

Abstract
Multiple sclerosis (MS) is a demyelinating inflammatory disorder of the central nervous system (CNS), which involves autoimmune responses to myelin antigens. Studies in experimental autoimmune encephalomyelitis (EAE), an animal model for MS, have provided convincing evidence that T cells specific for self-antigens mediate pathology in these diseases. Until recently, T helper type 1 (Th1) cells were thought to be the main effector T cells responsible for the autoimmune inflammation. However more recent studies have highlighted an important pathogenic role for CD4+ T cells that secrete interleukin (IL)-17, termed Th17, but also IL-17-secreting γδ T cells in EAE as well as other autoimmune and chronic inflammatory conditions. This has prompted intensive study of the induction, function and regulation of IL-17-producing T cells in MS and EAE. In this paper, we review the contribution of Th1, Th17, γδ, CD8+ and regulatory T cells as well as the possible development of new therapeutic approaches for MS based on manipulating these T cell subtypes.
Keywords: experimental autoimmune encephalomyelitis, multiple sclerosis, Th1 cell, Th17 cell, Treg cell
Introduction
Multiple sclerosis (MS)
MS is a chronic, progressive inflammatory disorder of the brain and spinal cord. The inflammatory plaque, whether determined histopathologically or using magnetic resonance imaging (MRI), is the pathological hallmark of MS [1]. Studies demonstrating the presence of inflammatory cells and their products in the brain lesions of MS patients, in addition to reports from animal models, has led to the generally accepted hypothesis that disease is mediated by pathogenic T cell responses against myelin antigens, followed by a broader neurodegenerative process [2]. The autoreactive T cells migrate across the blood–brain barrier (BBB) and mediate damage against the central neurones and their myelin sheaths, in particular, but also their axons. The key morphological feature of MS is primary demyelination of nerve axons leading to signal conduction block or conduction slowing at the site of demyelination. Neurological symptoms develop when conduction block occurs simultaneously in a significant proportion of fibres within a given pathway [3]. During clinical recovery, inflammation and oedema in the central nervous system (CNS) resolves and it is thought that there is restoration of CNS conduction due to glial ensheathment and remyelination. In contrast, axonal loss is irreversible and is most probably an important cause of neurological dysfunction in chronic MS.
The most common form of MS, termed relapsing–remitting MS (RRMS), is associated with acute inflammatory episodes resulting in a reduction in neurological function. Patients may experience some recovery between relapses, but 80% of patients with RRMS evolve to a more progressive form, termed secondary progressive MS (SPMS), which is associated with gradual loss of neurological function and ascending paralysis and is thought to be independent of inflammation.
Currently available therapies for MS are aimed primarily at reducing the number of relapses and slowing the progression of disability. The standard immunomodulatory therapies (IMTs) include interferon (IFN)-β and glatiramer acetate. However, these are only modestly effective first-line therapies, and about one-third of RRMS patients on IMTs will develop recurrent relapses and/or an increase in sustained disability; others can develop neutralizing antibodies to IFN-β. Patients who are truly suboptimal responders to first-line IMTs are considered currently for treatment with natalizumab as the first-choice second-line therapy. Natalizumab (Tysabri) is a humanized monoclonal antibody that targets the α4-chain of α4β1 integrin (VLA-4), and inhibits leucocytes binding to vascular cell adhesion molecule (VCAM)-1 and fibronectin and thus reduces leucocyte traffic across the BBB. The efficacy of natalizumab is significantly greater than the first-line therapies, emphasizing the importance of leucocyte trafficking in the pathogenesis of MS [4].
Experimental autoimmune encephalomyelitis (EAE)
EAE is a demyelinating disease of the CNS that shares clinical and pathological features with MS and is used as a model for the human disease. EAE is induced in susceptible animals by immunization with one of a number of myelin antigens emulsified in complete Freund's adjuvant (CFA) [5]. EAE is mediated by myelin-specific T cells, which are activated in the periphery and translocate into the CNS followed by permeabilization of the BBB [6,7]. Upon entering the CNS, the T cells are reactivated by local and infiltrating activated antigen-presenting cells (APC), which present major histocompatibility complex (MHC) class II-associated peptides, resulting in subsequent inflammatory processes and eventually in demyelination and axonal damage. Depending upon the immunization protocol and background of mice used, EAE can take an acute, chronic progressive or relapsing–remitting course [8]. EAE can also be induced by adoptive transfer of activated myelin-specific CD4+ T cells from mice with EAE into naive recipient mice [9]. The EAE model has provided a useful tool for developing a better understanding of the inflammatory processes throughout the course of disease, and has been invaluable for the development of new therapies against MS, including glatiramer acetate [10] and natalizumab [11]. However, there are differences between MS and EAE, and these have been highlighted by the fact that certain therapies have had opposite outcomes in these two diseases. For example, administration of interferon (IFN)-γ or anti-tumour necrosis factor (TNF) was protective in EAE, but exacerbated MS.
T helper type 1 (Th1) and Th17 cells
Th1 cells were thought originally to be the main pathogenic T cells in EAE and MS. This conclusion was based partly on the observation that IL-12p40-defective (IL-12p40−/−) mice were resistant to EAE, because IL-12 is required for differentiation of Th1 cells. Furthermore, treatment of MS patients with IFN-γ exacerbated disease [12]. However, IFN-γ−/− or signal transducer and activator of transcription-1 (STAT1)−/− mice lacking Th1 cells were found to develop more severe EAE [13,14]. These paradoxes were resolved partly following the discovery of IL-23, which is related structurally to IL-12. IL-23 shares the p40 chain with IL-12, which is associated with either a separate p19 or p35 chain for IL-23 and IL-12, respectively. IL-23p19−/−, like the IL-12p40−/−, mice were found to be resistant to EAE, whereas IL-12p35−/− mice were susceptible [15]. IL-23 was shown to be necessary to drive the induction or expansion of CD4+ T cells that secrete IL-17, termed Th17 cells [16]. Th17 cells have now been defined as a distinct subset of CD4+ T cells, which produce IL-17A, IL-17F, IL-21, IL-9, IL-22 and TNF-α, promote inflammation and are pathogenic in many autoimmune disorders (Fig. 1) [17]. Initial reports suggested that TGF-β and IL-6 were required for differentiation of murine Th17 cells, whereas IL-23 is required for their expansion [18]. However, it now appears that TGF-β functions to suppress Th1 and Th2 cells, the products of which can inhibit the differentiation of Th17 cells [19]. Although not yet resolved fully, it seems that IL-23 may not be required for lineage commitment, but is essential for further Th17 development. Furthermore, IL-1 signalling is required for Th17 development in vivo; IL-1RI−/− mice have defective Th17 responses and are resistant to the induction of EAE [20]. IL-1, IL-6, IL-21, IL-23 and TGF-β have all been shown to play a role in the differentiation of human Th17 cells in vitro[21–23]. However, it has been shown recently that TGF-β is dispensable for the differentiation of human Th17 cells; although analogous to the situation in mice, it favours their expansion indirectly relative to Th1 cells [24].
