

Abstract
With a view toward developing a rugged microflow cytometer, a sheath flow system was micromachined in hard plastic (polymethylmethacrylate) for analysis of particles and cells using optical detection. Six optical fibers were incorporated into the interrogation region of the chip, in which hydrodynamic focusing narrowed the core stream to ∼35 μm×40 μm. The use of a relatively large channel at the inlet as well as in the interrogation region (375 μm×125 μm) successfully minimized the risk of clogging. The device could withstand pressures greater than 100 psi without leaking. Assays using both coded microparticles and cells were demonstrated using the microflow cytometer. Multiplexed immunoassays detected nine different bacteria and toxins using a single mixture of coded microspheres. A549 cancer cells processed with locked nucleic acid probes were evaluated using fluorescence in situ hybridization.
Keywords: Flow cytometry, Microflow cytometer, Hydrodynamic focusing
Introduction
Portable microflow cytometers are under development for a wide variety of point-of-use applications in environmental and clinical analyses. Indeed at least six particle counters for very specific applications are already on the market [1, 2]. However, microflow cytometers capable of more generic analyses are still under development. While the greatest focus to date has been on microfluidic sheathing and integration of optics, several systems have been reported that also integrate pumps, valves, or reagent reservoirs [3, 4].
While the earliest microflow cytometers used channels etched in glass or silicon [5–7], the complexity of achieving focusing in both the horizontal and vertical directions encouraged more complex devices that were easier to fabricate using soft lithography. Thus for the past decade, the majority of pressure-driven microflow cytometers used polydimethylsiloxane (PDMS) and lithography technology to fabricate the fluidic channels and frequently to serve as the substrate for integration of the optical elements. However, PDMS has several disadvantages: replication of PDMS structures for mass manufacturing is not reliable, PDMS deforms under pressure, inflows under different pressures can generate pulsing in microchannel walls, and PDMS swells in the presence of solvents. Furthermore, PDMS seals are generally meant only for low pressure operations, and PDMS devices alternately exposed to wet and dry conditions for extended periods usually develop leaks.
Using PDMS, Ligler, Howell, and colleagues demonstrated a microflow cytometer in which passive hydrodynamic focusing was employed to completely surround the sample stream with sheath fluid and focus it in the laser beam [8–10]. In this device, the hydrodynamic forces were generated by chevron grooves strategically imbedded on the walls of the microchannel. The sheath solution was introduced by one pump into two inlets, one on either side of the sample inlet, to minimize pulsing. The two sheath streams confined the sample stream laterally in the middle of the channel. When the solutions reached the chevrons, the sheath solution flowing through the grooves generated a hydrodynamic lift that caused the sample stream to be squeezed in the vertical direction. Grooves on the top and bottom surfaces of the microchannel displaced the sample stream toward the center of the channel from both the top and bottom walls. As the number of chevron grooves increased, the height of the core sample decreased while the width increased.
Recently, several groups have fabricated microflow cytometers with two-dimensional sample stream focusing out of harder materials. The engineers at Translume have fabricated a sheath flow channel in glass based on the chevron design of Howell et al. [8] using direct-write laser illumination to both prepare the channels for etching and to create the waveguides [11]. This system is very attractive, but no reports of assay performance have yet appeared. Kummrow et al. [12] used hot embossing of polymethylmethacrylate (PMMA) to produce microflow cytometers with a single sheath stream introduced in four places along the channel for focusing the sample stream and with grooves for five optical fibers to measure light scatter and extinction. The mold insert for this system was fabricated by the same group using ultraprecision milling. An interesting approach using hybrid soft/hard devices has also been described by the Vellekoop group, beginning with an on-chip Coulter counter [13, 14] and evolving into a flow cytometer [13]. The flow channels are formed using a patterned SU-8 layer sandwiched between an etched silicon layer and a glass top layer. All of these devices could potentially solve the problems of pulsing of the channel walls, reproducible manufacturing, long-term monitoring, and extension of the types of fluids that can be analyzed. However, there is minimal data on assay performance, especially with fluorescence analysis, and no data on survival of the cytometers under the kinds of pressures that might be generated during cleaning procedures or in marine applications [15–17].
In selecting the design for a robust microflow cytometer, we made several choices that came with no guarantee of success. In avoiding PDMS due to the disadvantages mentioned above, we selected a plastic, PMMA, as the hard substrate material rather than glass or silicon. The cytometer design could be etched in glass or silicon, but PMMA provided the opportunity for rapid milling of prototype devices and resistance to breakage. The choice of milling, while it expedited prototype production, also limited the feature size and generated roughness at the channel walls. Since the core does not directly touch the channel walls as it passes through the interrogation region, the latter disadvantage was insignificant. However, the limitation on feature size influenced the size and placement of the chevron grooves. We had to consider the impact of this limitation during the design phase. In order to prevent clogging, we eliminated the tapered entrance of the sample channel into the interrogation channel, risking back pressure effects as the volumetric sheath flow rate was much higher than the sample flow rate. We angled the sheath flow inlets to minimize momentum effects that would contribute to such a back pressure, but only testing would determine whether we could achieve sufficiently high flow-rate ratios for confinement of the sample stream. If the sample stream was unstable, measurements would not be reproducible and the individual bead sets would not be distinguishable in the coded bead arrays.
To evaluate the performance of the hard microflow cytometer, we compared the analysis of both coded bead arrays and cell assays to data obtained using benchtop commercial systems. The samples were prepared and split into two aliquots for measurement in the hard microflow cytometer and the conventional systems. The data demonstrate that the hard flow cytometer has similar sensitivity and reproducibility for two- and three-color fluorescence analysis of cells and microspheres to that obtained using larger commercial systems.
