveforms represent typical lamellipodial protrusion and withdrawal events). B: A Box and whisker plot comparing the actual protrusion velocities between control and Pfn1-siRNA treated HUVEC. These data are based on analyses of 25 control and 22 Pfn1-siRNA transfected cells pooled from three independent experiments (“n” represents the total number of protrusion events analyzed in each case). The inset shows Pfn1 downregulation in HUVEC 96 h after siRNA treatment where GAPDH immunoblot serves as the loading control. C: A box and whisker plot comparing the frequency of lamellipodial withdrawal between control and Pfn1-depleted HUVEC (“n” represents the total number of kymographs analyzed in each case). The asterisk marks represent P < 0.05.
Silencing Pfn1 expression enhances VASP localization at the leading edge
Stable lamellipodial protrusion is one of the characteristic features of smooth and gliding type of cell movement. It has been recently shown that fish keratocytes (an extremely fast migrating epithelial cell type) migrate faster and in a smooth, gliding fashion when Ena/VASP family proteins are enriched at their leading edge (Lacayo et al., 2007). Leading edge targeting of Ena/VASP has also been shown to result in faster membrane protrusion (Bear et al., 2002). We therefore asked whether sub-cellular distribution of Ena/VASP in MDA-231 cells is altered by Pfn1-depletion. We focused on VASP because it is the most well-characterized member of the protein family. Since VASP is recruited to the leading edge in a protrusion-dependent manner, we performed costaining of VASP and phalloidin (to identify the leading edge of lamellipodia) of MDA-231 cells under different siRNA transfection conditions. Only those cells which formed prominent lamellipodia (identified from phalloidin-stained images) were selected for comparison of VASP distribution between the different treatment groups (Fig. 5A). VASP was localized at the leading edge of lamellipodia (arrowheads), focal adhesions (thick arrows) and occasionally, in dorsal ruffles (thin arrow). VASP signal at the very leading edge of lamellipodia was found to be conspicuously higher in Pfn1-depleted cells. Fluorescence intensity analyses of line scans across the leading edge showed that the average signal intensity of VASP at the very leading edge is almost fourfold higher in Pfn1-depleted cells than in control-siRNA treated cells (Fig. 5B). Immunoblot analyses confirmed that silencing Pfn1 expression does not alter the overall expression level of VASP in MDA-231 cells (Fig. 5C). Since VASP binds to barbed ends of actin filaments (Bear et al., 2002) and previous studies have shown that cofilin, an F-actin severing protein, plays a key role in generating barbed ends of actin filaments in breast cancer cells (Chan et al., 2000), we also compared the expression level of cofilin between control and Pfn1-depleted cells. Immunoblot data showed that cofilin expression in MDA-231 cells is not altered by Pfn1 depletion (Fig. 5C). It is, however, noteworthy here that Pfn1 depletion results in a slight reduction in the total actin level in MDA-231 cells as demonstrated previously by our group (Zou et al., 2007). Finally, to determine whether stronger VASP localization at the leading edge as a result of Pfn1 depletion is a reproducible feature in other cell types, we performed similar immunostaining experiments with HUVEC and normal HMEC. Interestingly, both cell lines also showed stronger VASP signal at the leading edge in the absence of Pfn1 expression therefore substantiating our findings (Fig. 5D).
Fig. 5.