Fig. 1.
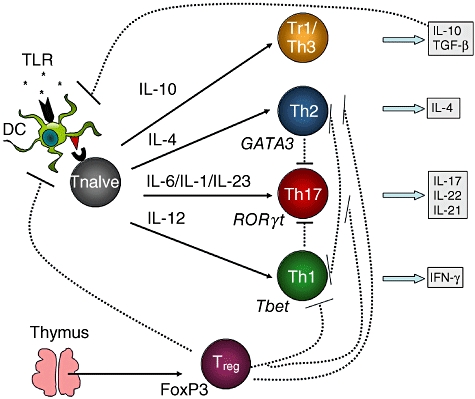
The differentiation and regulation of CD4+ T cell subsets. Naive T cells primed by antigen-presenting cells (APC) such as dendritic cells (DC) can differentiate into T regulatory-1 (Tr1)/T helper type 3 (Th3), Th1, Th2 or Th17 cells depending upon the cytokine environment. Priming in the presence of interleukin (IL)-10/transforming growth factor (TGF)-β, IL-12, IL-4 or combinations of IL-6/IL-1/IL-23 promotes the differentiation of Tr1/Th3, Th1, Th2 or Th17 cells, respectively [113]. Th17 cells can be regulated negatively by Th1 or Th2 cells. Tr1 and Th3 cells secrete IL-10 and TGF-β which can suppress effector cell responses, primarily by suppressing APC function. Natural regulatory T cells (Treg) cells are derived from the thymus (although they may also be converted in the periphery) and can suppress effector T cell responses directly or via the APC [72].
A number of studies have highlighted the central role of Th17 cells in the development and pathogenesis of EAE. Langrish et al. demonstrated that proteolipid protein (PLP)-specific T cells cultured in the presence of IL-23 generated Th17 cell lines that induced EAE following passive transfer into naive SJL mice, whereas Th1 cells lines generated by in vitro culture with IL-12 failed to induce EAE [16]. Furthermore, administration of neutralizing anti-IL-17 antibody reduced the severity of EAE, while blocking IFN-γ exacerbated disease [16]. Similarly, neutralization of IL-17 in the myelin–oligodendrocyte–glycoprotein (MOG)-induced EAE model during the course of the disease improved clinical symptoms modestly [25]. In one study IL-17−/− mice had very mild EAE, with delayed-onset, reduced disease scores and early recovery [26]. However, in another study deletion of IL-17F and simultaneous blockade of IL-17A, which are both related Th17 signature cytokines, had only very marginal benefit [27]. The almost complete and partial reduction in symptoms of EAE observed with blockade of IL-23 and IL-17, respectively, suggested that other Th17-derived cytokines might also be involved, although IL-21 and IL-22 appear to be dispensable [28,29]. However, neutralization of IL-9 attenuated EAE, suggesting a role for IL-9 in mediating Th17-mediated diseases [30]. Although there are some contradictory data on the relative importance of IL-17 and other Th17-derived cytokines in EAE, the consensus view that is emerging is that the IL-17 family of cytokines have a clear pathogenic role in EAE and MS. Although early studies on Th17 cells dismissed a role for Th1 cells, recent studies have suggested that both cell types may play distinct roles in pathology. It has been reported that adoptive transfer of highly purified Th1 could induce EAE, whereas Th17 cells, devoid of IFN-γ-producing cells, could not induce disease [7]. It was suggested that only Th1 cells could access the CNS initially, and that this facilitated subsequent recruitment of Th17 cells [7]. Evidence that recruited host Th17 cells contributed to disease was provided by the demonstration that the incidence of disease was lower in IL-17−/− compared with wild-type mice following transfer of Th1 cells [31].
Interestingly, although Th1 or Th17 cells can induce EAE, both the clinical signs and pathological features may differ. IL-12-polarized T cells promoted expression of monocyte attracting chemokines and macrophage-rich infiltrates into the spinal cord, whereas IL-23-polarized T cells activated neutrophil-attracting chemokines and promoted neutrophils, especially in the brain [32]. In another model, IL-17 was again shown to promote CD4 T cell infiltration into the brain parenchyma, which resulted in a clinically atypical EAE [33]. Thus, both Th1 and Th17 cells may play complementary roles in the pathogenesis of EAE. It is not clear how this might relate to a role of Th17 cells in MS, where the infiltrating T cells and monocytes predominate.
T cells producing both IL-17 and IFN-γ, which express T-box expressed in T cells (Tbet) and retinoid-related orphan receptors (ROR)γt, are also recruited to the CNS during EAE, and transferred Th17 cells can switch to IFN-γ production, indicating that there is plasticity within these populations [34,35]. It has also been suggested that the expression of Tbet defines the encephalogenicity of T cells rather than their cytokine profile [36], and inhibition of Tbet was shown to ameliorate EAE by inhibiting both Th1 and Th17 cells [37].
While most researchers favour a role for IL-23 in expansion of Th17 cells or stabilization of the Th17 phenotype, a recent report showed a similar course of EAE following transfer of MOG-specific T cells into either wild-type or IL-23−/− mice, suggesting that IL-23 was required only for the induction but not effector phase of disease [38]. It also appears that the function of IL-23 may extend beyond the activation of Th17 cells and this might explain the different levels of susceptibility in IL-23−/− and IL-17−/− mice. It was demonstrated that IL-23 is required for the homing of MOG-specific T cells to the CNS during EAE, but not for their expansion in peripheral organs [39]. The importance of CNS homing in the development of EAE was confirmed by the fact that deficiency in C-C type chemokine receptor 6 (CCR6), a chemokine receptor expressed on Th17 cells, conferred resistance to EAE, although CCR6−/− mice still developed peripheral Th17 responses. Susceptibility was restored by transfer of wild-type cells, after which effector cells were recruited independently of CCR6 [40].