Experimental
Assay reagents
The following reagents were kindly provided by Kirkgaard Perry Laboratories (Gaithersburg, MA): heat-deactivated bacteria; Escherichia coli O157:H7, Salmonella typhimurium, Campylobacter jejuni, Listeria monocytogenes, as well as affinity-purified antibodies; goat anti-E. coli, goat anti-Listeria, goat anti-Salmonella, and the streptavidin–phycoerythrin. Additional antibodies were purchased from Meridian Life Science (Saco, ME): Goat anti-Campylobacter and Rabbit anti-Shigella, The Mab-T14 anti-Francisella tularensis (tracer) was the kind gift of Dr. Peter Sveshnikov, Research Center for Molecular and Diagnostic Therapy (Moscow, Russia; Mab-T14 can also be obtained from HyTest Ltd. (Turku, Finland). Cholera toxin was purchased from Calbiochem/EMD Chemicals (Gibbstown, NJ). The rabbit anti-cholera toxin (CTX) was obtained from AbD Serotec (Raleigh, NC). Staphylococcal enterotoxin B (SEB) was purchased from Toxin Technology. Rabbit anti-SEB (tracer) and MAb 3b2a anti-SEB (capture) and goat anti-F. tularensis (capture) were kindly provided by the Naval Medical Research Center (Silver Spring, MD). The ricin was purchased from Vector (Burlingame, CA). The llama anti-ricin (tracer) and llama anti-SEB (tracer) were purified at NRL as described previously from plasma obtained llamas immunized using ricin or SEB toxoid at Triple J Farms (Bellingham, WA) under contract for production of custom polyclonal antibodies. Chicken IgY was from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Bovine serum albumin (BSA) and Tween-20 were obtained from Sigma Aldrich (St Louis, MO). 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride, N-hydroxysulfosuccinimide (sulfo-NHS), and NHS-LC-biotin were purchased from Pierce (Rockford, IL). Carboxyl xMAP microsphere sets (Luminex Corp., Austin, TX) were conjugated with capture antibodies using manufacturer's instructions for a two-step carbodiimide coupling reaction. Upon reaction completion, microspheres were washed by centrifugation and stored in phosphate-buffered saline containing 0.05% Tween-20 and 1 mg/mL bovine serum albumin (PBSTB) at 4 °C in the dark. Unless otherwise noted, the same antibodies were also utilized as both the capture as well as the tracer. The tracer antibodies were prepared by the addition of biotin. The antibodies were reacted with biotin-LC-LC-NHS (Pierce, Rockford, IL) at a 20:1 ratio for 1 h in PBS and then separated using gel filtration on a Bio-gel P10 column (Bio-Rad, Hercules, CA)
For the fluorescence in situ hybridization (FISH) experiments, two biotinylated locked nucleic acid (LNA)-modified DNA oligonucleotides were used. The first LNA probe (β-actin LNA), which is complementary to β-actin mRNA, was purchased from Exiqon (Woburn, MA, USA) and has the sequence 5′-biotin-ctcattgtagaaggtgtggtgcca-3′ with a proprietary LNA spiking pattern. The second probe is a nonspecific LNA probe (control LNA) purchased from IDT (Coralville, IA, USA) with the sequence 5′-biotin-gtGtaAcaCgtCtaTacGccCa-3′ (with LNA monomers in uppercase letters). A549 cancer cells (American Type Culture Collection, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (Cellgro, Herndon, VA), supplemented with 10% (v/v) fetal calf serum and 1% (v/v) penicillin/streptomycin (Sigma, St. Louis, MO). Detachment from culture flasks was performed using trypsin-ethylenediaminetetraacetic acid (Cellgro, Herndon, VA). Paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in 5% acetic acid in phosphate-buffered saline (PBS) was used for flow-FISH experiments.
Cytometer fabrication
The microflow cytometer was made in two halves (shown in Fig. 1) out of transparent pieces of polymethylmethacrylate (Plexiglas®; Arkema Inc, Philadelphia, PA). Milling was chosen as the manufacturing method due to the ease and quick turnaround capability in low volume prototyping. A computer numerical control (CNC) milling system (Mini Mill, Haas Automation Inc, Oxnard, CA) was used. The 3D model of the cytometer device was designed using Autodesk Inventor (Autodesk, Inc., San Rafael, CA) which was imported to Edgecam (Planit CAD/CAM Software, Charlotte, NC) to generate the CNC codes. The fluidic channel with integrated chevron grooves and fiber channels were machined into the base piece while the top piece had only the corresponding chevron grooves. The fluidic inlets and waste outlet were made in the top piece. The fluidic channel was 375 μm×125 μm (width×depth) and the fiber channels were 125 μm×125 μm. The two halves of the device had corresponding clearance holes for machine screws for device assembly. For the device used in this work, three sets of chevrons, 250 μm×125 μm (width×depth) were used. Because of the nature of the fabrication technique used, the leading edge of each chevron was designed for a radius of 125 μm, the radius of the smallest end mill tool available (0.01″ or 254 μm diameter). All tool bits were purchased from McMaster-Carr. Once the basic features of the fluidic system were finished, the two halves were aligned under a microscope and secured using a few screws. Then, threaded holes for the optical fiber channels were drilled for securing the fibers in place. Once all the machining was completed, the two halves of the cytometer were cleaned thoroughly. The two halves were then aligned and assembled on a base plate on which the optical fibers were secured. Optical fibers were prepared by removing buffer and jacket and cleaving the ends. The fibers were then inserted and secured using coned HPLC fittings (F-125, 1-PIECE MicroTight®, Upchurch Scientific (Oak Harbor, WA). Except for the portion of fiber that was laid down in the fiber channel, the buffer was left on to facilitate the mechanical locking using the cone nut. The lock nuts also acted as plugs for the fiber channels, preventing fluidic leakage. Polyetheretherketone (PEEK) tubing (Upchurch Scientific, Oak Harbor, WA; 400 μm internal diameter) was then inserted and secured in the fluidic inlets and outlet using 5-min epoxy (ITW Devcon, Danvers, MA). The sheath solution was first pumped through silicone tubing into a homemade pulse dampener, made from a 50-ml centrifuge tube, which was then connected to the two inlets in the cytometer using a Tee fitting (Cole-Parmer, Vernon Hills, IL.) A miniature peristaltic pump (Instech P625/275.143 with 0.031″ tubing (787 μm), Instech Laboratories, Inc., Plymouth Meeting, PA) was utilized for the sheath solution. The sample solution was injected using a precision syringe pump (CAVRO XE 1000, Tecan Systems, Inc., San Jose, CA.)
