

Abstract
Genome copy number changes (copy number variations: CNVs) include inherited, de novo and somatically acquired deviations from a diploid state within a particular chromosomal segment. CNVs are frequent in higher eukaryotes and associated with a substantial portion of inherited and acquired risk for various human diseases. CNVs are distributed widely in the genomes of apparently healthy individuals and thus constitute significant amounts of population-based genomic variation. Human CNV loci are enriched for immune genes and one of the most striking examples of CNV in humans involves a genomic region containing the chemokine genes CCL3L and CCL4L. The CCL3L–CCL4L copy number variable region (CNVR) shows extensive architectural complexity, with smaller CNVs within the larger ones and with interindividual variation in breakpoints. Furthermore, the individual genes embedded in this CNVR account for an additional level of genetic and mRNA complexity: CCL4L1 and CCL4L2 have identical exonic sequences but produce a different pattern of mRNAs. CCL3L2 was considered previously as a CCL3L1 pseudogene, but is actually transcribed. Since 2005, CCL3L-CCL4L CNV has been associated extensively with various human immunodeficiency virus-related outcomes, but some recent studies called these associations into question. This controversy may be due in part to the differences in alternative methods for quantifying gene copy number and differentiating the individual genes. This review summarizes and discusses the current knowledge about CCL3L–CCL4L CNV and points out that elucidating their complete phenotypic impact requires dissecting the combinatorial genomic complexity posed by various proportions of distinct CCL3L and CCL4L genes among individuals.
Keywords: chemokines, copy number variation, human, polymorphism
Copy number variation: from the global genomic variability to the implications in the immune system
In the last decade, many studies showed that a major component of the differences between individuals is variation in the copy number of segments of the genome [copy number variation (CNV) or copy number polymorphism (CNP)]. CNVs are distributed widely in the genomes of healthy individuals and thus constitute significant amounts of population-based genomic variation [1–7]. CNV seems to be at least as important as single nucleotide polymorphisms (SNPs) in determining the differences between individual humans [8]. CNV also seems to be a major driving force in evolution, especially in the rapid evolution that has occurred, and continues to occur, within the human and great ape lineage. Compared with other mammals, the genomes of humans and other primates show an enrichment of CNVs. Primate lineage-specific gene CNV studies reveal that almost one-third of all human genes exhibit a copy-number change in one or more primate species [9–12]. To date, almost 58 000 human CNVs from approximately 14 500 regions (CNVRs) have been identified (data from Database of Genomic Variants, http://projects.tcag.ca/variation/). These CNVRs may cover 5–15% of the human genome and encompass hundreds of genes [4,13], and their abundance underscores their substantial contribution to genetic variation and genome evolution [14]. CNVs can arise both meiotically and somatically, because identical twins can have different CNVs [15]. Furthermore, repeated sequences from the same individual can vary in copy number in different organs and tissues [16]. The general mechanisms that lead to changes in copy number include homologous recombination and non-homologous repair mechanisms [17].
Changes in copy number might alter the expression levels of genes included in the CNVR. For example, the salivary amylase gene, AMY1, shows CNV in human populations, and the amount of salivary amylase is directly proportional to the copy number of AMY1[18]. More importantly, CNVs shape tissue transcriptomes on a global scale [19]. Additional copies of genes also provide redundancy that allows some copies to evolve new or modified functions while other copies maintain the original function.
CNVs can represent benign polymorphic variations or convey clinical phenotypes by mechanisms such as altered gene dosage and gene disruption. CNV can be responsible for sporadic birth defects [20], other sporadic traits, Mendelian diseases and complex traits including autism, schizophrenia, epilepsy, Parkinson disease, Alzheimer disease, human immunodeficiency virus (HIV) infection and mental retardation [21–23].
Interestingly, the set of genes that vary in copy number seems to be enriched for genes involved in olfaction, immunity and secreted proteins [24]. The following diseases are associated with CNVs of the immune genes: (i) CNVs of FCGR3B and FCGR2C (encoding different Fcγ receptors) have been associated with a range of autoimmune diseases, including systemic lupus erythematosus (SLE), polyangiitis, Wegener's granulomatosis and idiopathic thrombocytopenic purpura [25–27]. (ii) CNVs of the complement genes CFHR1 and CFHR3, which belong to the complement factor H protein family, have been associated with age-related macular degeneration and atypical haemolytic-uraemic syndrome [28–30]. Complement C4 gene copy number has been related directly with systemic lupus erythematosus (SLE) [31]. (iii) On chromosome 8, a unit of seven β-defensin genes, which encode anti-microbial peptides with other diverse functions such as chemokine activity [32], has variability in its copy number [33]: low copy number has been associated with Crohn's disease [34,35], and high copy number with predisposition to psoriasis [36]. (iv) In this review, we will examine one of the most striking examples of CNV in the human genome, the chemokine genes CCL3L and CCL4L.
Copy number variation in chemokine superfamily: the CCL3L–CCL4L case
Chemokines are a large superfamily of small structurally related cytokines that regulate cell trafficking of various types of leucocytes to areas of injury, and play key roles in both inflammatory and homeostatic processes. Chemokines are classified into four families based on the arrangement of the first two cysteines of the typically conserved four cysteines: CXC, CC, C and CX3C (where X is any amino acid) [37]. The chemokine superfamily constitutes an extremely revealing case of a complex network of genes that has acquired a very diverse set of related functions through evolution [38]. Many chemokine genes are clustered in defined chromosomal locations [39]. Two main clusters encode the essential inflammatory chemokines: the CXC cluster located in chromosome 4q12–21 and the CC cluster located in chromosome 17q11.2–q12. A potential explanation for this chromosomal arrangement is found in the evolutionary forces that have shaped the genome into gene superfamilies [40]. Over the course of evolution, gene duplication has been a common event, affecting most gene families [41]. Once a duplication occurs, the two copies can evolve independently and develop specialized functions. This phenomenon explains the origin of chemokine clusters. An important characteristic of a chemokine cluster is that their genes code for many ligands that interact with a few receptors. Therefore, chemokine clusters act as single entities based on their overall function.
The cluster of proinflammatory CC chemokines contains 16 genes localized to a 2·06 Mb interval at 17q11.2–q12 on genomic contig NT_010799 (Fig. 1a). Four of these genes comprise the two closely related, paralogous pairs CCL3–CCL3L and CCL4–CCL4L[42]. Members within each pair share 95% sequence identity at both the genomic and the amino acid levels. Among all human chemokine genes, a singular characteristic of CCL3L and CCL4L, is that they are present in variable copy numbers in the human genome. The CNV affecting CCL3L–CCL4L has been studied extensively since 2002 (when Towson et al. reported the first data about the extent of CCL3L–CCL4L CNV in the Caucasian population [43]), although two groups had identified the existence of CCL3L–CCL4L as non-allelic copies of CCL3–CCL4 and as copy number variable genes 20 years ago [44,45].
Fig. 1.