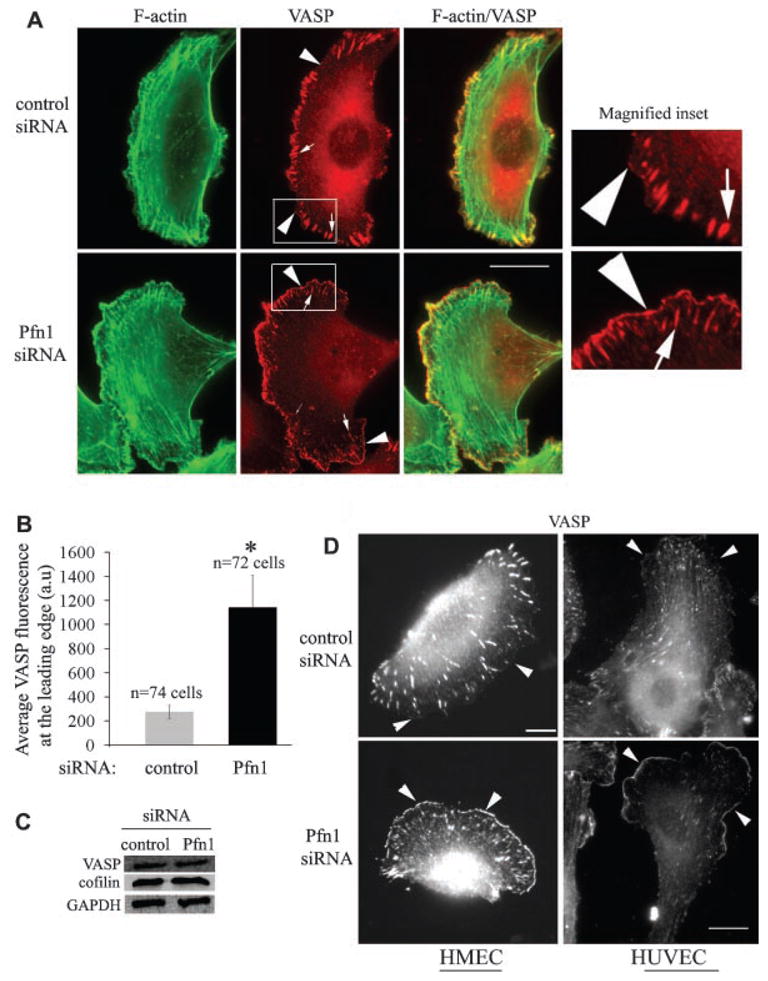
Silencing Pfn1 expression enhances leading edge localization of VASP. A: VASP (red) and phalloidin (green) costaining of MDA-231 cells show stronger leading edge (arrowhead) localization of VASP in Pfn1-deficient cells (VASP localization at focal adhesion and ruffles are marked by thick and thin arrows, respectively; scale—10 μm). Magnified forms of the insets (outlined by the boxes) are displayed in the right most parts. B: A bar graph comparing the relative intensity of VASP staining at the lamellipodial edge between control and Pfn1-depleted MDA-231 cells (a.u., arbitrary units; data summarized from two independent experiments and the asterisk mark denotes P < 0.05). C: Silencing Pfn1does not alter the total expression level of VASP and cofilin in MDA-231 cells (GAPDH blot serves as the loading control). D:VASP immunostaining of HMEC and HUVEC show similar VASP enhancement at the leading edge as a result of Pfn1 depletion. Scale bars in all images represent 20 μm.
Ena/VASP is responsible for hypermotile phenotype of Pfn1-deficient MDA-231 cells
Inhibition of Ena/VASP function leads to cell-type specific changes in cell migration. Leading edge targeting of VASP correlates with faster migration of fish keratocytes (Lacayo et al., 2007), but in the case of fibroblasts, inhibition or deletion of Ena/VASP results in increased motility (Bear et al., 2000). To determine whether lamellipodial targeting of VASP is responsible for creating hyper-motile phenotype of Pfn1-deficient MDA231 cells, we adopted a previously described strategy to inhibit Ena/VASP function by expressing Ena-VASP homology-1 (EVH1) binding motifs of ActA fused to a mitochondrial membrane anchor (EGFP-FPPPP-mito: referred to as “FP4-mito here”). This particular construct effectively depletes all Ena/VASP proteins from their normal sites of function and sequesters them on mitochondria (Bear et al., 2000). A similarly structured construct deficient in binding to Ena/VASP proteins (EGFP-APPPP-mito: referred to as “AP4-mito”) was used as a control. To demonstrate the efficacy of this construct in our cell system, we transfected Pfn1-siRNA treated MDA-231 cells with either AP4-mito or FP4-mito construct and then examined VASP localization by immunostaining (Supplemental Fig. S2). As expected, VASP distribution was not affected by expression of the AP4-mito construct: Pfn1-siRNA transfected cells bearing this construct still displayed strong VASP staining at their leading edges. However, cells expressing FP4-mito showed no VASP signal at the leading edge or in focal adhesions and instead exhibited the expected mitochondrial sequestration of VASP (specificity and efficacy of these two constructs were also confirmed for control-siRNA treated cells in a similar manner—data not shown).