Fig. 1.
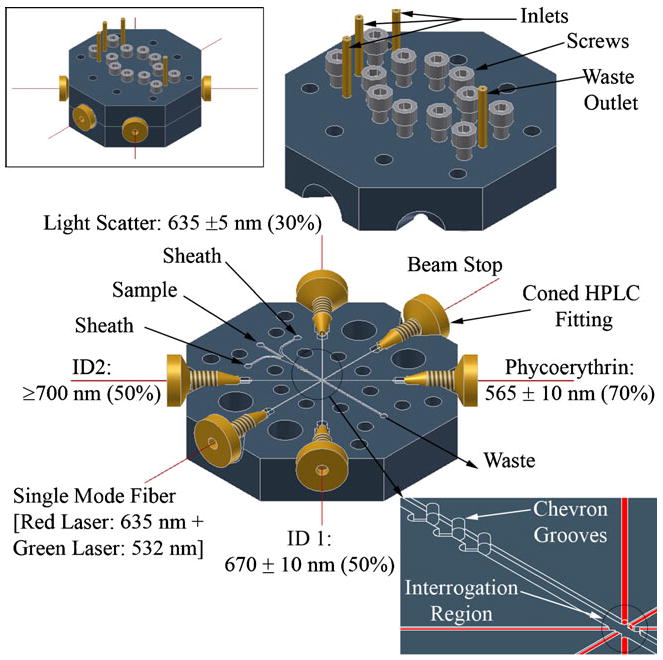
Overview of the hard PMMA microflow cytometer. Top inset assembled cytometer. Bottom inset details of chevrons grooves and optical fibers assembly at the interrogation region
Optical system
The layout of the optics was similar to that implemented in the soft (PDMS) microflow cytometer previously used for multiplexed assays with coded microspheres and has been described in detail elsewhere [10]. Unlike the previous optical configuration, light from diode lasers at 532 nm (GM32-10H, 10 mW, Intelite, Inc. Minden, NV) and 635 nm (LAS-200-635-15, 15 mW, Lasermax Inc., Rochester, NY) was combined into a single-mode fiber using a custom 532/635 nm WDM fiber coupler (Gould Fiber Optics, Millersville, MD), which provided excitation light normal to the fluid flow. A multimode fiber was positioned opposite the excitation fiber to guide excess light away from the channel and thus minimize scatter off the microchannel walls. Four multimode fibers (Fiber Instrument Sales, Inc., Oriskany, NY) collected the light signals from microspheres passing through the interrogation region. Large-angle scatter (45°) from the microspheres was collected at 635±5 nm. A 670±10 nm bandpass filter (665DF20, Omega Optical, Inc., Brattleboro VT) and a 700 nm long-pass (LL700, Corion Corp., Franklin MA) filter were used for microsphere identification, and a 565±10 nm bandpass filter was used for detection of phycoerythrin fluorescence (565WB20, Omega Optical, Inc., Brattleboro, VT). The outputs of the photomultiplier tubes (PMTs) were recorded using an analog-to-digital converter (NI USB-6251M, National Instruments, Austin, TX). Data acquisition and data analysis software was written in LabWindows/CVI (National Instruments, Austin, TX).
Multiplexed microsphere assays
Microsphere sets with attached capture antibodies were mixed, then washed and re-suspended in PBSTB. The microsphere set-antibody combinations used were 100-anti-Shigella, 98-anti-Campylobacter, 96-BSA, 92-anti-SEB, 81-anti-CTX, 79-chicken IgY, 77-anti-Tularemia, 71-anti-ricin, 58-anti-Salmonella, 56-anti-E. coli, and 50-anti-Listeria. In a 96-well filter plate, serial dilutions of positive control were prepared in 100 μL of PBSTB per well: 107, 106, 105, 104, 103, 102, and 101 cells/mL for bacteria and 1000, 200, 40, 8, 1.6, 0.32, and 0.064 ng/mL for toxins. An aliquot of washed microspheres were added to each well, approximately 200 microspheres of each set (2,200 total), and incubated for 30 min at room temperature in the dark. Next, each well was washed with 100 μL of PBSTB and 50 μL of tracer antibody at 10 μg/mL was added. The single tracer antibody of the assay being tested was used for the dose–response curves. The full mixture of tracer antibodies was used for multiplex assay confirmation at the limit of detection determined by the dose response curves. After 30 min of incubation, the tracer antibody containing buffer was removed and streptavidin–phycoerythrin (10 μg/mL) was added for 15 min before analysis. Aliquots of each sample were tested using both the hard microflow cytometer and the Luminex 100 (www.luminexcorp.com). The Luminex system is specifically designed only for performing assays using the coded microspheres.