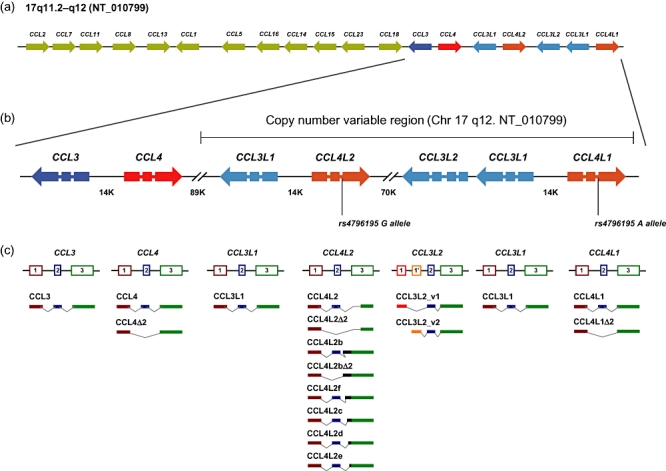
Genomic organization and mRNA products of human CCL3–CCL4 and CCL3L–CCL4L genes. (a) Map of the CC chemokine cluster in the 17q11.2–q12 region, based on the genomic sequence NT_010799. The orientation of each gene is shown by an arrow. (b) Genomic organization of human CCL3–CCL4 and CCL3L–CCL4L genes based on the genomic sequence NT_010799. Distances between genes are expressed in Kb. The nucleotide change [single nucleotide polymorphism (SNP) rs4796195] that leads to CCL4L1 (A allele) or CCL4L2 (G allele) is shown. (c) Transcription pattern of human CCL3–CCL4 and CCL3L–CCL4L genes. mRNAs derived from each individual gene are shown.
The CNVR that includes CCL3L and CCL4L genes (and other non-related loci) seems to have been generated through a segmental duplication of a genomically unstable stretch of about 120 kb located on this region of chromosome 17 [43–48]. In fact, the q arm of chromosome 17 of humans has multiple regions of genomic instability where gene duplications, chromosomal rearrangements and copy number variation are common [49,50]. Furthermore, the human CCL3L–CCL4L region shows evidence of complex homologous recombination events. For example, high-resolution CNV data reveal extensive architectural complexity in the CCL3L–CCL4L region, which includes smaller CNVs embedded within larger ones and interindividual variation in breakpoints [5,49]. One of the consequences of this complexity is that individuals may vary not only in the total copy number of CCL3L and CCL4L genes, but also their individual components. Underscoring this, although the copy number of CCL3L correlates with CCL4L, individuals average more copies of CCL3L than CCL4L[43,51,52]. Currently, gene copy numbers in humans range from 0 to 14 for CCL3L and from 0 to 10 for CCL4L with a strong population structure. Sub-Saharan African populations display the highest number of CCL3L–CCL4L copies (median 6 for CCL3L and 4 for CCL4L), whereas Europeans present the lowest copy numbers (median 2 for CCL3L and CCL4L). The number of individuals without CCL3L or CCL4L is always below 5% in all continental regions [52,53].
The duplicated region encoding human CCL3L–CCL4L genes has an ancestral correlate in non-human primates. The CCL3L–CCL4L copy numbers are much higher in non-human primates than in human populations [53–55]. Gonzalez et al. determined the gene copy numbers of the chimpanzee (Pan troglodytes) CCL3L orthologues from 83 animals. The CCL3L copies range from 6 to 17 per diploid genome (median 9; mean 9·3) [53]. Similarly, Degenhardt et al. observed extensive variation in copy number of the CCL3L region among 57 samples of rhesus macaque (Macaca mulatta): copy number estimates range from 5 to 31 copies per diploid genome (median 10; mean 11·1) [54].
Genes and nomenclature in the CCL3L–CCL4L cluster
Currently, the official symbols of the genes included in the CCL3L–CCL4L cluster are based on the public human genome sequence which contains, by chance, three CCL3L copies and two CCL4L copies. CCL3L and CCL4L have been numbered based on their position from the more centromeric to the more telomeric. Thus the official symbols for CCL3L genes are CCL3L1 (GeneID: 6349), CCL3L2 (GeneID: 390788) and CCL3L3 (GeneID: 414062). The official symbols for CCL4L genes are CCL4L1 (GeneID: 9560) and CCL4L2 (GeneID: 388372). However, we believe that the nomenclature criterion should consider whether the genes are really different rather than solely their copy number. Although CCL3L1 and CCL3L3 are separate genes, both have three identical exons and encode identical proteins [42,47], and therefore they are denoted together here as CCL3L1 (Fig. 1). CCL3L2 (known previously as LD78γ or GOS19-3) was identified initially as a pseudogene, as it contains two exons that are homologous to exons 2 and 3 of the CCL3L1 gene and appeared to contain a 5′ truncation compared with CCL3L1[46]. However, Shostakovich-Koretskaya et al. recently identified novel 5′ exons for CCL3L2 which give rise to two alternatively spliced transcripts by bioinformatics and mRNA profiling (Fig. 1c) [51]. These alternatively transcribed mRNA species contain chemokine-like domains but are not predicted to encode classical chemokines (data not shown [51]).
Regarding CCL4L genes, CCL4L1 and CCL4L2 share 100% sequence identity in the coding regions. However, a fixed mutation at the intron–exon boundary of some CCL4L genes results in the production of aberrantly spliced transcripts [48]. We proposed the name of the originally described gene (corresponding to GeneID: 388372) as CCL4L1 and CCL4L2 (GeneID: 9560) as the gene that contains the mutation at the intron–exon boundary [38,48,52,56]. We use this nomenclature in this review (view Fig. 1) and we note that the same concept has been applied recently by others [51].
Functional aspects of CCL3–CCL4-, CCL3L–CCL4L-derived chemokines
To understand more clearly the role of CCL3L–CCL4L CNV in normal host-protective inflammatory responses as well as in disease-associated physiopathology, it is important to consider the functional differences among the chemokines encoded by CCL3L and CCL4L genes and also the CCL3 and CCL4 genes.
Functional differences between CCL3–CCL4
As aforementioned, CCL3 and CCL4 are two structurally and functionally related CC chemokines. CCL3 and CCL4 were both discovered in 1988, when Wolpe et al. purified a protein doublet from the supernatant of lipopolysaccharide (LPS)-stimulated murine macrophages [57]. Because of its inflammatory properties in vitro as well as in vivo, the protein mixture was called macrophage inflammatory protein-1 (MIP-1). Further biochemical separation and characterization of the protein doublet yielded two distinct, but highly related proteins, MIP-1α and MIP-1β[58]. From 1988 to 1991, several groups reported independently the isolation of the human homologues of MIP-1α and MIP-1β[59–61]. As a consequence, alternate designations were used for MIP-1α (LD78α, AT464·1, GOS19-1) and MIP-1β (ACT-2, AT744·1), similar to other members of chemokine superfamily. In an attempt to clarify the confusing nomenclature associated with chemokines and their receptors, a new nomenclature was introduced by Zlotnik and Yoshie in 2000 [37]. MIP-1α and MIP-1β were renamed as CCL3 and CCL4. The non-allelic copies of CCL3 and CCL4 were designated as CCL3L (previously LD78β, AT 464·2, GOS19-2) and CCL4L (previously LAG-1, AT744·2).