Next, we performed time-lapse imaging of control and Pfn1-siRNA bearing MDA-231 cells following transfection with either AP4-mito or FP4-mito construct and compared the migration speed of EGFP-negative (only carries siRNA) and EGFP-positive (expresses either AP4-mito or FP4-mito against siRNA background) cells. Figure 6A shows that for either of the siRNA treatment groups, there was no significant difference in the average speed of migration between EGFP-negative and the corresponding AP4-mito bearing cells. Expression of FP4-mito reduced the average speed of migration of control siRNA treated cells by a nominal 15% although this difference was not statistically significant. The average speed of AP4-mito expressing Pfn1-depleted cells was 50% higher compared to the corresponding control siRNA-treated cells. The FP4-mito construct, however, reduced the speed of Pfn1-deficient cells by nearly 65%; the average speed of these cells was in fact 40% less than that of FP4-mito bearing control siRNA treated cells. Overall these data suggest that Ena/VASP compensates for the loss of Pfn1 and further augments the overall motility of MDA-231 cells when Pfn1 expression is silenced.
Fig. 6.

Mitochondrial sequestration of Ena/VASP dramatically impairs themotility and lamellipodial protrusion ofPfn1-deficient MDA-231 cells. A: A box-whisker plot summarizes the relative speed of control and Pfn1-siRNA treated cells with (FP4-mito) or without (either AP4-mito or EGFP-negative) mitochondrial sequestration of Ena/VASP (all data are normalized to AP-mito expressing control siRNA treated cells; “n” represents the number of cells analyzed for each group pooled from three independent 2-h long time-lapse experiments). The single and double asterisks represent P < 0.05 and P < 0.01, respectively. B: Representative kymographs showing leading edge traces (white arrows) of Pfn1-depleted MDA-231 cells with or without Ena/VASP inhibition. C: A box and whisker plot comparing the net protrusion velocities between the different treatment groups of Pfn1-deficient cells (data normalized to AP4-mito expressing cells and “n” indicates the number of cells in each group pooled from three-independent experiments; *P < 0.05).
Since Ena/VASP sequestration in Pfn1-deficient cells leads to a dramatic inhibition in motility, we speculated that lamellipodial protrusion of these cells might be compromised. To confirm this, we studied the leading edge dynamics of Pfn1-depleted MDA-231 cells with or without Ena/VASP sequestration. We found that the kymograph patterns of EGFP-negative and AP4-mito expressing cells were very similar; however FP4-mito expressing cells exhibited significant reduction in protrusion (as judged from a near-flat profile—Fig. 6B). Data analyses showed that Ena/VASP inhibition by FP4-mito resulted in nearly 70% reduction in the net protrusion velocity of Pfn1-defiicient MDA-231 cells (Fig. 6C), suggesting that lamellipodial targeting of Ena/VASP plays a major role in the protrusive activity of these cells.
Ena/VASP proteins bind to the barbed ends of actin filaments and support actin polymerization in the presence of actin filament capping proteins (Barzik et al., 2005; Pasic et al., 2008). This has been proposed to be the mechanism underlying Ena/VASP-driven membrane protrusion. Leading edge localization of VASP in fibroblasts and keratocytes requires free barbed ends of growing actin filaments (Bear et al., 2002; Lacayo et al., 2007). In our case, when we treated Pfn1-depleted MDA-231 cells with 100 nM cytochalasin D [CD; under these conditions CD works mainly as a pharmacological barbed-end capper without causing significant depolymerization of actin filaments (Bear et al., 2002)] for 30 min, VASP became completely delocalized from the leading edge while treatment with carrier DMSO had not effect at all (Supplemental Fig. S3). Phalloidin counterstaining confirmed that 100 nM CD treatment did not cause any major depolymerization of actin filaments; also the characteristic fan-shaped lamellipodial structure (arrow) was seen to be preserved. Antagonistic action of CD on lamellipodial targeting of VASP is consistent with VASP’s binding to the barbed ends of actin filaments as expected.