Samples were introduced into the microflow cytometer at 10 μL/min using the CAVRO pump. Sheath fluid was pumped at 500 μL/min to focus the microspheres in the interrogation region. Data was acquired at 250 kHz and analyzed using custom LabWindows programs. The cytometer analyzes every microsphere passing through the interrogation region, taking 6–10 measurements for every event, and produces an average fluorescence measurement per microsphere. Each analyzed population set, containing approximately 200 microspheres, generated a mean phycoerythrin fluorescence measurement. The mean of means from multiple experiments was used to calculate the standard error to show the level of variation in the data.
Dose–response curves were generated using serial dilutions of the target analytes. Using the zero concentration fluorescence measurement as a background, three times the standard error was added to the value to establish a minimum limit of detection. From this number, we determined a conservative limit of detection quoted as the lowest concentration actually measured that was above the mean of the values without analyte plus three times the standard error of the mean.
Fluorescence in situ hybridization assays
Flow cytometry-FISH was performed using previously described methods [18, 19]. Briefly, A549 cells were fixed in a solution of 4% paraformaldehyde and 5% acetic acid in phosphate-buffered saline (PBS) for 10 min at room temperature. After fixation, the cells were washed twice in PBS. The cells were then permeabilized for 30 min at 37 °C in 0.1 μg/mL Proteinase K (Ambion/Applied Biosystems) in TE buffer and washed twice in PBS. The cells were then separated into aliquots containing 1.5×106 cells each. Next, a 30 min pre-hybridization step was carried out in hybridization buffer (50% formamide, 10% dextran sulfate, 50 mM NaPi (pH=7.0), 2X saline–sodium citrate buffer, and 10 μg sheared salmon sperm DNA.) Following the pre-hybridization, a solution of 0.1X saline–sodium citrate (SSC) buffer was added to reduce the viscosity, followed by centrifugation (2,500 rcf) and removal of the supernatant. Hybridization was then carried out for 90 min. at 60°C in 100 μl of hybridization buffer containing 20 pmol of β-actin LNA.
Following hybridization, the cells were washed twice at 65 °C with 50% formamide and 2X SSC buffer for 10 min each, and twice with 0.1X SSC buffer for 20 min each. Negative controls were subjected to the same conditions as above and consisted of either the control LNA or no LNA instead of the β-actin LNA. Following posthybridization washes, the cells were blocked with 1X in situ hybridization blocking buffer (Vector Laboratories, Burlingame, CA, USA) for 30 min at room temperature and then stained for 15 min with 10 μg/mL phycoerythrin (PE)-conjugated streptavidin. The cells were washed twice with 0.1X SSC buffer and twice with PBS for 5 min each.
To measure the cell autofluorescence, a sample of cells was subjected to the flow-FISH conditions above, containing β-actin LNA but without the PE tracer. Other controls included no LNA but with PE tracer to measure nonspecific PE binding to the cells and scrambled LNA sequence with PE tracer as a control for the specificity of the LNA hybridization. Flow cytometry analysis was performed on aliquots of each sample using both the hard microflow cytometer and a commercial cytometer (Accuri C6, Ann Arbor, MI) equipped with a 488 nm laser and emission filters for PE (www.accuricytometers.com).
Results and Discussion
Simulations
In order to optimize the system stability and sensitivity, it was necessary to focus the sample stream to a narrow diameter in the center of the channel to ensure that the particles pass through the very center of the optical interrogation region. To that end, the COMSOL Multiphysic® finite element package was used to simulate the fluid flow and to determine a design that would give the best possible signal. The simulation was carried out with preferred experimental conditions, such as channel size (large enough to prevent clogging and to be compatible with available endmill tool bit sizes), relative flow rates between the sheath and the sample solutions (maximum ratio compatible with pumps and stable sample flow), and also the overall flow velocity for a given channel size. The flow velocity or overall volumetric flow rate is a critical parameter as the data acquisition system has an absolute maximum speed at which it can acquire data on particles passing through the interrogation region.
To save computation time, we took advantage of the symmetry in channel design and only one half of the system was simulated. A non-slip boundary condition was applied to the channel walls, and the symmetry condition was applied at the vertical symmetry plane through the center of the channel. The simulation was carried out using a model with different numbers of chevron-shaped grooves on both the top and bottom of the channel [8] and the cross-section images were obtained at desired positions. The images were then reflected using Photoshop to render the full cross-section of the channel. Figure 2 shows the cross-sections (size and shape) of the sample fluid as a function of the sheath-to-sample flow-rate ratio and the numbers of integrated chevron grooves. Ideally, the sample should be a very tight square or circle. In lieu of that, the best option for the optical detection system being used in this work is to have the sample better focused in the vertical direction (wide and short as oppose to narrow and tall) such that the particles would always pass through the center of the interrogation region.
Fig. 2.