The relative roles of Th1 and Th17 cells in MS and other human autoimmune diseases are still not clear. Th17 cells have been detected in tissues from a variety of human autoimmune and inflammatory disorders, including Crohn's disease, systemic lupus erythematosus (SLE), rheumatoid arthritis and psoriasis (reviewed in [41]). IL-17 and IFN-γ production by T cells have been associated with disease activity in MS patients, and are also expressed in brain lesions. Microarray analyses have detected IL-17 expression in MS brain lesions [42,43], and enrichment of IL-17 producing cells, which included glial cells, CD4+ and CD8+ T cells, were identified in the active rather than inactive areas of MS brain lesions [44]. Elevated frequencies of IL-17-producing cells have been associated with disease activity in the peripheral blood of MS patients [45,46]. However, it has also been reported that although IL-17 and IFN-γ were elevated during very early in disease, enhancement of IFN-γ alone was associated with relapse [47]. Human Th17 cells migrate more efficiently than Th1 cells (based on an in vitro BBB migration assay), and display cytotoxic activity against neurones [48]. Interestingly, a recent study has identified an enrichment of T cells expressing both IL-17 and IFN-γ in MS brain tissue, suggesting that IL-17+ IFN-γ+ T cells may be involved in pathology [49].
Clearly, much of the focus in recent literature has been on the relative role of Th17 and Th1 cells in EAE and MS; however, as reviewed above this question is not clear-cut and still far from being resolved. While EAE can be induced by adoptive transfer of either Th1 or Th17 cells, there appears to be no absolute requirement for either of the Th1 or Th17 signature cytokines. The instability of the Th17 phenotype has, no doubt, contributed to the difficulties in defining the role of Th17 cells. On the other hand, IL-23, IL-6 and IL-1 are required for the differentiation and expansion of Th17 cells and development of EAE, as is the expression of Tbet (Table 1). Although the precise role of these factors in EAE is still not clear, the evidence from knock-out mice and antibody therapy suggests that they contribute to pathology. Studies in MS patients and normal human donors have provided evidence that Th1 and Th17 cells are active or expanded during diseases, but their contribution to pathology has, understandably, been more difficult to define.
Table 1.
Experimental evidence of a role for T helper type 1 (Th1) and Th17 cells in experimental autoimmune encephalomyelitis (EAE).
| Molecule/cell type* | Manipulation† | Background‡ | Induction method§ | Effect on EAE¶ | Reference |
|---|---|---|---|---|---|
| IL-12p40 | Genetic deletion | C57BL/6 | MOG/CFA | Resistant | [15] |
| IL-12p35 | Genetic deletion | C57BL/6 | MOG/CFA | Susceptible | [15] |
| IL-23p19 | Genetic deletion | B6×129 | MOG/CFA | Resistant | [15,38] |
| IL-23p19 | α-IL-23p19 pre-onset | SJL | PLP/CFA | Resistant | [114] |
| IL-23p19 | α-IL-23p19 post-onset | SJL | PLP/CFA | Prevented relapse | [114] |
| IL-23p19 | Genetic deletion (recipient only) | C57BL/6 | WT T cell transfer | Susceptible | [38] |
| IL-23p19 | Genetic deletion (donor only) | C57BL/6 | p19−/− T cell transfer | Delayed-onset reduced severity | [38] |
| IL-6 | Genetic deletion | 129Sv×C57BL/6 | MOG/CFA | Resistant | [115] |
| IL-6R | T cell conditional deletion of gp130 | C57BL/6 | MOG/CFA | Resistant | [116] |
| IL-1R | Genetic deletion | C57BL/6 | MOG/CFA | Resistant | [20] |
| IFN-γ | Genetic deletion | B10.PL | MBP/CFA | Increased mortality delayed resolution? | [13] |
| Tbet | Genetic deletion | C57BL/6 | MOG/CFA | Resistant | [14] |
| STAT1 | Genetic deletion | C57BL/6 | MOG/CFA | Increased severity | [14] |
| IL-17 | Genetic deletion | C57BL/6 | MOG/CFA | Delayed-onset reduced severity | [26,27]** |
| IL-25 | Genetic deletion | C57BL/6 | MOG/CFA | Accelerated-onset enhanced severity | [117] |
| IL-21/IL-21R | Genetic deletion | C57BL/6 | MOG/CFA | Susceptible | [28] |
| IL-22 | Genetic deletion | C57BL/6 | MOG/CFA | Susceptible | [29] |
| IL-9/IL-9R | Neutralization/genetic deletion | C57BL/6 | MOG/CFA | Delayed-onset/attenuated disease | [30] |
| Th1 | Adoptive transfer | C57BL/6 | Th1 transfer | Induced disease | [7] |
| Th1 | Adoptive transfer | SJL | Th1 transfer | Failed to induce disease | [16] |
| Th1 | Adoptive transfer | SJL | Th1 transfer | Induced disease | [32] |
| Th17 | Adoptive transfer | C57BL/6 | Th17 transfer | Failed to induce disease | [7] |
| Th17 | Adoptive transfer | SJL | Th17 transfer | Induced disease | [16,32] |
Molecule or cell type of interest that was manipulated in each study.
Approaches taken to manipulate cells or molecules include gene deletion, administration of neutralizing antibody or adoptive transfer of T cell lines.
Mouse strain background.
Methods used to induce EAE include active induction with myelin-antigens plus complete Freund's adjuvant (CFA) or passive induction by adoptive transfer of T cell lines.
Effect of manipulation on the clinical symptoms of EAE.
Found only marginal contribution after deletion of interleukin (IL)-17A and IL-17F. IFN: interferon; MOG: myelin–oligodendrocyte–glycoprotein; PLP: proteolipid protein; STAT1: signal transducer and activation of transcription-1; WT: wild-type.
CD8+ T cells
MS has been viewed historically as a CD4+ T cell-mediated autoimmune disease, due in part to the genetic association of MS with MHC class II alleles; however, an association with MHC class I alleles has also been reported [50]. Furthermore, the frequency of CD8+ T cells is greater than that of CD4+ T cells in inflamed plaques, and CD8+ T cells show oligoclonal expansion in plaques, CSF and blood, all suggestive of a pathogenic role in MS [51].