Membrane recruitment of Lamellipodin (Lpd), a member of MRL (Mig10/Rap1-interacting adaptor molecule/lamellipodin) family of adaptor proteins, has been identified as one of the potential mechanisms for targeting Ena/VASP to the leading edge (Krause et al., 2004). Lpd staining of Pfn1-siRNA treated MDA-231 cells showed its localization at the lamellipodial tip (arrows) and ruffles (arrowheads—Fig. 7A). There was also a very strong colocalization of Lpd and VASP at the leading edge (Fig. 7A). As shown previously in fibroblasts (Krause et al., 2004), Lpd targeting to the leading edge in our cells is not VASP-dependent since expressing FP4-mito that sequesters Ena/VASP to mitochondria still preserves strong leading edge staining of Lpd (Fig. 7B). Lpd targets to the cell membrane via anchoring of its PH (pleckstrin-homology) domain to PI(3,4)P2 [phosphatidylinositol-3,4-bisphosphate (Krause et al., 2004)]. PI(3,4)P2 is primarily generated by dephosphorylation of PIP3 by 5′-phosphatases. Since generation of PIP3 requires the action of PI3-Kinase (PI3K), we postulated that membrane targeting of VASP, if caused by Lpd, should also be sensitive to PI3K function. Indeed, when Pfn1-silenced cells were treated with 25 μM of LY 294002 (a specific inhibitor of PI3K) for 30 min, VASP delocalized from the leading edge (VASP localization at the focal adhesions remained intact after LY treatment and cells treated with diluent control DMSO still showed strong staining of VASP at the leading edge as expected—Fig. 7C). These sets of experimental observation are consistent with a model in which Lpd is involved in leading edge targeting of VASP in MDA-231 cells.
Fig. 7.
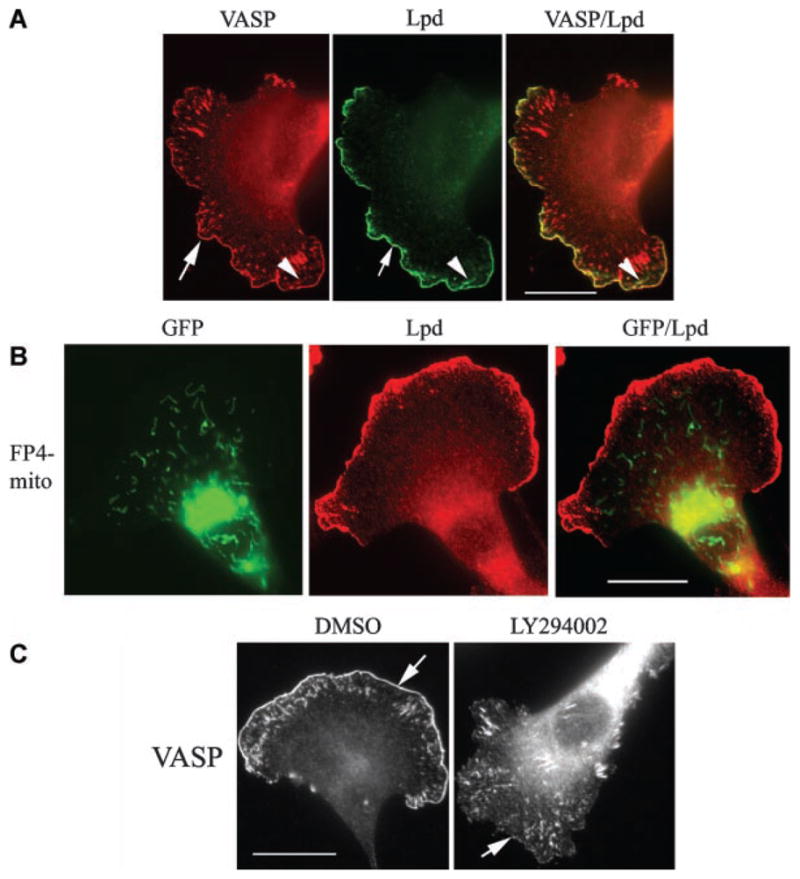
VASP is colocalized with Lpd at the leading edge in Pfn1-deficient cells. A: Immunostaining shows complete colocalization of VASP (red) and Lpd (green)at the leading edge (arrows) in Pfn1 depleted MDA-231 cells (appears yellow in the merged image).B:Pfn1-silenced MDA-231 cells expressing FP4-mito (green) maintain strong leading edge targeting of Lpd (red). The merged image in the right most part shows no overlapping of these two fluorescence signals. C: LY294002 treatment dramatically delocalizes VASP from the leading edge (arrows), while DMSO (diluent control) treated cells show strong lamellipodial distribution of VASP as expected. The scale bar in all images represents 20 μm.