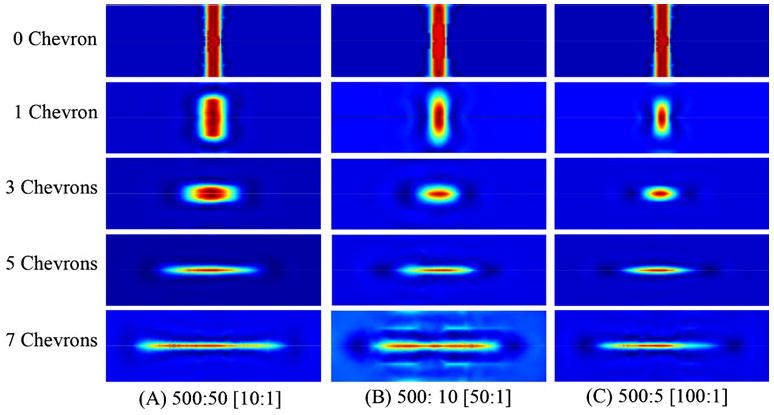
COMSOL simulation results. The cross-sections show the channel 2 mm past the grooves at a sheath (BLUE) to core (RED) flow ratio of (A) 500:50, (B) 500:10, and (C) 500:5 for different numbers of chevron grooves. The channel is 375 μm×125 μm
Based on the simulations, a design with three-chevron grooves in both the top and bottom of the channel, with a flow-rate ratio of 500:10, was selected. This should provide a focused sample stream with dimensions of 45 μm×20 μm (width×height). Running the experiment at 500:5 (100:1) would tighten the sample cross-section a bit, but not enough to sway the design decision as the syringe pump can inject the sample more reliably at 10 μl/min than at 5 μl/min. It is possible to maintain the sample flow rate at 10 μl/min and increase the sheath flow rate to 1000 μl/min to achieve the 100:1 flow ratio. However, this would double the linear velocity of the particles, requiring an even faster data acquisition system.
Characterization of the three-chevron hard microflow cytometer
The basic design of the current hard microflow cytometer is an adaptation of the PDMS microflow cytometer previously reported by our group [9, 10]. The PDMS cytometer used a 390 μm×130 μm (width×height) channel with four chevron grooves, and it had a tapered (reduced width) sample inlet. The current hard microflow cytometer, on the other hand, uses 375 μm×125 μm channel with three-chevron grooves. The sample inlet in this new design maintains the same size as the main channel (375 μm×125 μm). By making the sample inlet the same size as the main channel, larger particles can be easily introduced to the system. It is more difficult for debris, either from the sample or sheath solution, to clog the system. The larger inlet also reduced the required pressure to pump the sample and generates a more stable sample stream. Furthermore, any clog in the hard microflow cytometer can be flushed with a high pressure which the PDMS cytometer may not be able to withstand. Another important distinction between the PDMS and the hard microflow cytometer is the geometry of the chevron grooves. In the PDMS cytometer, the chevrons are 70 μm×50 μm (width×height) and the leading edges are sharp. With the hard microflow cytometer, the chevrons grooves are 250 μm×125 μm (width×height) and the leading edges are rounded. The chevron width of 250 μm was selected so that the same end mill tool bit can make the grooves as well as the main channel. Smaller chevrons can be made if desired. The more critical changes in the hard cytometer chevrons are their depth and the roundedness of the leading edge. Both of these parameters cause the sample stream to flatten (in the vertical direction) more quickly compared to shallower and sharp chevrons such as those used in the PDMS cytometer. Device fabrication using CNC milling is compatible with a wider selection of materials for chemical/biocompatibility, and thus it is an attractive method for a low volume production environment. Although the PMMA channels are easier to clean than PDMS, PMMA is not compatible with most organic solvents.
For the optical detection system, the red and green lasers were coupled together into a single-mode fiber as the excitation source where two separate fibers were used in the original PDMS cytometer. In the PDMS cytometer, the green laser was positioned at 45° from the sample stream, which limited the fibers useable for PE detection. By coupling the two source fibers into one, more fibers are available for detecting signals of interest, enabling the detection of higher numbers of signals with better detection limits.
To verify the sheathing process with the hard microflow cytometer, a dye solution was used as sample for visualization. A stereomicroscope equipped with a CMOS camera (Moticam 1000, Motic Inc., Canada) was used to capture the images. To obtain images of the sheathing process from the side, a narrow cytometer with identical fluidic channel was fabricated. Since surfaces from the milling processes were not smooth, and thus not transparent, the channel could not be viewed directly from the side. To render the surface smooth and transparent, the surface was covered with a drop of PDMS and cured at 80°C for 1 h. The PDMS filled all the pits created on the surface during the milling process, making it transparent.
Figure 3 shows the images from the top and side of the channel. The sheath-to-sample flow ratio was 50:1 in these experiments. From the microscope images, the sheathed sample is approximately 35 μm×40 μm (width×height) while it is estimated to be 45 μm×20 μm from the COMSOL simulation result. Variation between the simulation and the experimental data may be due to the actual dimension achieved in the fabrication process where as the simulation used the exact dimension. During the device fabrication, the substrate is secured flat on the mill's work table and the vertical position of each tool bit used was measured using an electronic edge finder. Considering the accuracy of the gauge and the warping of the plastic, the depth of the channel can vary by as much as 10–20 μm. Imperfect surface finishes might also contribute to variation between the simulations and experimental result; it is difficult to improve the current milling process using such a small tool bit. A better alternative for improving the cross-sectional shape of the sample stream would be to add more chevron grooves to flatten the sample stream (increasing the width while reducing the height). The particles would then travel closer to the center of the source excitation laser path, which in turn should produce better signal.[8] However, we consider the agreement achieved excellent considering the resolution of the milling process.
Fig. 3.