A few studies involving cell transfer have suggested that CD8+ T cells are pathogenic in EAE (reviewed in [52]. However, there is also evidence of a regulatory role for CD8+ T cells in EAE; EAE is more severe in mice deficient in or depleted of CD8+ T cells and disease severity has been correlated inversely with the frequency of CD8+ T cells [52]. A recent study demonstrated that CD8+ regulatory T cells (Treg) cells suppressed EAE via a TGF-β-dependent mechanism [53].
γδ T cells
γδ T cells are thought to provide a first line of defence against infection, especially at mucosal sites, through the production of IFN-γ. γδ T cells directly recognize ligands induced by stress, inflammation or infection, including non-peptide antigens, alkylamines, phosphoantigens, heat-shock proteins and non-classical MHC class I molecules, such as MHC class I chain-related molecule (MIC A) and MIC B. γδ T cells can act as a source of innate cytokine and have the potential to influence adaptive immune responses.
For the last 10–15 years γδ T cells have been thought to play a role in both MS and EAE. Clonally expanded γδ T cells were found in acute MS brain lesions [54] and in the cerebrospinal fluid of MS patients with recent disease onset [55], suggesting that γδ T cells contribute to neuroinflammation. In the EAE model, however, both protective [56–58] and pathogenic [59–63] roles have been ascribed to γδ T cells. A number of factors might explain some of the conflicting results in these studies, which have used a number of different mouse strains in combination with either depleting antibodies or genetic manipulation of γδ T cells. First, it has been suggested the UC7-13D5 anti-γδ antibody which accelerates, rather than depleting, the onset of EAE, may activate γδ T cells by cross-linking the receptor [56]. Secondly, the frequency of γδ subtypes varies between mouse strains, and as different functions have been ascribed to various subtypes, this could account for many of the discordant results in different mouse strains. Finally, the Tcrd−/− mice used in some studies are deficient only in the δ T cell receptor (TCR) gene and thus the γδ T cells are still present, although unable to respond to TCR stimulation [64]. This suggests that TCR-independent activation of γδ T cells could still occur in these mice, potentially confounding the interpretation of experiments.
It has been shown recently that IL-17 is produced by γδ T cells as well as αβ T cells during infection [65,66], raising the possibility that γδ T cell-derived IL-17 may contribute to CNS pathology in EAE and MS. Indeed, recent data from our laboratory have shown that IL-17-producing γδ T cells are present at high frequency in the brains of mice with EAE [67]. Furthermore, disease was less severe in Tcrd−/− mice, supporting a pathogenic role for γδ T cells. MOG-specific IL-17 production by conventional T cells was reduced in Tcrd−/− mice, and this was consistent with the demonstration that γδ T cell-derived IL-17 promoted further IL-17 production by CD4+ T cells [67]. γδ T cells were shown to produce IL-17 in response to IL-1 and IL-23 in the absence of TCR engagement, and these cells increased susceptibility to EAE. Thus, in addition to being an important source of IL-17 early in the EAE disease course, γδ T cells also served to amplify IL-17 production by CD4+ T cells [67]. Overall, the consensus from the various studies suggests a pathogenic role for γδ T cells in EAE, particularly early in the disease course, although they may also have a regulatory role during disease resolution in some models [57,58,67].
Treg
Treg cells include both natural and adaptive (also termed inducible) Treg cells. Natural Treg (nTreg) cells express cell surface CD25 and the transcription factor forkhead box P3 (FoxP3), which is essential for directing regulatory function [68,69]. nTreg cells were identified originally through their function in controlling autoimmunity by mediating immunological tolerance to self-antigens [70]. Adaptive Treg cells, including T regulatory-1 (Tr1), Th3 and various subsets of CD8+ Treg cells, are derived in the periphery from uncommitted naive T cells stimulated by antigen under the influence of the immunosuppressive cytokines IL-10 and TGF-β, but also retinoic acid (RA). Tr1 cells exert their suppressive function primarily via IL-10 secretion [71], whereas Th3 cells secrete high levels of TGF-β.
Fully differentiated nTreg cells are derived from the thymus, when CD4 T cells with higher than normal affinity for self-antigens are selected positively [72]. Such self-antigen-specific nTreg cells are thought to play a dominant role in preventing autoimmunity, whereas inducible Treg cells are more likely to be generated in response to foreign antigens during infection. However, the lines between natural and inducible Treg cells are somewhat blurred by the fact that peripheral conversion of nTreg cells can also occur, especially under the influence of TGF-β and RA (reviewed in [73]). Furthermore, although suppression involving IL-10 and TGF-β are the hallmark of inducible Treg cells, the suppressive function of nTreg cells in vivo is often cytokine-dependent. Therefore, the lack of markers, apart from IL-10 or TGF-β secretion, to identify Tr1 or Th3 cells, respectively, can hamper the interpretation of many experiments. Studies on human Treg cells, particularly those in disease settings, can be compromised by the fact that many Treg cell markers, including CD25 and FoxP3, may be up-regulated on activated effector cells. For this reason, CD127lo is now often used in addition to CD25 and FoxP3 to identify human nTreg cells [74].
There is no doubt that the various types of Treg cells play a crucial role in maintaining immune homeostasis, and both natural and adaptive Treg cells are likely to regulate autoimmune inflammation during MS and EAE (Fig. 2). Potentially autoreactive T cells specific for myelin antigens are present in healthy individuals, suggesting that these cells are constrained by regulatory mechanisms which may be impaired in MS. Indeed, Tr1 responses have been shown to be reduced in MS patients, reflected in reduced levels of IL-10 [75,76]. While there have also been reports of reduced frequency of nTreg cells in MS patients [77], the majority of studies have found a similar frequency to that observed in normal individuals [78,79]. One study found a reduced frequency of nTreg cells in RRMS but not in SPMS [80], perhaps reflecting the fact that inflammation is thought to play a role in RRMS, but not in the progressive phase. Interestingly, an increased frequency of CD25+ FoxP3+ nTreg cells has been found in the CSF but not the blood of MS patients [78].
Fig. 2.