Discussion
Gain-of-motility of certain types of adenocarcinoma cells induced by loss of Pfn1 expression has been previously reported by us and others (Wu et al., 2006; Zou et al., 2007); but, the mechanisms underlying this effect have not been delineated yet. This has been challenging particularly since Pfn1 has been traditionally viewed as a molecule required for cell migration. This study was the first attempt to identify a possible mechanism of how reduced Pfn1 expression could actually promote migration of breast cancer cells.
Our time-lapse data revealed that silencing Pfn1 expression leads to faster motility of both MDA-231 breast cancer cells and normal HMEC. This is in contrast to the behavior of HUVEC which exhibit significantly reduced motility after Pfn1 depletion as shown by our group in a previous study (Ding et al., 2006). To better understand the physical basis for cell-specific difference in the overall motility response resulting from Pfn1 depletion, we studied the lamellipodial dynamics of HUVEC and MDA-231 cells and found that in both cell types, the actual protrusion velocity is reduced when Pfn1 expression is suppressed. This is the first direct demonstration of the effect of Pfn1 depletion on the actual protrusion velocity of cells, and is consistent with previously shown slower intracellular propulsion of bacterial pathogens in the absence of Pfn1 (Loisel et al., 1999; Mimuro et al., 2000). Interestingly, Pfn1-deficient MDA-231 cells showed less withdrawal of protrusion, both in terms of its frequency and magnitude, compared to control cells which eventually resulted in a more effective net protrusion and an overall increase in motility.
We found a striking enrichment of VASP at the very leading edge of MDA-231 cells as a consequence of Pfn1 depletion, and this finding was also reproducible in both HMEC and HUVEC. We further showed that specifically Pfn1-deficient but not control MDA-231 cells become severely impaired in motility when Ena/VASP function is inhibited by mitochondrial sequestration. These data imply that (1) Pfn1 by itself still acts as a pro-migratory molecule in MDA-231 cells as literature has demonstrated in other cell types (Haugwitz et al., 1994; Ding et al., 2006), and (2) it is the enhanced Ena/VASP localization at the leading edge which rescues and further augments the overall motility of MDA-231 cells when Pfn1 expression is silenced. Although we have only studied VASP localization in this study, the other two Ena/VASP proteins, Mena and EVL have overlapping functions and localization with VASP. Therefore, it is likely that Mena also contributes to the hypermotile phenotype of Pfn1-deficient cells as it is expressed robustly in MDA-231 cells (unpublished observation in Gertler laboratory). The mitochondrial sequestering construct used in this study is effective against all three members of Ena/VASP protein family; whether there are any differences in the contributions of Mena or VASP to the phenotype of Pfn1-deficient cells remains to be determined.
Ena/VASP proteins have a well-established role in promoting membrane protrusion (Rottner et al., 1999). Previous studies showed that Ena/VASP increases the speed of intracellular actin-driven propulsion of Listeria monocytogenes (Laurent et al., 1999; Geese et al., 2002) and leading edge targeting of VASP correlates with faster protrusion in cells (Bear et al., 2002; Lacayo et al., 2007). Mitochondrial sequestration of Ena/VASP leading to reduced membrane protrusion in Pfn1-deficient MDA-231 cells is consistent with those findings. Ena/VASP can bind Pfn1-actin complexes in an orientation favorable for transfer of monomer to growing barbed ends. Ena/VASP is known to facilitate actin polymerization by antagonizing the action of F-actin capping proteins (Bear et al., 2002); the anti-capping activity does not require Pfn, but is enhanced by the presence of Pfn (Barzik et al., 2005). In the present case, leading edge targeting of VASP facilitates lamellipodial protrusion in a near absence of Pfn1 expression. In a previous study, we failed to detect Pfn2 expression in MDA-231 cells at least by immunoblotting (Zou et al., 2007). Although it is possible that very low levels of residual Pfn1 or Pfn2 enhance VASP-dependent actin polymerization, the contribution should be minimal. Therefore, it is likely that VASP drives membrane protrusion via Pfn-independent anti-capping action.