Visualizing the sheathing process for groove-generated sheath flow in a hard microflow cytometer with a sheath-to-sample flow ratio of 50:1. a Top view of the hard microflow cytometer showing focused dye solution (green), and b side view, showing the vertical displacement of the sample solution (green) as it passes the chevrons. Note the step-wise reduction of the solution height as it passes each chevron. Two lines are added to the side view image (b) to indicate the bottom and top of the channel. The channel cross-section was designed to be 375 μm×125 μm
Pressure test
For assay and microsphere array experiments performed in this study, the fluids were introduced through PEEK tubing secured in place by epoxy. For high-pressure applications, HPLC nuts and fittings can be used to secure the PEEK tubing for improved pressure resistance. With a view toward high pressure cleaning or underwater applications, a pressure test system was prepared to determine how much pressure the hard microflow cytometer itself can withstand. The two sheath inlets and the outlet were plugged using HPLC nuts. The fiber channels and all the edges of the cytometer where the top and bottom pieces interfaced were sealed with epoxy. The sample inlet was then connected to a computer-controlled pressure regulator connected to an air cylinder. A DAQ board (ADR 2000, Ontrak Control Systems) recorded the applied pressure and the actual pressure measured by an electronic pressure meter (X1009, Ashcroft). The cytometer was then placed in a container filled with water. Pressure was gradually increased to a set value and maintained for 5 min. Leakage in the system was visually inspected by observing bubbles caused by leaking air. The pressure was increased up to 100 psi, the maximum pressure set at the air cylinder using a manual regulator. At 100 psi, no leakage in the system was observed. Similar testing on a PDMS microflow channel showed leakage starting at 50 psi.
Assay performance and comparison to other cytometers
The hard microflow cytometer was capable of distinguishing 11 sets of dye-coded microspheres, which is a significant improvement over the six sets previously identified using a similar PDMS microflow cytometer [10]. Overall alignment of chevrons in the microchannel and fibers in the interrogation region may have improved compared to the PDMS cytometer, which improves microsphere identification. In the PDMS cytometer, there were two optical detection fibers that needed to be split into the four PMTs. In the hard microflow cytometer, there are four optical detection fibers that can each be directly connected to the four PMTs, with less loss of light. Additionally, the data was acquired at a sampling rate of 250 kHz, which is faster than the 150 kHz rate used with the PDMS cytometer (Fig. 4).
Fig. 4.
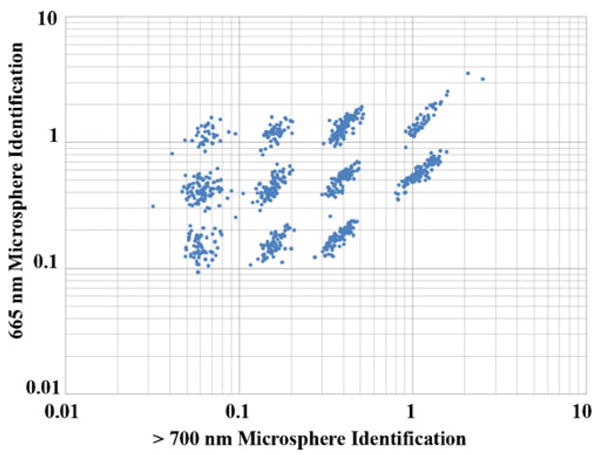
Hard microflow cytometer analysis of coded bead arrays. Scattergrams of the individual coded beads showed excellent resolution of 11 bead sets based on the intensities of red (>700 nm, x axis) and orange (665±10 nm, y axis) fluorescence. Luminex bead sets 50, 54, 58, 71, 75, 79, 81, 92, 96, 98, and 100 were mixed for this analysis
Nine of the bead sets were modified with antibodies for the multiplexed immunoassay. The two additional bead sets provided internal controls: BSA-coated beads provided a negative control for nonspecific adsorption (false-positive response), while the chicken IgY-coated beads bound the anti-chicken IgY tracer antibody to produce a positive control. Dose–response curves were created for each immunoassay using a combination of coded microspheres detecting Shigella, Campylobacter, SEB, cholera toxin, Tularemia, ricin, Salmonella, E. coli, and Listeria. The concentrations tested were 107, 106, 105, 104, 103, 102, and 101 cells/mL for bacteria and 1000, 200, 40, 8, 1.6, 0.32, and 0.064 ng/mL for toxins. Figure 5 shows the dose–response curves of each assay evaluated. The reagent additions were performed in a single microtiter filter plate before the microspheres were divided for evaluation using both the Luminex and the microflow cytometer. The results of the bacterial assays are plotted separately from the toxin assays as they have different x axes. Figure 5 panels a and b show the results obtained using the Luminex 100 while panels c and d show the results obtained using the microflow cytometer. Simple inspection, comparing the results shows a very good correspondence between both sets of data.
Fig. 5.

Dose–response curves for bacterial and toxin samples analyzed using the hard microflow cytometer. Standard error bars are shown at each point, but usually fall within the limits of the symbols on the graph
A more critical evaluation of the data compared the sensitivity for each assay as determined by each instrument from the dose–response curves. The lowest concentration detected was established as the lowest measured concentration greater than the background fluorescence plus three times the standard error. The background fluorescence was the mean PE fluorescence measured from the assay without antigen. For the hard microflow cytometer, the PE fluorescence of each microsphere was sampled six to ten times as the microsphere passed through the interrogation region, then averaged. A population of microspheres was selected based on the amounts of the two coding dyes and light scatter, and their PE fluorescence was averaged. Thus the value for each population in an assay is a mean of means, and variance is determined using standard error. The Luminex xPONENT software outputs a number of parameters such as median, mean, trim mean, and standard deviation; to evaluate the data in a similar manner as the microflow cytometer, the data was graphed using the mean and standard error.