CCL3 and CCL4 precursors and mature proteins share 58% and 68% identical amino acids, respectively (Fig. 2). Both chemokines are expressed upon stimulation by monocytes/macrophages, T and B lymphocytes and dendritic cells (although they are inducible in most mature haematopoietic cells). Functionally, CCL3 and CCL4 are potent chemoattractants of monocytes, T lymphocytes, dendritic cells and natural killer cells [47]. Despite these similarities, CCL3 and CCL4 differ in the recruitment of specific T cell subsets: CCL3 preferentially attracts CD8 T cells while CCL4 preferentially attracts CD4 T cells [62]. Interestingly, Bystry and co-workers demonstrated that B cells and professional antigen-presenting cells (APCs) recruit CD4+CD25+ regulatory T cells via CCL4 [63]. This role of CCL4 in immune regulation was reinforced later by Joosten et al. [64], who identified a human CD8+ regulatory T cell subset that mediates suppression through CCL4 but not CCL3. CCL3 and CCL4 also differ in their effect on stem cell proliferation: CCL3 suppresses proliferation of haematopoietic progenitor cells [65]. CCL4 has no suppressive or enhancing activity on stem cells or early myeloid progenitor cells by itself, but has the capacity to block the suppressive actions of CCL3 [66].
Fig. 2.

Alignment of human CCL3–CCL4 and CCL3L–CCL4L derived proteins. Signal peptides are depicted in grey. Cysteines are depicted in red. Basic amino acids, which are involved in the binding of chemokines to the glycosaminoglycans are depicted in blue. The S/G swap shared between CCL3–CCL3L1 and CCL4–CCL4L1/L2 proteins is depicted in green.
A different receptor usage may help to explain, at least in part, why these molecules have overlapping, but not identical, bioactivity profiles: CCL3 signals through the chemokine receptors CCR1 and CCR5. In contrast, CCL4 signals mainly through the CCR5 [47], although it can also induce moderate chemotaxis in CCR1 and CCR3-expressing cells [67] (Table 1). Additionally, CCL4 is cleaved in vivo by CD26, which is a dipeptidyl–peptidase that cuts dipeptides from the NH2 terminus of regulatory peptides with a proline or alanine residue in the penultimate position [68]. The truncated form of CCL4, CCL4(3–69), lacks the two first amino acids [69]. Functional studies of the purified truncated protein revealed that CCL4(3–69) also signals through CCR5 and exhibits enhanced biological activity through CCR1 compared to the full-length CCL4. It also has a novel binding specificity for CCR2b (Table 1) [70]. CCL4(3–69) appears to be produced only by activated T cells; it has not been detected in culture supernatants of monocytes or macrophages.
Table 1.
Receptor usage of CCL3–CCL4- and CCL3L–CCL4L-derived proteins.
| CCR1 | CCR2b | CCR3 | CCR5 | Anti-HIV activity | |
|---|---|---|---|---|---|
| CCL3/CCL3L1 | |||||
| CCL3(1–70) | Yes/+++ | No | No | Yes/++ | Yes/++ |
| CCL3(5–70) | Yes/++++ | No | No | Yes/+++ | Yes/++ |
| CCL3L1(1–70) | Yes/++ | No | Yes/+++ | Yes/++++ | Yes/++++ |
| CCL3L1(3–70) | Yes/++++ | No | Yes/+ | Yes/+++++ | Yes/++++ |
| CCL3L1(5–70) | Yes/+++ | No | No | Yes/+++ | Yes/++ |
| CCL3L2 | ? | ? | ? | ? | ? |
| CCL4/CCL4L1/CCL4L2 | |||||
| CCL4 | Yes/+ | No | Yes/+ | Yes/+++ | Yes/+++ |
| CCL4(3–69) | Yes/+++ | Yes/+++ | No | Yes/+++ | Yes/+++ |
| CCL4L1 | Yes/+ | No | Yes/+ | Yes/+++ | Yes/+++ |
| CCL4L2 | ? | ? | ? | ? | ? |
HIV: human immunodeficiency virus.
Functional differences between CCL3–CCL3L1
The CCL3 and CCL3L1 mature proteins differ in three amino acids: CCL3L1 has a proline (P) in position 2 instead of the serine (S) in CCL3, and the other two changes are reciprocal S/G (glycine) swaps in the region between cysteines 3 and 4 (Fig. 2). The CCL3L1 receptor usage includes CCR5 and CCR1 but, unlike CCL3, CCL3L1 also binds efficiently to CCR3 (Table 1) [71]. CCL3L1 is significantly more potent in inducing intracellular Ca2+ signalling and chemotaxis through the CCR5 than CCL3 (and CCL5). CCL3L1's binding affinity to CCR5 is sixfold higher than CCL3's affinity. Furthermore, CCL3L1 antagonizes HIV-1 entry through CCR5 to a significantly greater extent than CCL3 [72–75]. In fact, CCL3L1 is consistently better at HIV-1 antagonism than CCL5, described previously as the most potent CCR5-dependent HIV-1 entry inhibitor. This enhanced activity of CCL3L1 is due to the presence of the proline residue at position 2 of the mature protein [74], and supports the importance of the NH2-terminal regions of both CXC and CC chemokines for their biological activity [76]. Interestingly, truncated forms of CCL3L1 are found in vivo: CCL3L1(3–70) and CCL3L1(5–70). (i) CCL3L1(3–70) results from processing full-length CCL3L1 by CD26. Compared with full-length CCL3L1, CCL3L1(3–70) has an increased binding affinity for CCR1 and CCR5 and shows a reduced interaction with CCR3 (Table 1). Its enhanced CCR1 and CCR5 affinity converted CCL3L(3–70) into a highly efficient monocyte and lymphocyte chemoattractant [77]. The high affinity of this truncated molecule for CCR5 explains its highly potent blocking of HIV-1 infection [71,77]. (ii) CCL3L1(5–70) interacts more strongly with CCR1 than intact CCL3L1, but its reduced affinity for CCR5 decreases its anti-viral activity significantly (Table 1) [74]. Although CCL3L1(5–70) could potentially derive from CD26 proteolysis of CCL3L1(3–70) (with a penultimate alanine), only a limited further truncation of CCL3L1(3–70) was detected after prolonged incubation with CD26 [77]. This suggests that other aminopeptidases may be involved in the further degradation of CCL3L1(3–70) chemokine to CCL3L1(5–70).
Finally, natural sources contain the full-length protein, CCL3(1–70), and a truncated form lacking the first four amino acids, CCL3(5–70)[77]. Compared to the full-length CCL3, CCL3(5–70) shows enhanced binding affinity to CCR1 and CCR5 (Table 1) [74].
Functional differences between CCL4–CCL4L1
CCL4 and CCL4L1 mature proteins differ only in one amino acid: a conservative S to G change at amino acid 47 of the mature protein (Fig. 2) [48,78]. Few studies have been compared the functions of CCL4 and CCL4L1. Modi et al. reported a functional redundancy of the human CCL4 and CCL4L1 chemokines: their competitive binding assays, cell motility and anti-HIV-1 replication experiments revealed similar activities of the CCL4 and CCL4L1 proteins [67]. However, structural analysis of the CCL4 and CCL4L1 proteins revealed the importance of amino acid 47 of the mature protein: this amino acid (S) in CCL4 protein forms a hydrogen bond with amino acid Thr44, thus conferring structural stability to the loop defined by the β-turn between the second and third strands of the β-sheet [79]. However, the glycine (G) at that position in the CCL4L1 protein cannot form this hydrogen bond. This loop is believed to be essential for the binding of CCL4 to the glycosaminoglycans (GAGs) [80]. It has been suggested that the immobilization of chemokines on GAGs forms stable, solid-phase chemokine foci and gradients crucial for directing leucocyte trafficking in vivo. Their higher effective local concentration increases their binding to cell surface receptors and influences chemokine T1/2in vivo[81–84]. Hence, the destabilization of this loop could reduce the stability of CCL4L1 binding to GAGs and therefore modify their functional features in vivo. It is important to note that the available data about functional studies of CCL4 and CCL4L1 were obtained by in vitro experiments, where the binding of these chemokines to GAGs is neglected. The apparent functional redundancy of CCL4 and CCL4L1 in vitro warrants further in vivo studies examining their GAG binding capabilities.