The effect of Ena/VASP perturbation on the overall cell motility is cell-type dependent. This apparent discrepancy between different model systems is not completely surprising. Although the actual protrusion velocity increases when VASP is targeted to the leading edge, the overall persistence of protrusion and the net cell movement decrease in the case of slow-moving fibroblasts. This is because VASP-mediated protrusions in fibroblasts tend to be unstable and are prone to withdrawal (Bear et al., 2002). The lamellipodial dynamics of rapidly migrating cell types such as fish epithelial keratocytes show much less oscillatory behavior when compared to fibroblasts, and therefore, the overall motility response of these cells to leading edge targeting of VASP can be quite different. Interestingly, our data showed that protrusions generated by Pfn1-deficient MDA-231 cells are less susceptible to withdrawal therefore implying enhanced stability of these protrusions, and this is likely to be a result of better protrusion-adhesion coupling. Although the underlying mechanism is not clear, one simple explanation could be that since Pfn1-silenced cells protrude slower, these protrusions may have a better chance of engagement to the underlying substrate via adhesion receptors.
A key question remaining to be investigated in detail is how absence of Pfn1 expression leads to VASP enrichment at the leading edge. One possibility is that both control and Pfn1-depleted cells have comparable efficiency of targeting VASP to the membrane during protrusion. Since Pfn1-depleted cells show evidence of less withdrawal of protrusion, it is possible that VASP-rich protruding membrane is better sustained and represented in fixed-cell preparations, as in immunostaining experiments. However, a comparable efficiency of targeting VASP to the leading edge is unlikely to result in a fourfold difference in membrane staining of VASP between control and Pfn1-depleted MDA-231 cells. An alternative explanation that we favor is that silencing Pfn1 expression actually increases the membrane targeting efficiency of VASP. Since a deletion mutant of VASP lacking its polyproline domain (a region that binds to Pfn1) targets normally to the leading edge (Loureiro et al., 2002), Pfn1 is unlikely to have a direct effect on Ena/VASP targeting to the leading edge. Therefore, we think loss of Pfn1 expression enhances leading edge targeting of VASP through an indirect mechanism, likely involving Lpd (a strong colocalization between Lpd and VASP at the leading edge is clearly consistent with this possibility).
How might loss of Pfn1 expression promote Lpd/VASP targeting to the leading edge? Since Lpd targets to cell membrane via its interaction with PI(3,4)P2, we propose a model (Fig. 8) that Pfn1 downregulation creates a hypermotile phenotype of breast cancer cells by augmenting PI(3,4)P2-directed signaling. At least two possibilities can be considered in this model. First, since Pfn1 is capable of binding to PI(3,4)P2 and affinity of Pfn1 for PI(3,4)P2 is, in fact, slightly higher than other phosphoinositides, such as PI(4,5)P2 and PI(3,4,5)P3 (Lu et al., 1996), Pfn1 can potentially compete with Lpd/VASP complex for binding to this lipid, and this inhibition is likely to be released when Pfn1 expression is knocked down. Second, Pfn1 has also been implicated in phospholipid turnover (Goldschmidt-Clermont et al., 1990). We have recently shown that overexpression of Pfn1 in MDA-231 cells dramatically suppresses PI(3,4,5)P3 generation (Das et al., 2009), Therefore, perturbing Pfn1 expression may also modulate membrane targeting of Lpd/VASP by altering PI(3,4)P2 content in cells. These possibilities need to be explored in the future for a more definitive mechanistic link between loss of Pfn1 expression and enhanced VASP targeting to the membrane ultimately leading to increased cell motility.
Fig. 8.

Proposed model of how loss of Pfn1 expression augments breast cancer cell motility.
In conclusion, we have now identified a possible mechanism of how loss of expression of a pro-migratory molecule like Pfn1 can result in increased migration of breast cancer cells through enhanced lamellipodial targeting of Ena/VASP proteins. We will need to determine the generalizability of our findings by expanding our studies to other breast cancer cell lines and other types of adenocarcinoma that have reduced Pfn1 expression compared to their normal counterparts.