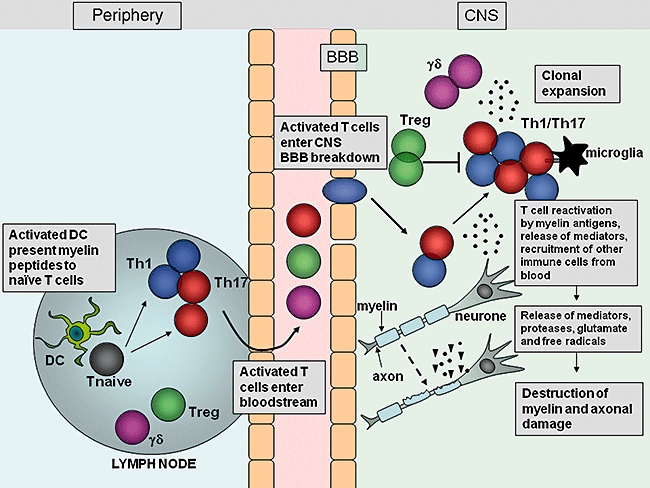
Migration and effector function of T cells in the central nervous system (CNS) during experimental autoimmune encephalomyelitis (EAE). After immunization with myelin antigens, complete Freund's adjuvant (CFA) and pertussis toxin, dendritic cells (DC) are activated in the lymph nodes by Toll-like receptor (TLR) agonists within the mycobacterium tuberculosis component of CFA, and present myelin antigen to naive T cells. The activated myelin-specific T cells enter the bloodstream and traffic to and enter the CNS. Breakdown of the blood–brain barrier (BBB) occurs, allowing recruitment of other inflammatory cells into the CNS. T cells entering the CNS encounter their cognate myelin antigens and become reactivated by local APC. T cells expand and release inflammatory mediators which help recruit other immune cells to the site of inflammation. Activation of local microglial cells and infiltrating cells results in production of proteases, glutamate, reactive oxygen species and other cytotoxic agents which promote myelin breakdown. Damage to the myelin sheath surrounding axons is followed by axonal damage and neurological impairment.
While there appears to be no numerical deficit in Treg cells in MS, a number of functional studies using in vitro suppression assays have documented impairments in Treg cells from MS patients [77–79,81–87], which was reversed after therapy with IFN-β[86,88], glatiramer acetate [89] or steroids [90]. The discrepancy between the studies on Treg cells from MS patients may be due, at least in part, to the fact that Treg cell markers, such as CD25 and FoxP3, can be induced on effector T cells after activation and may therefore confound both phenotypic and functional studies in inflammatory settings. In the case of functional studies, sorting on the basis of CD25hi alone could have led to the inclusion of activated effector T cells, which would reduce the suppressive effect of the regulatory population. Indeed, a more recent and very thorough study by Michel et al. revealed that when CD127lo was used in addition to CD25 to sort Treg cells, there was no reduction in the suppressive function of Treg cells from MS patients compared with controls [91]. The functional studies employed proliferation and/or production of IFN-γ as readouts for suppression of responder T cells, but did not examine the ability of Treg cells from MS patients to suppress IL-17 production by responder T cells. However, we have shown recently that a subset of Treg cells expressing CD39 were able to suppress both IFN-γ and IL-17 in healthy controls; however, CD39+ Treg cells from RRMS patients had an impaired ability to suppress IL-17 [92], suggesting that there is defective regulation of IL-17 by nTreg cells from these patients.
Adoptive transfer and depletion experiments in mice have provided more definitive evidence that Treg can control the development and severity of EAE. Early studies in transgenic mice expressing TCR specific for myelin antigen, that develop spontaneous EAE, revealed that non-transgenic CD4+ T cells could prevent spontaneous disease, suggesting a role for CD4+ Treg cells [93,94]. In MOG-induced EAE transfer of CD4+CD25+ T cells reduced disease severity, and these cells were shown to be capable of suppressing MOG-specific T cell responses in vitro[95]. The protective effects of CD4+CD25+ Treg cells appear to be mediated by IL-10, as Treg cells from IL-10−/− failed to confer protection [96]. However, we have found that regulatory responses induced by helminth infection suppress MOG-specific Th17 responses and protect against EAE by a TGF-β-dependent mechanism [97]. In the PLP-induced model, the susceptibility of different mouse strains to EAE correlates inversely with the frequency of PLP-specific CD4+CD25+ Treg cells [98]. Furthermore, depletion of CD25+ cells rendered the resistant B10 mice susceptible to EAE [98] and increased susceptibility to suboptimally induced EAE [99]. These studies suggest that Treg cells influence the susceptibility to and threshold of disease in the EAE models. While this review has focused largely upon nTreg cells, it should be noted that IL-10 or TGF-β-secreting CD4+ adaptive Treg cells, as well as CD8, γδ T cells and natural killer (NK) T cells, also have regulatory activity and may help constrain pathogenic T cells in EAE and MS.
Interplay between Treg and effector T cells
In addition to their influence on susceptibility of the host to demyelinating disease, Treg cells also appear to play a role in mediating the recovery from actively induced EAE, which is usually self-limiting. Treg cells accumulate in the CNS during the recovery phase [100–102], and their depletion inhibits recovery [102]. Recovery has been associated with induction/activation of TGF-β[103] and IL-10 [102]. IL-10 may be derived from both nTreg cells as well as FoxP3- CD4+ T cells, including Tr1 cells [102]. It has also been shown that neurones may be capable of converting effector T cells to Treg cells via a TGF-β-dependent mechanism [101]. Interestingly, Korn et al. demonstrated that, although Treg cells were present in the CNS at the peak of disease, they were incapable of suppressing CNS-derived effector T cells [100]. This was attributed to the fact that the effector T cells were resistant to suppression as a result of their highly inflammatory milieu. Taken together, these studies suggest that autoimmune inflammation depends upon a fine balance between regulatory and inflammatory responses, so that during the onset and peak of disease inflammatory responses dominate but then contract in favour of Treg cells during resolution. Indeed, in MS, relapse and remission have been correlated with relative decreases and increases in the frequency of Treg cells. Thus, it will important for future studies to determine the factors that control the balance between Treg and effector T cells (Teff).