Additionally, regulation of CCL4 and CCL4L1 expression appears different. Lu et al. reported an independent expression of the CCL4 and CCL4L1 genes in monocytes and B lymphocytes [85]. This observation suggests that differential expression of these proteins in different cells provides an advantage to the host and that these proteins might have different functions in vivo.
Both CCL4 and CCL4L1 genes produce alternatively spliced mRNAs that lack the second exon, which give rise to the CCL4Δ2 and CCL4L1Δ2 variants (Figs 1c and 2) [48,78]. The predicted CCL4Δ2 and CCL4L1Δ2 proteins of only 29 aa would only maintain the first two amino acids from the CCL4 and CCL4L1 proteins, lacking three of the four cysteine residues critical for intramolecular disulphide bonding. Therefore, CCL4Δ2 and CCL4L1Δ2 may not be structurally considered chemokines. Despite the difficulty in predicting protein folding, these variants do not seem to be able to bind to CCR5 and thus may have no CCL4/CCL4L1 activity [48].
Finally, we note that CCL4L1, CCL4Δ2 and CCL4L1Δ2 are also potential targets of CD26 and their cleavage by this dipeptidyl–peptidase may produce truncated forms. However, this prediction has not yet been demonstrated.
Increased complexity of CCL4L genes: CCL4L1 versus CCL4L2
As mentioned, although human CCL4L1 and CCL4L2 share 100% sequence identity in the coding regions, a fixed mutation at the intron–exon boundary of CCL4L2 results in the production of aberrantly spliced transcripts. Specifically, CCL4L2 show one base substitution (rs4796195 in dbSNP) at the acceptor splice site of intron 2 [48]. According to the canonical splicing pattern [86], the donor splice site of the second intron in CCL4L1 has GT immediately after exon 2, and the acceptor site has AG just before the point where intron 2 sequence is cleaved. In CCL4L2, the canonical sequence of the acceptor splice site (AG) has changed to GG and the spliceosome is unable to recognize the mutated acceptor site (GG). Instead, alternative acceptor sites around the original one are selected, and a minimum of eight different mRNAs are generated (Fig. 1c) [48]. The most abundant of these mRNAs derived from CCL4L2 corresponds to the CCL4L2 variant, which accounts for 80% of total mRNA expression [48]). CCL4L2 is generated by the use of an acceptor splice site located 15 nucleotides downstream of the original site. The predicted CCL4L2 mature protein has 64 amino acids and lacks the initial five amino acids encoded by the third exon (Phe42, Gln43, Thr44, Lys45 and Arg46), but the rest of the sequence remains unchanged (Fig. 2). The functional consequences of deleting these five amino acids in CCL4L2 are unknown and, to date, there are no published functional studies involving CCL4L2. However, some computational data suggest the importance of these five amino acids: (i) critical analysis of the conserved amino acids in CC chemokines show that Phe42, Thr44 and to a lesser degree Lys45, are highly conserved residues in this subfamily. (ii) CCL4 (as well as CCL3 and CCL5) tends to self-associate and form homodimers, tetramers or high molecular mass aggregates in vitro, and possibly in vivo under certain conditions, in a process that involves residues Lys45 and Arg46[87]. Furthermore, naturally occurring CCL4/CCL3 heterodimers are present at physiological concentrations [88]. Therefore, the deletion of these five amino acids could have a negative effect on the ability of CCL4L2 to form self-aggregates or heterodimers with CCL3 or CCL3L1. (iii) Additionally, due to the fact that Lys45 and Arg46 are also critical residues in the CCL4 binding to GAGs [80], it is expected that the GAG binding of CCL4L2 will be seriously reduced, if not abrogated.
The remaining CCL4L2 mRNA variants occur at very low abundance, and the folding prediction and the functional features of their putative proteins are difficult to establish. The biological relevance of these proteins (if effectively produced) is unknown and may be influenced by their low expression level.
CCL3L and CCL4L gene expression: copies count
Since the beginning of the CNV discovery, one of the most intriguing questions has been its consequences on gene expression. To date, the global impact of CNV on gene expression phenotypes varies depending upon the gene [89], as increased copy number can be correlated positively [90] or negatively [91] with gene expression levels. Focusing upon CCL3L, gene copy number regulates the production of CCL3L1 both at mRNA and protein level: specifically, increasing CCL3L copy number was associated positively with CCL3L1 mRNA production and protein secretion [43,53,92]. The relationship between CCL4L copy number and the amount of CCL4L1 mRNA or protein expression has some, but still no conclusive, data. Although Townson and co-workers demonstrated that high CCL3L copy number correlates with increased chemokine production [43], this study also analysed the CCL4L gene and failed to detect any consistent increase in CCL4L1 mRNA production from samples with a high CCL4L copy number. However, they found that individuals with only one copy of CCL4L had a consistently lower expression of CCL4L1 than those with a higher copy number. We note that at the time of its 2002 publication, Townson et al. were not aware of the existence of the CCL4L2 variant, which produces transcripts and proteins distinct to CCL4L1[48], and their need to be quantified independently. The assumption that all the CCL4L copies that they quantified corresponded to CCL4L1 could explain the lack of a consistent correlation between CCL4L gene copy number and CCL4L1 mRNA production in this study. More recently, a study by Melzer et al. reported a new cis-effect of a SNP located near the CCL4L1 gene (227 kb) on CCL4L1 protein production [93]. They hypothesize that the effect is caused by the CCL4L CNV in linkage disequilibrium with the analysed SNP. Although CCL4L copy number probably influences mRNA/protein production, further studies are needed to assess the effect of CCL4L copies on gene expression. Future studies in this direction should analyse CCL4L1 and CCL4L2 copies independently to assess precisely the effect of the total CCL4L copies on gene expression (a general approach to discriminate CCL4L1 and CCL4L2 from the total CCL4L copies has been described [52]).
CNV and disease: the role of CCL3L and CCL4L
If CNV affects entire genes, especially those with important effects on biological function, CNV would naturally be expected to affect susceptibility to disease. Concerning this review, CCL3L–CCL4L CNV has been associated with a variety of diseases, with viral infections and autoimmune diseases being the most represented categories. In Table 2, we summarized the disease association studies involving CCL3L and/or CCL4L CNV, including both positive and negative results. The most extensively studied and controversial association involves CCL3L CNV and HIV infection. The first data appeared in 2005, when a paper reported effects of CCL3L1 copy number variation on HIV-1 acquisition, viral load and disease progression [53]. This study was followed by several publications investigating clinically correlated phenotypes in a largely overlapping set of HIV-positive individuals [94–97]. Other independent studies have confirmed different aspects of this association in different human populations [51,98–102]. In theory, the higher the copy number, the higher the ligand concentration, which should protect the host from HIV infection or disease progression. Chimpanzees with higher copies do not develop acquired immune deficiency syndrome (AIDS); this association suggests biological significance. CNV of CCL3L genes also affects the rate of progression to AIDS in rhesus macaques [54]. However, two recent large studies dispute these previous findings by showing the absence of any substantial effect of CCL3L1 CNV on HIV-1 infection, viral load or disease progression [92,103]. This controversy may be due in part to the differences in alternative methods for quantifying CCL3L1 copy number and differentiating this gene from its prototype CCL3 and from the neighbouring CCL3L2 (excellently discussed in [104]). To study the experimental aspects of CCL3L1 copy number quantification in depth, Field et al. [105] evaluated the CCL3L1 copy numbers in more than 10 000 British individuals and documented differences between the results generated by TaqMan assay and by an alternative assay called the paralogue ratio test (PRT). More recently, Shrestha et al. [106] evaluated the different assays used to measure gene copy numbers of CCL3L1 and indicated that some of the inconsistencies in these association studies could be due to assays that provide heterogenous results.