The performance of the hard microflow cytometer was equivalent to the commercial Luminex cytometer in most instances. Table 1 shows the lowest concentration measured that was over the calculated detection limits. The differences between the two cytometers, if any, were less than a single order of magnitude, indicating that the prototype hard microflow cytometer already approaches the sensitivity obtained by the commercial instrument.
Table 1.
Sensitivity of the hard microflow cytometer as indicated by the lowest concentration detected
| Cytometer | Campylobacter | E. coli | Listeria | Salmonella | Shigella | Tularemia | Cholera toxin | Ricin | SEB |
|---|---|---|---|---|---|---|---|---|---|
| Luminex | 103 | 103 | 104 | 105 | 104 | 103 | 0.32 | 1.6 | 0.32 |
| Microflow cytometer | 103 | 103 | 104 | 105 | 104 | 103 | 1.6 | 1.6 | 1.6 |
The lowest concentration detected is reported as the lowest actually measured value that was at least three standard errors above the mean of the sample values without target present. Values for bacteria are reported as cells/mL. Toxin concentrations are in ng/mL.
In addition to performing multiplexed immunoassays with coded microspheres, the utility and versatility of the hard microflow cytometer was demonstrated using an assay for RNA expression in mammalian cells. The LNA flow-FISH method is a technique to detect messenger RNA in cells using LNA-modified oligonucleotide probes. A549 epithelial cells were fixed and permeabilized before hybridization with a biotinylated LNA probe targeting β-actin mRNA. LNA bound to β-actin are stained with streptavidin–phycoerythrin before detection by flow cytometry. Negative controls were the LNA probe without streptavidin–phycoerythrin, streptavidin–phycoerythrin without LNA probe, and scrambled LNA probe with streptavidin–phycoerythrin.
The microflow cytometer produced 45° light scatter and phycoerythrin measurements to yield relative size and fluorescence data similar to that of the Accuri C6 cytometer. Figure 6 shows a comparison of LNA flow-FISH data measured in the microflow cytometer and the commercial Accuri C6 cytometer. The β-actin LNA probe, as expected, produced the brightest fluorescence in both systems. The microflow cytometer also produced similar relative fluorescence measurements for the negative controls (d>c>b>a).
Fig. 6.
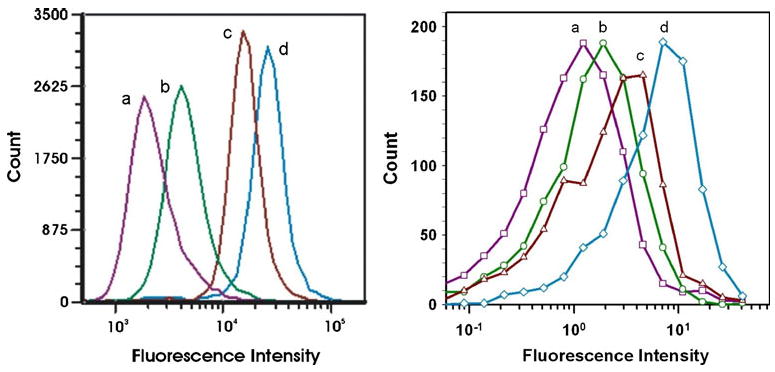
Fluorescence in situ hybridization assays. FISH analysis was performed using an Accuri C6 cytometer (left) and the hard microflow cytometer (right). Samples run on both cytometers were cells containing a β-actin LNA without PE tracer b no LNA c scrambled LNA, and d β-actin LNA
We performed the LNA flow-FISH experiment primarily as a challenge to our system and to test its ability to measure cells. LNA flow-FISH is an interesting but uncommon technique that often produces relatively lower levels of fluorescence than many cellular labeling techniques. Figure 6 shows that the microflow cytometer and commercial flow cytometer produce similar results. While the relative fluorescence units differ between the two systems, in both cases the fluorescence is detectable at the lowest intensity and increases in the different cell populations in the order no tracer, control sequence with tracer, scrambled sequence (partial match), and matching sequence. Differences in the degree of peak separation could be due to differences between the hard microflow cytometer and the Accuri C6 with regard to the efficiency of focusing of the sample stream in the laser beam, the resolution of the PMTs used for signal discrimination, or the data processing software.
Conclusions
Though CNC milling was employed in making the prototype hard microflow cytometer used in the experiments, it is not the most economical for mass production. Extrusion, molding, and embossing techniques are more suitable for mass production when plastic or polymeric devices are to be made. With these mass production methods, the reproducibility in the channel dimensions and the surface finishes can be dramatically improved. Advantages that the milling technique does provide are: (1) dimensions for feature sizes that can be readily translated to molding or embossing and (2) the possibility of making the cytometer out of a variety of plastics or metals, to provide testable prototypes for applications that may require use of high pressure or organic solvents.
The hard microflow cytometer performed at least as well as a similar PDMS microflow cytometer in the analysis of microparticle immunoassays. The hard microflow cytometer distinguished 11 dye-coded microsphere sets compared to the six sets previously distinguished using the PDMS chip. Consequently, we were able to perform greater assay multiplexing, advancing from a sixplex to a nineplex immunoassay with two sets of microspheres used as positive and negative controls. With this nineplex, we determined the lowest concentration detected as a significant signal above background for bacteria (Campylobacter, E. coli, Listeria, Salmonella, Shigella, and Tularemia) and toxins (cholera toxin, ricin, and SEB), while using BSA-coated and chicken IgY-coated particles as controls. Sensitivity was comparable to the PDMS microflow cytometer and the commercial cytometer. By optimizing the shape and size of the sample stream further, the system performance is expected to be enhanced with a possible increase in multiplexing capability.