Studies in the passive EAE model have shown that the inflamed CNS drives specifically the proliferation of Treg cells [104]. Treg cells were found to accumulate in the CNS but not in the lymphoid organs, and were derived from expansion of existing host Treg cells rather than conversion of non-Treg cells [104]. Consistent with this Korn et al., using cell transfer from FoxP3-GFP mice, showed that Treg cells in the CNS were expanded from pre-existing Treg cells rather than converted from non-Treg cells [100]. This is in contrast to an earlier study suggesting that Treg cells could be converted in the CNS [101]. Interestingly, CNS-derived Treg cells were able to suppress MOG-specific IFN-γ but not IL-17 [104], suggesting that Th17 cells might be more resistant to suppression. Alternatively, these results might be explained by the recent observation that FoxP3+ Treg cells can produce IL-17, thereby masking any suppressive effect on Th17 cells. We demonstrated that CD4+CD25+FoxP3+CD39+ Treg cells did suppress IL-17, whereas total CD4+CD25+FoxP3+ human Treg cells failed to suppress IL-17 by responder T cells, due to the fact that the CD39-CD4+CD25+FoxP3+ subset produced IL-17, which masked the suppressive effect of the Treg population as a whole [92]. Therefore, it appears that Treg cells can suppress Th17 cells but the suppressive Treg cells cannot be defined on the basis of FoxP3 expression alone.
Conclusions and prospectus for new therapies
The overwhelming evidence of a role for T cells in the pathogenesis of EAE and MS makes them an attractive target for therapeutic intervention. Indeed, the two most commonly used drugs used to treat MS, IFN-β and glatiramir acetate, can increase Treg cells and IL-10 and thereby suppress proinflammatory T cell cytokines, although they probably also act on other cell types. More deliberate strategies to manipulate T cells involve either targeting inflammatory T cells or their effector functions, or boosting the function of Treg cells. The anti-VLA-4 antibody natalizumab, which inhibits the entry of T cells into the CNS, is relatively effective compared to other licensed drugs. However, because of the associated risk of developing potentially fatal progressive multi-focal leucoencephalopathy, it is now available only to patients who have failed other therapies or those with aggressive disease.
Other therapies in clinical trials include fingolimod and alemtuzumab. Fingolimod prevents sphingosine-1-phosphate (S1P) binding to its receptor (a process that facilitates the migration of lymphocytes from the lymph nodes), thereby preventing lymphocyte recruitment from lymph nodes to sites of inflammation. Alemtuzumab (formerly CAMPATH-1H) kills T cells and other target cells by binds to CD52. It selectively depletes CD4+ T cells for a median of 60 months [105]. A Phase II study, with a 2-year follow-up, demonstrated that 85% of patients who received low-dose and 90% who received high-dose alemtuzumab were relapse-free compared to 60% of the patients who were treated with IFN-β[106].
Strategies to either induce Treg cells in vivo or expand ex vivo are also being considered for the treatment of MS. A recent study provided proof-of-principle for nTreg cell-based therapy in the EAE model. Transfer of myelin basic protein (MBP)-reactive CD4+CD25+ Treg cells from TCR transgenic mice prevented disease when given prior to immunization, and prevented relapses when administered after the onset of disease [107]. Of crucial importance for future therapies in humans, the effect was seen only when relevant myelin antigen-specific Treg cells were transferred, but not with polyclonal Treg cells. This could represent a significant obstacle to the possible use of Treg therapy in MS, where the relevant antigens are not well defined.
While clinical trials have demonstrated clearly marked suppression of inflammatory disease activity and reduction in disability progression, many second-line therapies for MS are accompanied by the risk of life-threatening complications [9]. This is partly a reflection of the global immunosuppressive effects of these drugs. Efforts are now focused upon targeting Th17 cells and associated cytokines, including anti-IL-17 antibodies and antibodies or antagonists to cytokines that drive Th17 cells, such as IL-6 and IL-23. Although Th17 cells are involved in protection against infection, evidence to date suggests that selective suppression of Th17, but not Th1 cells, may confer protection against MS without serious side effects. Antibodies against IL-17, IL-12p40 and IL-23 are already in clinical trials for a number of autoimmune and chronic inflammatory diseases. The data appear very encouraging for rheumatoid arthritis and psoriasis [108–110]. However, a Phase II trial with ustekinumab, an IL-12/IL-23 p40 monoclonal antibody, was not effective in preventing new lesion formation in MS [111], although this may be attributed to the inclusion of patients with advanced disease, and it has been suggested that the drug may be more effective in patients with very early disease [112]. Thus, while the IL-17/IL-23 axis is clearly an important drug target for the treatment of MS, its potential as a front-line therapy awaits further clinical evaluation [113].
Disclosure
Kingston Mills is a co-founder and shareholder in Opsona Therapeutics Ltd, a start-up company involved in the development of anti-inflammatory drugs.
References
- 1.Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354:942–55. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.McDonald WI, Sears TA. The effects of experimental demyelination on conduction in the central nervous system. Brain. 1970;93:583–98. doi: 10.1093/brain/93.3.583. [DOI] [PubMed] [Google Scholar]
- 4.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 5.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–19. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- 6.Furtado GC, Marcondes MC, Latkowski JA, Tsai J, Wensky A, Lafaille JJ. Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol. 2008;181:4648–55. doi: 10.4049/jimmunol.181.7.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Connor RA, Prendergast CT, Sabatos CA, et al. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–4. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinman L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron. 1999;24:511–14. doi: 10.1016/s0896-6273(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 9.Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1952–60. doi: 10.1038/nprot.2006.284. [DOI] [PubMed] [Google Scholar]
- 10.Teitelbaum D, Meshorer A, Hirshfeld T, Arnon R, Sela M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur J Immunol. 1971;1:242–8. doi: 10.1002/eji.1830010406. [DOI] [PubMed] [Google Scholar]
- 11.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–6. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 12.Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893–5. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 13.Ferber IA, Brocke S, Taylor-Edwards C, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 14.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 16.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 18.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 19.Das J, Ren G, Zhang L, et al. Transforming growth factor {beta} is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206:2407–16. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–91. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–2. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 24.Santarlasci V, Maggi L, Capone M, et al. TGF-beta indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur J Immunol. 2009;39:207–15. doi: 10.1002/eji.200838748. [DOI] [PubMed] [Google Scholar]