Table 2.
Disease association studies involving CCL3L–CCL4L copy number variations (CNV).
| Gene | Population/cohort | Cases | Controls | Association | Type of association | Ref. |
|---|---|---|---|---|---|---|
| HIV infection | ||||||
| CCL3L | WHMC (EA, AA, HA) Argentinean children | 1127 407 | 2379 395 | Yes | Disease association Clinical aspects | [53] |
| CCL3L | WHMC (EA, AA, HA) MGH UCSF | 1132 98 65 | Yes* | Clinical aspects | [95] | |
| CCL3L | WHMC (EA, AA, HA) UCSF UCSD | 445 209 174 | Yes* | Clinical aspects | [94] | |
| CCL3L | WHMC (EA, AA, HA) | 1103 | Yes* | Clinical aspects | [96] | |
| CCL3L | WHMC (EA, AA, HA) | 1103 | Yes* | Clinical aspects | [97] | |
| CCL3LCCL4L | Ukraine | 178 | 120 | Yes | Disease association Clinical aspects | [51] |
| CCL3L | South Africa | 79 | 235 | Yes | Disease association Clinical aspects | [100] |
| CCL3L | South Africa | 46 | 74 | Yes | Disease association | [102] |
| CCL3L | Estonia | 208 | 166 | Yes | Disease association | [98] |
| CCL3L | Japan | 95 | 205 | Yes | Disease association | [101] |
| CCL3L | South Africa | 71 | Yes | Clinical aspects | [99] | |
| CCL3L | Rhesus macaque | 57 | Yes | Clinical aspects | [54] | |
| CCL3LCCL4L | AA, HA, EA | 227 | 184 | No | Disease association Clinical aspects | [108] |
| CCL3L | Euro-CHAVI TACC MACS | 1042 277 451 | 195 | No | Disease association Clinical aspects | [92] |
| CCL3L | MACS (EA, AA) | 580 | 437 | No | Disease association Clinical aspects | [103] |
| Type 1 diabetes | ||||||
| CCL3LCCL4L | British | 5771 | 6854 | No | Disease association | [105] |
| CCL3L | New Zealand (Caucasian) | 252 | 282 | No† | Disease association | [109] |
| Chronic hepatitis C | ||||||
| CCL3L | Germany | 254 | 210 | Yes | Disease association | [110] |
| Systemic lupus erythematosus (SLE) | ||||||
| CCL3L | San Antonio SLE cohort Colombian SLE cohort Ohio SLE cohort | 134 143 192 | 60 421 134 | Yes* | Disease association Clinical aspects | [111] |
| CCL3L | Colombia (Spanish ancestry) | 146 | 409 | Yes‡ | Disease association | [112] |
| Rheumatoid arthritis | ||||||
| CCL3L | New Zealand (Caucasian) British (Caucasian) | 834 302 | 933 255 | Yes/No§ | Disease association | [109] |
| Lung transplantation acute rejection | ||||||
| CCL4L | Spain (Caucasian) | 161 | Yes | Clinical aspects | [56] | |
| Kawasaki disease | ||||||
| CCL3L | United States | 164¶ | Yes* | Disease association | [113] | |
| Primary Sjögren's syndrome | ||||||
| CCL3L | Colombia (Spanish ancestry) | 61 | 409 | Yes‡ | Disease association | [112] |
For conjoint effects of CCL3L–CCR5.
The authors state that there was evidence for association of CCL3L copy number in the T1D cohort, but they reported a non-significant result (odds ratio 1·46, 95% confidence interval 0·98–2·20, P = 0·064).
For conjoint effects of FCGR3B–CCL3L.
CCL3L copy number was a risk factor for rheumatoid arthritis (RA) in the New Zealand cohort but not in the smaller UK RA cohort.
Cohort of 164 children with Kawasaki disease and their biological parents (transmission disequilibrium test).
WHMC: Wilford Hall Medical Center; MGH: Massachusetts General Hospital; UCSF: University of California San Francisco; UCSD: University of California San Diego; TACC: Tri-Service AIDS Clinical Consortium; MACS; Multicenter AIDS Cohort Study; EA: European American; AA: African American; HA: Hispanic American. HIV: human immunodeficiency virus.
Concluding remarks and future perspectives
The CCL3L–CCL4L CNVR is a model of extensive architectural complexity, which exhibits smaller CNVs embedded within larger ones and interindividual variation in breakpoints [5]. This degree of complexity is also highlighted by recent sequence data showing that the most extreme copy number variation corresponds to genes that are embedded within segmental duplications [107], such as the CCL3L–CCL4L genes [42,55]. Although there is a high degree of correlation between the copy number of CCL3L and CCL4L genes, most individuals contain more copies of CCL3L than CCL4L[43,51,52]. Additionally, this CNVR contains the following additional tiers of genetic and mRNA complexity: (i) CCL3L2, which was considered previously as a pseudogene, contains novel 5′ exons that produce two alternatively spliced transcripts [51]. (ii) Although CCL4L1 and CCL4L2 have identical exonic sequences, an (A→G) transition in the acceptor splice site in intron 2 of CCL4L2 generates aberrantly spliced CCL4L2 transcripts [48].
Therefore, dissecting the combinatorial genomic complexity posed by varying proportions of distinct CCL3L and CCL4L genes among individuals is required to elucidate the complete phenotypic impact of this locus. Available sequence information that determines the CNV of these four genes separately (CCL3L1, CCL3L2, CCL4L1 and CCL4L2) would allow testing of whether their association with the pathogenesis of a human disease or phenotype is affected by an individual gene or by a combination of these genes. In fact, a few published studies already tackle this approach: Shostakovich-Koretskaya et al. [51] determined the influence of the combinatorial content of distinct CCL3L and CCL4L genes on HIV/AIDS susceptibility. They developed two separate assays to quantify the total copy number of all CCL3L or CCL4L genes, and separate assays each for the individual components of CCL3L (CCL3L1 and CCL3L2) and CCL4L (CCL4L1 and CCL4L2). This study confirms and amplifies the results of previous studies which showed that a low dose of CCL3L genes is associated with an increased risk of acquiring HIV and progressing rapidly to AIDS. Their results also demonstrate that a low CCL4L gene dose has similar associations. Furthermore, they show that the balance between the copy numbers of the genes that transcribe classical (CCL3L1 and CCL4L1) versus aberrantly spliced (CCL3L2 and CCL4L2) mRNA species influences HIV/AIDS susceptibility: a higher gene content of CCL4L2 or a lower content of CCL3L1 and CCL4L1 increased the risk of transmission and an accelerated disease course. A similar negative influence of CCL4L2 on HIV acquisition was shown previously [48]. We also have shown that CNV in the CCL4L gene is associated with susceptibility to acute rejection in lung transplantation [56]. After specifically quantifying the CCL4L1 and CCL4L2 copies, we demonstrated that the correlation between CCL4L copy number and risk of acute lung transplant rejection was explained mainly by the number of copies of the CCL4L1 gene. These two studies imply that the assessment of global CCL4L dose requires capturing the sum of two genes (CCL4L1 and CCL4L2) with inversely related copy number frequencies [51,52] and differential effects. Thus, the true phenotypic impact of CCL4L1 and CCL4L2 cannot be made exclusively using the CCL3L copy number as a proxy for CCL4L or by evaluation of the composite CCL4L. This might explain, in part, why previous studies may not have found an association between CCL4L copy number and HIV disease [108]. Similarly, accounting for this genomic complexity, including CCL3L2 copy number may be crucial for full interpretation of association studies.