Using fluorescence in situ hybridization with locked nucleic acid probes for RNA analysis, we also confirmed the applicability of the hard microflow cytometer for analysis of cells. Using light scatter to detect cells and fluorescence levels to distinguish cells exposed to the nucleic acid probe for β-actin, the hard microflow cytometer showed that the cells expressing the target RNA had a higher level of fluorescence than controls. The data obtained using the microflow cytometer correlated well with the data acquired using a commercial flow cytometer.
We have demonstrated a rugged prototype, with dimensions readily convertible to mass manufacturing methods, that provides performance in multiplexed coded-microsphere immunoassays and cellular analysis comparable to commercial flow cytometers. The hard microflow cytometer is amenable to on-site repetitive measurements for both environmental and clinical analyses, especially with its resistance to clogging and capacity for high pressure cleaning.
Acknowledgments
ALT and KLR were Postdoctoral Fellows of the National Research Council and JSK was a Postdoctoral Fellow of the American Society of Engineering Education. VL was an undergraduate summer intern in the Naval Research Enterprise Internship Program. This work was supported by ONR 6.2 work unit 6336 and NIH grant UO1 A1075489. The views are those of the authors and do not represent opinion or policy of the US Navy, Department of Defense, National Institutes of Health or Department of Health and Human Services.
References
- 1.Shapiro HM. The evolution of cytometers. Cytom A. 2004;58A(1):13–20. doi: 10.1002/cyto.a.10111. [DOI] [PubMed] [Google Scholar]
- 2.A history of flow cytometry and sorting. In: Shapiro H, editor; L FS, Kim JS, editors. The microflow cytometer. Pan Stanford; Singapore: 2010. pp. 1–24. [Google Scholar]
- 3.Ateya DA, et al. The good, the bad, and the tiny: a review of microflow cytometry. Anal Bioanal Chem. 2008;391(5):1485–1498. doi: 10.1007/s00216-007-1827-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golden JP, et al. Multi-wavelength microflow cytometer using groove-generated sheath flow. Lab Chip. 2009;9(13):1942–1950. doi: 10.1039/b822442k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClain MA, et al. Microfluidic devices for the high-throughput chemical analysis of cells. Anal Chem. 2003;75(21):5646–5655. doi: 10.1021/ac0346510. [DOI] [PubMed] [Google Scholar]
- 6.Blankenstein G, Larsen UD. Modular concept of a laboratory on a chip for chemical and biochemical analysis. Biosens Bioelectron. 1998;13(3–4):427–438. [Google Scholar]
- 7.Schrum DP, et al. Microchip flow cytometry using electrokinetic focusing. Anal Chem. 1999;71(19):4173–4177. doi: 10.1021/ac990372u. [DOI] [PubMed] [Google Scholar]
- 8.Howell PB, Jr, et al. Two simple and rugged designs for creating microfluidic sheath flow. Lab Chip. 2008;8(7):1097–1103. doi: 10.1039/b719381e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golden JP, et al. Multi-wavelength microflow cytometer using groove-generated sheath flow. Lab Chip. 2009;9:1942–1950. doi: 10.1039/b822442k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JS, et al. Multiplexed detection of bacteria and toxins using a microflow cytometer. Anal Chem. 2009;81(13):5426–5432. doi: 10.1021/ac9005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dugan M, Said AA, Haddock T, Bado P, Bellouard Y. Laser-based fabrication of microflow cytometers with integrated optical waveguides. In: L FS, Kim KS, editors. The Microflow Cytometer. Pan Stanford; Singapore: 2010. pp. 287–310. [Google Scholar]
- 12.Kummrow A, et al. Microfluidic structures for flow cytometric analysis of hydrodynamically focussed blood cells fabricated by ultraprecision micromachining. Lab Chip. 2009;9(7):972–981. doi: 10.1039/b808336c. [DOI] [PubMed] [Google Scholar]
- 13.Hairer G, Vellekoop MJ. An integrated flow-cell for full sample stream control. Microfluid Nanofluid. 2009;7(5):647–658. [Google Scholar]
- 14.Kostner S, Vellekoop M. Cell analysis in a microfluidic cytorneter applying a DVD pickup head. Sens Actuators, B. 2008;132(2):512–517. [Google Scholar]
- 15.Dubelaar GBJ, et al. Design and first results of CytoBuoy: a wireless flow cytometer for in situ analysis of marine and fresh waters. Cytometry. 1999;37(4):247–254. [PubMed] [Google Scholar]
- 16.Cunningham A, et al. Fine-scale variability in phytoplankton community structure and inherent optical properties measured from an autonomous underwater vehicle. J Mar Syst. 2003;43(1–2):51–59. [Google Scholar]
- 17.Thyssen M, Garcia N, Denis M. Sub meso scale phytoplankton distribution in the North East Atlantic surface waters determined with an automated flow cytometer. Biogeosciences. 2009;6(4):569–583. [Google Scholar]
- 18.Robertson KL, Thach DC. LNA flow-FISH: a flow cytometry-fluorescence in situ hybridization method to detect messenger RNA using locked nucleic acid probes. Anal Biochem. 2009;390(2):109–114. doi: 10.1016/j.ab.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 19.Baerlocher GM, et al. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1(5):2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]