- 25.Hofstetter HH, Ibrahim SM, Koczan D, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–30. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–73. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 27.Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–9. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting edge: IL-21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. 2008;180:7097–101. doi: 10.4049/jimmunol.180.11.7097. [DOI] [PubMed] [Google Scholar]
- 29.Kreymborg K, Etzensperger R, Dumoutier L, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–104. doi: 10.4049/jimmunol.179.12.8098. [DOI] [PubMed] [Google Scholar]
- 30.Nowak EC, Weaver CT, Turner H, et al. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med. 2009;206:1653–60. doi: 10.1084/jem.20090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lees JR, Iwakura Y, Russell JH. Host T cells are the main producers of IL-17 within the central nervous system during initiation of experimental autoimmune encephalomyelitis induced by adoptive transfer of Th1 cell lines. J Immunol. 2008;180:8066–72. doi: 10.4049/jimmunol.180.12.8066. [DOI] [PubMed] [Google Scholar]
- 32.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–41. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–42. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abromson-Leeman S, Bronson RT, Dorf ME. Encephalitogenic T cells that stably express both T-bet and RORgammat consistently produce IFNgamma but have a spectrum of IL-17 profiles. J Neuroimmunol. 2009;215:10–24. doi: 10.1016/j.jneuroim.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi G, Cox CA, Vistica BP, Tan C, Wawrousek EF, Gery I. Phenotype switching by inflammation-inducing polarized Th17 cells, but not by Th1 cells. J Immunol. 2008;181:7205–13. doi: 10.4049/jimmunol.181.10.7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Weiner J, Liu Y, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206:1549–64. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gocke AR, Cravens PD, Ben LH, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–8. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- 38.Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–98. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- 39.Gyulveszi G, Haak S, Becher B. IL-23-driven encephalo-tropism and Th17 polarization during CNS-inflammation in vivo. Eur J Immunol. 2009;39:1864–9. doi: 10.1002/eji.200939305. [DOI] [PubMed] [Google Scholar]
- 40.Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–23. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 41.Sallusto F, Lanzavecchia A. Human Th17 cells in infection and autoimmunity. Microbes Infect. 2009;11:620–4. doi: 10.1016/j.micinf.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 43.Montes M, Zhang X, Berthelot L, et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol. 2009;130:133–44. doi: 10.1016/j.clim.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matusevicius D, Kivisakk P, He B, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 46.Durelli L, Conti L, Clerico M, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. 2009;65:499–509. doi: 10.1002/ana.21652. [DOI] [PubMed] [Google Scholar]
- 47.Frisullo G, Nociti V, Iorio R, et al. IL17 and IFNgamma production by peripheral blood mononuclear cells from clinically isolated syndrome to secondary progressive multiple sclerosis. Cytokine. 2008;44:22–5. doi: 10.1016/j.cyto.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 48.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing T(H)17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 50.Sawcer S, Ban M, Maranian M, et al. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet. 2005;77:454–67. doi: 10.1086/444547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friese MA, Fugger L. Pathogenic CD8(+) T cells in multiple sclerosis. Ann Neurol. 2009;66:132–41. doi: 10.1002/ana.21744. [DOI] [PubMed] [Google Scholar]
- 52.Weiss HA, Millward JM, Owens T. CD8+ T cells in inflammatory demyelinating disease. J Neuroimmunol. 2007;191:79–85. doi: 10.1016/j.jneuroim.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 53.Chen ML, Yan BS, Kozoriz D, Weiner HL. Novel CD8(+) regulatory T cells suppress experimental autoimmune encephalomyelitis by TGF-beta- and IFN-gamma-dependent mechanisms. Eur J Immunol. 2009;39:3423–35. doi: 10.1002/eji.200939441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci USA. 1992;89:4588–92. doi: 10.1073/pnas.89.10.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimonkevitz R, Colburn C, Burnham JA, Murray RS, Kotzin BL. Clonal expansions of activated gamma/delta T cells in recent-onset multiple sclerosis. Proc Natl Acad Sci USA. 1993;90:923–7. doi: 10.1073/pnas.90.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi Y, Kawai K, Ito K, Honda H, Sobue G, Yoshikai Y. Aggravation of murine experimental allergic encephalomyelitis by administration of T-cell receptor gammadelta-specific antibody. J Neuroimmunol. 1997;73:169–74. doi: 10.1016/s0165-5728(96)00187-7. [DOI] [PubMed] [Google Scholar]
- 57.Ponomarev ED, Dittel BN. Gamma delta T cells regulate the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanism. J Immunol. 2005;174:4678–87. doi: 10.4049/jimmunol.174.8.4678. [DOI] [PubMed] [Google Scholar]
- 58.Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol. 2004;173:1587–95. doi: 10.4049/jimmunol.173.3.1587. [DOI] [PubMed] [Google Scholar]
- 59.Rajan AJ, Gao YL, Raine CS, Brosnan CF. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J Immunol. 1996;157:941–9. [PubMed] [Google Scholar]
- 60.Rajan AJ, Klein JD, Brosnan CF. The effect of gammadelta T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J Immunol. 1998;160:5955–62. [PubMed] [Google Scholar]
- 61.Spahn TW, Issazadah S, Salvin AJ, Weiner HL. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33-35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR delta chain gene. Eur J Immunol. 1999;29:4060–71. doi: 10.1002/(SICI)1521-4141(199912)29:12<4060::AID-IMMU4060>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 62.Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol. 2004;173:682–94. doi: 10.4049/jimmunol.173.1.682. [DOI] [PubMed] [Google Scholar]
- 63.Cardona AE, Teale JM. Gamma/delta T cell-deficient mice exhibit reduced disease severity and decreased inflammatory response in the brain in murine neurocysticercosis. J Immunol. 2002;169:3163–71. doi: 10.4049/jimmunol.169.6.3163. [DOI] [PubMed] [Google Scholar]
- 64.Koenecke C, Chennupati V, Schmitz S, Malissen B, Forster R, Prinz I. In vivo application of mAb directed against the gammadelta TCR does not deplete but generates ‘invisible’ gammadelta T cells. Eur J Immunol. 2009;39:372–9. doi: 10.1002/eji.200838741. [DOI] [PubMed] [Google Scholar]
- 65.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 66.Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–72. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- 67.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 68.Hori S, Sakaguchi S. Foxp3: a critical regulator of the development and function of regulatory T cells. Microbes Infect. 2004;6:745–51. doi: 10.1016/j.micinf.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 69.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 70.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 71.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 72.Liston A, Rudensky AY. Thymic development and peripheral homeostasis of regulatory T cells. Curr Opin Immunol. 2007;19:176–85. doi: 10.1016/j.coi.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 73.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30:626–35. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 74.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–11. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinez-Forero I, Garcia-Munoz R, Martinez-Pasamar S, et al. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur J Immunol. 2008;38:576–86. doi: 10.1002/eji.200737271. [DOI] [PubMed] [Google Scholar]