In summary, for future studies involving CCL3L–CCL4L CNVR and, in general, from a broader perspective of relevance to the CNV field, to determine normal phenotypic variation or disease susceptibility it seems to be crucial to define precisely the genomic structure, taking into account the specific combination of the distinct genes within a CNVR. The use of incomplete data will be always a source of controversy, providing misleading information. Only a complete analysis will clarify the importance of CCL3L–CCL4L CNVR in disease.
Acknowledgments
This work was supported by grants from the FIPSE (Fundación para la Investigación y la Prevención del Sida en España) (Project 36487/05), FIS (Fondo de Investigaciones Sanitarias) (Project PI 07/0329) and PEI (Pla Estratègic d'Investigació) from BST (Banc de Sang i Teixits).
Disclosure
All authors declare no conflicts of interest.
References
- 1.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 3.Tuzun E, Sharp AJ, Bailey JA, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–32. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 4.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry GH, Ben-Dor A, Tsalenko A, et al. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet. 2008;82:685–95. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidd JM, Cooper GM, Donahue WF, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–12. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–46. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 9.Dumas L, Kim YH, Karimpour-Fard A, et al. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res. 2007;17:1266–77. doi: 10.1101/gr.6557307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee AS, Gutiérrez-Arcelus M, Perry GH, et al. Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum Mol Genet. 2008;17:1127–36. doi: 10.1093/hmg/ddn002. [DOI] [PubMed] [Google Scholar]
- 11.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–64. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 12.Marques-Bonet T, Kidd JM, Ventura M, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–81. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarroll SA, Kuruvilla FG, Korn JM, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–74. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 14.Reymond A, Henrichsen CN, Harewood L, Merla G. Side effects of genome structural changes. Curr Opin Genet Dev. 2007;17:381–6. doi: 10.1016/j.gde.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Bruder CE, Piotrowski A, Gijsbers AA, et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet. 2008;82:763–71. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piotrowski A, Bruder CE, Andersson R, et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29:1118–24. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 17.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–64. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry GH, Dominy NJ, Claw KG, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256–60. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henrichsen CN, Vinckenbosch N, Zöllner S, et al. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41:424–9. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 20.Lu XY, Phung MT, Shaw CA, et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–18. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–55. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 22.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–81. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wain LV, Armour JA, Tobin MD. Genomic copy number variation, human health, and disease. Lancet. 2009;374:340–50. doi: 10.1016/S0140-6736(09)60249-X. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen DQ, Webber C, Ponting CP. Bias of selection on human copy-number variants. PLoS Genet. 2006:2. doi: 10.1371/journal.pgen.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 26.Fanciulli M, Norsworthy PJ, Petretto E, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific. Autoimmunity. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breunis WB, van Mirre E, Bruin M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. 2008;111:1029–38. doi: 10.1182/blood-2007-03-079913. [DOI] [PubMed] [Google Scholar]
- 28.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–7. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 29.Zipfel PF, Edey M, Heinen S, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3:e41. doi: 10.1371/journal.pgen.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spencer KL, Hauser MA, Olson LM, et al. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum Mol Genet. 2008;17:971–7. doi: 10.1093/hmg/ddm369. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Chung EK, Wu YL, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–54. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niyonsaba F, Ogawa H, Nagaoka I. Human beta-defensin-2 functions as a chemotactic agent for tumour necrosis factor-alpha-treated human neutrophils. Immunology. 2004;111:273–81. doi: 10.1111/j.0019-2805.2004.01816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hollox EJ, Armour JA, Barber JC. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am J Hum Genet. 2003;73:591–600. doi: 10.1086/378157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fellermann K, Stange DE, Schaeffeler E, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–48. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bentley RW, Pearson J, Gearry RB, et al. Association of higher DEFB4 genomic copy number with Crohn's disease. Am J Gastroenterol. 2010;105:354–9. doi: 10.1038/ajg.2009.582. [DOI] [PubMed] [Google Scholar]
- 36.Hollox EJ, Huffmeier U, Zeeuwen PL, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–5. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 38.Colobran R, Pujol-Borrell R, Armengol MP, Juan M. The chemokine network. I. How the genomic organization of chemokines contains clues for deciphering their functional complexity. Clin Exp Immunol. 2007;148:208–17. doi: 10.1111/j.1365-2249.2007.03344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thornton JW, DeSalle R. Gene family evolution and homology: genomics meets phylogenetics. Annu Rev Genomics Hum Genet. 2000;1:41–73. doi: 10.1146/annurev.genom.1.1.41. [DOI] [PubMed] [Google Scholar]
- 41.Wagner A. Birth and death of duplicated genes in completely sequenced eukaryotes. Trends Genet. 2001;17:237–9. doi: 10.1016/s0168-9525(01)02243-0. [DOI] [PubMed] [Google Scholar]
- 42.Modi WS. CCL3L1 and CCL4L1 chemokine genes are located in a segmental duplication at chromosome 17q12. Genomics. 2004;83:735–8. doi: 10.1016/j.ygeno.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 43.Townson JR, Barcellos LF, Nibbs RJ. Gene copy number regulates the production of the human chemokine CCL3-L1. Eur J Immunol. 2002;32:3016–26. doi: 10.1002/1521-4141(2002010)32:10<3016::AID-IMMU3016>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 44.Nakao M, Nomiyama H, Shimada K. Structures of human genes coding for cytokine LD78 and their expression. Mol Cell Biol. 1990;10:3646–58. doi: 10.1128/mcb.10.7.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irving SG, Zipfel PF, Balke J, et al. Two inflammatory mediator cytokine genes are closely linked and variably amplified on chromosome 17q. Nucleic Acids Res. 1990;18:3261–70. doi: 10.1093/nar/18.11.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirashima M, Ono T, Nakao M, et al. Nucleotide sequence of the third cytokine LD78 gene and mapping of all three LD78 gene loci to human chromosome 17. DNA Seq. 1992;3:203–12. doi: 10.3109/10425179209034019. [DOI] [PubMed] [Google Scholar]