Supplementary Material
Acknowledgments
We wish to thank Dave Gau, Tuhin Das, Anna DiRienzo and William Veon for technical assistance. This work was supported by grants from National Cancer Institute (R01-CA108607) and American Heart Association (0665414U) to PR, VA merit award to AW, NIH grant GM58801 and funds from the Ludwig center at MIT to FG.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Literature Cited
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280:28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fassler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. doi: 10.1083/jcb.148.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Golub TR, Lander S, Hynes RO. Genomic analysis of metastasis reveals an essential role of RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- Das T, Bae YH, Wells A, Roy P. Profilin-1 overexpression upregulates PTEN and suppresses AKT activation in breast cancer cells. J Cell Physiol. 2009;218:436–443. doi: 10.1002/jcp.21618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Lambrechts A, Parepally M, Roy P. Silencing profilin-1 inhibits endothelial cell proliferation, migration and cord morphogenesis. J Cell Sci. 2006;119:4127–4137. doi: 10.1242/jcs.03178. [DOI] [PubMed] [Google Scholar]
- Ferron F, Rebowski G, Lee SH, Dominguez R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007;26:4597–4606. doi: 10.1038/sj.emboj.7601874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geese M, Loureiro JJ, Bear JE, Wehland J, Gertler FB, Sechi AS. Contribution of Ena/VASP proteins to intracellular motility of listeria requires phosphorylation and proline-rich core but not F-actin binding or multimerization. Mol Biol Cell. 2002;13:2383–2396. doi: 10.1091/mbc.E02-01-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont PJ, Machesky LM, Baldassare JJ, Pollard TD. The actin-binding protein profilin binds to PIP2 and inhibits its hydrolysis by phospholipase C. Science. 1990;247:1575–1578. doi: 10.1126/science.2157283. [DOI] [PubMed] [Google Scholar]
- Gronborg M, Kristiansen TZ, Iwahori A, Chang R, Reddy R, Sato N, Molina H, Jensen ON, Hruban RH, Goggins MG, Maitra A, Pandey A. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol Cell Proteomics. 2006;5:157–171. doi: 10.1074/mcp.M500178-MCP200. [DOI] [PubMed] [Google Scholar]
- Haugwitz M, Noegel AA, Karakesisoglou J, Schleicher M. Dictyostelium amoebae that lack G-actin-sequestering profilins show defects in F-actin content, cytokinesis, and development. Cell. 1994;79:303–314. doi: 10.1016/0092-8674(94)90199-6. [DOI] [PubMed] [Google Scholar]
- Holt MR, Koffer A. Cell motility: Proline-rich proteins promote protrusions. Trends Cell Biol. 2001;11:38–46. doi: 10.1016/s0962-8924(00)01876-6. [DOI] [PubMed] [Google Scholar]
- Janke J, Schluter K, Jandrig B, Theile M, Kolble K, Arnold W, Grinstein E, Schwartz A, Estevez-Schwarz L, Schlag PM, Jockusch BM, Scherneck S. Suppression of tumorigeniciy in breast cancer cells by the microfilament protein profilin 1. J Exp Med. 2000;191:1675–1685. doi: 10.1084/jem.191.10.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson L, Nystrom LE, Sundkvist I, Markey F, Lindberg U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol. 1977;115:465–483. doi: 10.1016/0022-2836(77)90166-8. [DOI] [PubMed] [Google Scholar]
- Krause M, Leslie JD, Stewart M, Lafuente EM, Valderrama F, Jagannathan R, Strasser GA, Rubinson DA, Liu H, Way M, Yaffe MB, Boussiotis VA, Gertler FB. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev Cell. 2004;7:571–583. doi: 10.1016/j.devcel.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Lacayo CI, Pincus Z, VanDuijn MM, Wilson CA, Fletcher DA, Gertler FB, Mogilner A, Theriot JA. Emergence of large-scale cell morphology and movement from local actin filament growth dynamics. PLoS Biol. 2007;5:e233. doi: 10.1371/journal.pbio.0050233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts A, Braun A, Jonckheere V, Aszodi A, Lanier LM, Robbens J, Colen IV, Vandekerckhove J, Fassler R, Ampe C. Profilin II is alternatively spliced, resulting in profilin isoforms that are differently expressed and hae distinct biochemical properties. Mol Cell Biol. 2000;20:8209–8219. doi: 10.1128/mcb.20.21.8209-8219.