- 76.Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest. 2006;116:3252–7. doi: 10.1172/JCI29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Venken K, Hellings N, Thewissen M, et al. Compromised CD4+ CD25(high) regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. 2008;123:79–89. doi: 10.1111/j.1365-2567.2007.02690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol. 2007;147:412–18. doi: 10.1111/j.1365-2249.2006.03271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haas J, Hug A, Viehover A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35:3343–52. doi: 10.1002/eji.200526065. [DOI] [PubMed] [Google Scholar]
- 80.Venken K, Hellings N, Hensen K, et al. Secondary progressive in contrast to relapsing-remitting multiple sclerosis patients show a normal CD4+CD25+ regulatory T-cell function and FOXP3 expression. J Neurosci Res. 2006;83:1432–46. doi: 10.1002/jnr.20852. [DOI] [PubMed] [Google Scholar]
- 81.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsaknaridis L, Spencer L, Culbertson N, et al. Functional assay for human CD4+CD25+ Treg cells reveals an age-dependent loss of suppressive activity. J Neurosci Res. 2003;74:296–308. doi: 10.1002/jnr.10766. [DOI] [PubMed] [Google Scholar]
- 83.Huan J, Culbertson N, Spencer L, et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res. 2005;81:45–52. doi: 10.1002/jnr.20522. [DOI] [PubMed] [Google Scholar]
- 84.Frisullo G, Nociti V, Iorio R, et al. Regulatory T cells fail to suppress CD4(+)T-bet(+) T cells in relapsing multiple sclerosis patients. Immunology. 2008;127:418–28. doi: 10.1111/j.1365-2567.2008.02963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Venken K, Thewissen M, Hellings N, et al. A CFSE based assay for measuring CD4+CD25+ regulatory T cell mediated suppression of auto-antigen specific and polyclonal T cell responses. J Immunol Methods. 2007;322:1–11. doi: 10.1016/j.jim.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 86.de Andres C, Aristimuno C, de Las Heras V, et al. Interferon beta-1a therapy enhances CD4+ regulatory T-cell function: an ex vivo and in vitro longitudinal study in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2007;182:204–11. doi: 10.1016/j.jneuroim.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 87.Kumar M, Putzki N, Limmroth V, et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. 2006;180:178–84. doi: 10.1016/j.jneuroim.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 88.Korporal M, Haas J, Balint B, et al. Interferon beta-induced restoration of regulatory T-cell function in multiple sclerosis is prompted by an increase in newly generated naive regulatory T cells. Arch Neurol. 2008;65:1434–9. doi: 10.1001/archneur.65.11.1434. [DOI] [PubMed] [Google Scholar]
- 89.Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci USA. 2005;102:6449–54. doi: 10.1073/pnas.0502187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu L, Xu Z, Xu M. Glucocorticoid treatment restores the impaired suppressive function of regulatory T cells in patients with relapsing-remitting multiple sclerosis. Clin Exp Immunol. 2009;158:26–30. doi: 10.1111/j.1365-2249.2009.03987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Michel L, Berthelot L, Pettre S, et al. Patients with relapsing-remitting multiple sclerosis have normal Treg function when cells expressing IL-7 receptor alpha-chain are excluded from the analysis. J Clin Invest. 2008;118:3411–19. doi: 10.1172/JCI35365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fletcher JM, Lonergan R, Costelloe L, et al. CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. 2009;183:7602–10. doi: 10.4049/jimmunol.0901881. [DOI] [PubMed] [Google Scholar]
- 93.Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 94.Olivares-Villagomez D, Wang Y, Lafaille JJ. Regulatory CD4(+) T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med. 1998;188:1883–94. doi: 10.1084/jem.188.10.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–16. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 96.Zhang X, Koldzic DN, Izikson L, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–56. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 97.Walsh KP, Brady MT, Finlay CM, Boon L, Mills KH. Infection with a helminth parasite attenuates autoimmunity through TGF-beta-mediated suppression of Th17 and Th1 responses. J Immunol. 2009;183:1577–86. doi: 10.4049/jimmunol.0803803. [DOI] [PubMed] [Google Scholar]
- 98.Reddy J, Illes Z, Zhang X, et al. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2004;101:15434–9. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stephens LA, Gray D, Anderton SM. CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc Natl Acad Sci USA. 2005;102:17418–23. doi: 10.1073/pnas.0507454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korn T, Reddy J, Gao W, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–31. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–25. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 102.McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–32. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- 103.Zhang X, Reddy J, Ochi H, Frenkel D, Kuchroo VK, Weiner HL. Recovery from experimental allergic encephalomyelitis is TGF-beta dependent and associated with increases in CD4+LAP+ and CD4+CD25+ T cells. Int Immunol. 2006;18:495–503. doi: 10.1093/intimm/dxh390. [DOI] [PubMed] [Google Scholar]
- 104.O'Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol. 2007;179:958–66. doi: 10.4049/jimmunol.179.2.958. [DOI] [PubMed] [Google Scholar]
- 105.Cox AL, Thompson SA, Jones JL, et al. Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur J Immunol. 2005;35:3332–42. doi: 10.1002/eji.200535075. [DOI] [PubMed] [Google Scholar]
- 106.Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786–801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- 107.Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol. 2009;39:1108–17. doi: 10.1002/eji.200839073. [DOI] [PubMed] [Google Scholar]
- 108.Lima XT, Abuabara K, Kimball AB, Lima HC. Briakinumab. Exp Opin Biol Ther. 2009;9:1107–13. doi: 10.1517/14712590903092188. [DOI] [PubMed] [Google Scholar]
- 109.Melnikova I. Psoriasis market. Nat Rev Drug Discov. 2009;8:767–8. doi: 10.1038/nrd2996. [DOI] [PubMed] [Google Scholar]
- 110.van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:549–53. doi: 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- 111.Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. [DOI] [PubMed] [Google Scholar]
- 112.Longbrake EE, Racke MK. Why did IL-12/IL-23 antibody therapy fail in multiple sclerosis? Expert Rev Neurother. 2009;9:319–21. doi: 10.1586/14737175.9.3.319. [DOI] [PubMed] [Google Scholar]
- 113.Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol. 2008;38:2636–49. doi: 10.1002/eji.200838535. [DOI] [PubMed] [Google Scholar]
- 114.Chen Y, Langrish CL, McKenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–26. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. 1998;28:2178–87. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 116.Korn T, Mitsdoerffer M, Croxford AL, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:18460–5. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kleinschek MA, Owyang AM, Joyce-Shaikh B, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–70. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]