- 47.Menten P, Wuyts A, Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002;13:455–81. doi: 10.1016/s1359-6101(02)00045-x. [DOI] [PubMed] [Google Scholar]
- 48.Colobran R, Adreani P, Ashhab Y, et al. Multiple products derived from two CCL4 loci: high incidence of a new polymorphism in HIV+ patients. J Immunol. 2005;174:5655–64. doi: 10.4049/jimmunol.174.9.5655. [DOI] [PubMed] [Google Scholar]
- 49.Cardone MF, Jiang Z, D'Addabbo P, et al. Hominoid chromosomal rearrangements on 17q map to complex regions of segmental duplication. Genome Biol. 2008;9:R28. doi: 10.1186/gb-2008-9-2-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–37. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 51.Shostakovich-Koretskaya L, Catano G, Chykarenko ZA, et al. Combinatorial content of CCL3L and CCL4L gene copy numbers influence HIV–AIDS susceptibility in Ukrainian children. AIDS. 2009;23:679–88. doi: 10.1097/QAD.0b013e3283270b3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colobran R, Comas D, Faner R, et al. Population structure in copy number variation and SNPs in the CCL4L chemokine gene. Genes Immun. 2008;9:279–88. doi: 10.1038/gene.2008.15. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–40. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 54.Degenhardt JD, de Candia P, Chabot A, et al. Copy number variation of CCL3-like genes affects rate of progression to simian-AIDS in rhesus macaques (Macaca mulatta) PLoS Genet. 2009;5:e1000346. doi: 10.1371/journal.pgen.1000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gornalusse G, Mummidi S, He W, Silvestri G, Bamshad M, Ahuja SK. CCL3L copy number variation and the co-evolution of primate and viral genomes. PLoS Genet. 2009;5:e1000359. doi: 10.1371/journal.pgen.1000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colobran R, Casamitjana N, Roman A, et al. Copy number variation in the CCL4L gene is associated with susceptibility to acute rejection in lung transplantation. Genes Immun. 2009;10:254–9. doi: 10.1038/gene.2008.96. [DOI] [PubMed] [Google Scholar]
- 57.Wolpe SD, Davatelis G, Sherry B, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med. 1988;167:570–81. doi: 10.1084/jem.167.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherry B, Tekamp-Olson P, Gallegos C, et al. Resolution of the two components of macrophage inflammatory protein 1, and cloning and characterization of one of those components, macrophage inflammatory protein 1 beta. J Exp Med. 1988;168:2251–9. doi: 10.1084/jem.168.6.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baixeras E, Roman-Roman S, Jitsukawa S, et al. Cloning and expression of a lymphocyte activation gene (LAG-1) Mol Immunol. 1990;27:1091–102. doi: 10.1016/0161-5890(90)90097-j. [DOI] [PubMed] [Google Scholar]
- 60.Zipfel PF, Balke J, Irving SG, Kelly K, Siebenlist U. Mitogenic activation of human T cells induces two closely related genes which share structural similarities with a new family of secreted factors. J Immunol. 1989;142:1582–90. [PubMed] [Google Scholar]
- 61.Lipes MA, Napolitano M, Jeang KT, Chang NT, Leonard WJ. Identification, cloning, and characterization of an immune activation gene. Proc Natl Acad Sci USA. 1988;85:9704–8. doi: 10.1073/pnas.85.24.9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taub DD, Conlon K, Lloyd AR, Oppenheim JJ, Kelvin DJ. Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 alpha and MIP-1 beta. Science. 1993;260:355–8. doi: 10.1126/science.7682337. [DOI] [PubMed] [Google Scholar]
- 63.Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol. 2001;2:1126–32. doi: 10.1038/ni735. [DOI] [PubMed] [Google Scholar]
- 64.Joosten SA, van Meijgaarden KE, Savage ND, et al. Identification of a human CD8+ regulatory T cell subset that mediates suppression through the chemokine CC chemokine ligand 4. Proc Natl Acad Sci USA. 2007;104:8029–34. doi: 10.1073/pnas.0702257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graham GJ, Wright EG, Hewick R, et al. Identification and characterization of an inhibitor of haemopoietic stem cell proliferation. Nature. 1990;344:442–4. doi: 10.1038/344442a0. [DOI] [PubMed] [Google Scholar]
- 66.Broxmeyer HE, Sherry B, Cooper S, et al. Macrophage inflammatory protein (MIP)-1 beta abrogates the capacity of MIP-1 alpha to suppress myeloid progenitor cell growth. J Immunol. 1991;147:2586–94. [PubMed] [Google Scholar]
- 67.Howard OM, Turpin JA, Goldman R, Modi WS. Functional redundancy of the human CCL4 and CCL4L1 chemokine genes. Biochem Biophys Res Commun. 2004;320:927–31. doi: 10.1016/j.bbrc.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 68.Gorrell MD, Gysbers V, McCaughan GW. CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol. 2001;54:249–64. doi: 10.1046/j.1365-3083.2001.00984.x. [DOI] [PubMed] [Google Scholar]
- 69.Guan E, Wang J, Norcross MA. Amino-terminal processing of MIP-1beta/CCL4 by CD26/dipeptidyl-peptidase IV. J Cell Biochem. 2004;92:53–64. doi: 10.1002/jcb.20041. [DOI] [PubMed] [Google Scholar]
- 70.Guan E, Wang J, Roderiquez G, Norcross MA. Natural truncation of the chemokine MIP-1 beta /CCL4 affects receptor specificity but not anti-HIV-1 activity. J Biol Chem. 2002;277:32348–52. doi: 10.1074/jbc.M203077200. [DOI] [PubMed] [Google Scholar]
- 71.Struyf S, Menten P, Lenaerts JP, et al. Diverging binding capacities of natural LD78beta isoforms of macrophage inflammatory protein-1alpha to the CC chemokine receptors 1, 3 and 5 affect their anti-HIV-1 activity and chemotactic potencies for neutrophils and eosinophils. Eur J Immunol. 2001;31:2170–8. doi: 10.1002/1521-4141(200107)31:7<2170::aid-immu2170>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 72.Menten P, Struyf S, Schutyser E, et al. The LD78beta isoform of MIP-1alpha is the most potent CCR5 agonist and HIV-1-inhibiting chemokine. J Clin Invest. 1999;104:R1–5. doi: 10.1172/JCI7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xin X, Shioda T, Kato A, Liu H, Sakai Y, Nagai Y. Enhanced anti-HIV-1 activity of CC-chemokine LD78beta, a non-allelic variant of MIP-1alpha/LD78alpha. FEBS Lett. 1999;457:219–22. doi: 10.1016/s0014-5793(99)01035-2. [DOI] [PubMed] [Google Scholar]
- 74.Nibbs RJ, Yang J, Landau NR, Mao JH, Graham GJ. LD78beta, a non-allelic variant of human MIP-1alpha (LD78alpha), has enhanced receptor interactions and potent HIV suppressive activity. J Biol Chem. 1999;274:17478–83. doi: 10.1074/jbc.274.25.17478. [DOI] [PubMed] [Google Scholar]
- 75.Aquaro S, Menten P, Struyf S, et al. The LD78beta isoform of MIP-1alpha is the most potent CC-chemokine in inhibiting CCR5-dependent human immunodeficiency virus type 1 replication in human macrophages. J Virol. 2001;75:4402–6. doi: 10.1128/JVI.75.9.4402-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Proost P, Struyf S, Van Damme J. Natural post-translational modifications of chemokines. Biochem Soc Trans. 2006;34:997–1001. doi: 10.1042/BST0340997. [DOI] [PubMed] [Google Scholar]