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Loisel TP, Harbeck B, Wehman A, Grobe L, Jockusch BM, Wehland J, Gertler FB, Carlier MF. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J Cell Biol. 1999;144:1245–1258. doi: 10.1083/jcb.144.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Loureiro JJ, Rubinson DA, Bear JE, Baltus GA, Kwiatkowski AV, Gertler FB. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Mol Biol Cell. 2002;13:2533–2546. doi: 10.1091/mbc.E01-10-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PJ, Shieh WR, Rhee SG, Yin HL, Chen CS. Lipid products of phosphoinositide 3-kinase bind human profilin with high affinity. Biochemistry. 1996;35:14027–14034. doi: 10.1021/bi961878z. [DOI] [PubMed] [Google Scholar]
- Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimuro H, Suzuki T, Suetsugu S, Miki H, Takenawa T, Sasakawa C. Profilin is required for sustaining efficient intra- and intercellular spreading of Shigella Flexneri. J Biol Chem. 2000;275:28893–28901. doi: 10.1074/jbc.M003882200. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Spengler BA, Kisselbach K, Evans AE, Biedler JL, Ross RA. The human non-muscle α-actinin protein encoded by the ACTN4 gene suppresses tumorigenicity of human neuroblastoma cells. Oncogene. 2000;19:380–386. doi: 10.1038/sj.onc.1203310. [DOI] [PubMed] [Google Scholar]
- Pasic L, Kotova T, Schafer DA. Ena/VASP proteins capture actin filament barbed ends. J Biol Chem. 2008;283:9814–9819. doi: 10.1074/jbc.M710475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner K, Behrendt B, Small JV, Wehland J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1999;1:321–322. doi: 10.1038/13040. [DOI] [PubMed] [Google Scholar]
- Roy P, Jacobson K. Overexpression of profilin reduces the migration of invasive breast cancer cells. Cell Motil Cytoskeleton. 2004;57:84–95. doi: 10.1002/cm.10160. [DOI] [PubMed] [Google Scholar]
- Suetsugu S, miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 1998;17:6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syriani E, Gomez-Cabrero A, Bosch M, Moya A, Abad E, Gual A, Gasull X, Morales M. Profilin induces lamellipodia by growth factor-independent mechanism. FASEB J. 2008;22:1581–1596. doi: 10.1096/fj.06-7654com. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Mullaur L, Ogiso Y, Fujita H, Moriya S, Furuuchi K, Harabayashi T, Shinohara N, Koyanagi T, Kuzumaki N. Gelsolin: A candidate for suppressor of human bladder cancer. Cancer Res. 1995;55:3228–3232. [PubMed] [Google Scholar]
- Verheyen EM, Cooley L. Profilin mutations disrupt multiple actin-dependent processes during drosophila development. Development. 1994;120:717–728. doi: 10.1242/dev.120.4.717. [DOI] [PubMed] [Google Scholar]
- Wang FL, Wang Y, Wong WK, Liu Y, Addivinola J, Liang P, Chen LB, Kantoff PW, Pardee AB. Two differentially expressed genes in normal human prostate tissue and in carcinoma. Cancer Res. 1996;56:3634–3637. [PubMed] [Google Scholar]
- Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14:461–469. doi: 10.1016/j.tcb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Wu N, Zhang W, Yang Y, Liang YL, Wang LY, Jin JW, Cai XM, Zha XL. Profilin 1 obtained by proteomic analysis in all-trans retinoic acid-treated hepatocarcinoma cell lines is involved in inhibition of cell proliferation and migration. Proteomics. 2006;6:6095–6106. doi: 10.1002/pmic.200500321. [DOI] [PubMed] [Google Scholar]
- Zou L, Jaramillo M, Whaley D, Wells A, Panchapakesa V, Das T, Roy P. Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br J Cancer. 2007;97:1361–1371. doi: 10.1038/sj.bjc.6604038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.