- 77.Proost P, Menten P, Struyf S, Schutyser E, De Meester I, Van Damme J. Cleavage by CD26/dipeptidyl peptidase IV converts the chemokine LD78beta into a most efficient monocyte attractant and CCR1 agonist. Blood. 2000;96:1674–80. [PubMed] [Google Scholar]
- 78.Modi WS, Bergeron J, Sanford M. The human MIP-1beta chemokine is encoded by two paralogous genes, ACT-2 and LAG-1. Immunogenetics. 2001;53:543–9. doi: 10.1007/s002510100366. [DOI] [PubMed] [Google Scholar]
- 79.Lodi PJ, Garrett DS, Kuszewski J, et al. High-resolution solution structure of the beta chemokine hMIP-1 beta by multidimensional NMR. Science. 1994;263:1762–7. doi: 10.1126/science.8134838. [DOI] [PubMed] [Google Scholar]
- 80.Koopmann W, Ediriwickrema C, Krangel MS. Structure and function of the glycosaminoglycan binding site of chemokine macrophage-inflammatory protein-1 beta. J Immunol. 1999;163:2120–7. [PubMed] [Google Scholar]
- 81.Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S. T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature. 1993;361:79–82. doi: 10.1038/361079a0. [DOI] [PubMed] [Google Scholar]
- 82.Ali S, Palmer AC, Banerjee B, Fritchley SJ, Kirby JA. Examination of the function of RANTES, MIP-1alpha, and MIP-1beta following interaction with heparin-like glycosaminoglycans. J Biol Chem. 2000;275:11721–7. doi: 10.1074/jbc.275.16.11721. [DOI] [PubMed] [Google Scholar]
- 83.Proudfoot AE, Handel TM, Johnson Z, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–90. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lortat-Jacob H. The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr Opin Struct Biol. 2009;19:543–8. doi: 10.1016/j.sbi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 85.Lu J, Honczarenko M, Sloan SR. Independent expression of the two paralogous CCL4 genes in monocytes and B lymphocytes. Immunogenetics. 2004;55:706–11. doi: 10.1007/s00251-003-0636-z. [DOI] [PubMed] [Google Scholar]
- 86.Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–75. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Czaplewski LG, McKeating J, Craven CJ, et al. Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES. Characterization of active disaggregated chemokine variants. J Biol Chem. 1999;274:16077–84. doi: 10.1074/jbc.274.23.16077. [DOI] [PubMed] [Google Scholar]
- 88.Guan E, Wang J, Norcross MA. Identification of human macrophage inflammatory proteins 1alpha and 1beta as a native secreted heterodimer. J Biol Chem. 2001;276:12404–9. doi: 10.1074/jbc.M006327200. [DOI] [PubMed] [Google Scholar]
- 89.Stranger BE, Forrest MS, Dunning M, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–53. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Somerville MJ, Mervis CB, Young EJ, et al. Severe expressive-language delay related to duplication of the Williams–Beuren locus. N Engl J Med. 2005;353:1694–701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee JA, Madrid RE, Sperle K, et al. Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann Neurol. 2006;59:398–403. doi: 10.1002/ana.20732. [DOI] [PubMed] [Google Scholar]
- 92.Urban TJ, Weintrob AC, Fellay J, et al. CCL3L1 and HIV/AIDS susceptibility. Nat Med. 2009;15:1110–12. doi: 10.1038/nm1009-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Melzer D, Perry JR, Hernandez D, et al. A genome-wide association study identifies protein quantitative trait loci (pQTLs) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ahuja SK, Kulkarni H, Catano G, et al. CCL3L1–CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nat Med. 2008;14:413–20. doi: 10.1038/nm1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dolan MJ, Kulkarni H, Camargo JF, et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV–AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol. 2007;8:1324–36. doi: 10.1038/ni1521. [DOI] [PubMed] [Google Scholar]
- 96.Kulkarni H, Agan BK, Marconi VC, et al. CCL3L1–CCR5 genotype improves the assessment of AIDS Risk in HIV-1-infected individuals. PLoS ONE. 2008;3:e3165. doi: 10.1371/journal.pone.0003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kulkarni H, Marconi VC, Agan BK, et al. Role of CCL3L1–CCR5 genotypes in the epidemic spread of HIV-1 and evaluation of vaccine efficacy. PLoS ONE. 2008;3:e3671. doi: 10.1371/journal.pone.0003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huik K, Sadam M, Karki T, et al. CCL3L1 copy number is a strong genetic determinant of HIV seropositivity in Caucasian intravenous drug users. J Infect Dis. 2010;201:730–9. doi: 10.1086/650491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shalekoff S, Meddows-Taylor S, Schramm DB, et al. Host CCL3L1 gene copy number in relation to HIV-1-specific CD4+ and CD8+ T-cell responses and viral load in South African women. J Acquir Immune Defic Syndr. 2008;48:245–54. doi: 10.1097/QAI.0b013e31816fdc77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kuhn L, Schramm DB, Donninger S, et al. African infants' CCL3 gene copies influence perinatal HIV transmission in the absence of maternal nevirapine. AIDS. 2007;21:1753–61. doi: 10.1097/QAD.0b013e3282ba553a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakajima T, Ohtani H, Naruse T, et al. Copy number variations of CCL3L1 and long-term prognosis of HIV-1 infection in asymptomatic HIV-infected Japanese with hemophilia. Immunogenetics. 2007;59:793–8. doi: 10.1007/s00251-007-0252-4. [DOI] [PubMed] [Google Scholar]
- 102.Meddows-Taylor S, Donninger SL, Paximadis M, et al. Reduced ability of newborns to produce CCL3 is associated with increased susceptibility to perinatal human immunodeficiency virus 1 transmission. J Gen Virol. 2006;87:2055–65. doi: 10.1099/vir.0.81709-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bhattacharya T, Stanton J, Kim EY, et al. CCL3L1 and HIV/AIDS susceptibility. Nat Med. 2009;15:1112–15. doi: 10.1038/nm1009-1112. [DOI] [PubMed] [Google Scholar]
- 104.Shrestha S, Tang J, Kaslow RA. Gene copy number: learning to count past two. Nat Med. 2009;15:1127–9. doi: 10.1038/nm1009-1127. [DOI] [PubMed] [Google Scholar]
- 105.Field SF, Howson JM, Maier LM, et al. Experimental aspects of copy number variant assays at CCL3L1. Nat Med. 2009;15:1115–17. doi: 10.1038/nm1009-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shrestha S, Nyaku M, Edberg JC. Variations in CCL3L gene cluster sequence and non-specific gene copy numbers. BMC Res Notes. 2010;3:74. doi: 10.1186/1756-0500-3-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–7. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shao W, Tang J, Song W, et al. CCL3L1 and CCL4L1: variable gene copy number in adolescents with and without human immunodeficiency virus type 1 (HIV-1) infection. Genes Immun. 2007;8:224–31. doi: 10.1038/sj.gene.6364378. [DOI] [PubMed] [Google Scholar]
- 109.McKinney C, Merriman ME, Chapman PT, et al. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2008;67:409–13. doi: 10.1136/ard.2007.075028. [DOI] [PubMed] [Google Scholar]
- 110.Grünhage F, Nattermann J, Gressner OA, et al. Lower copy numbers of the chemokine CCL3L1 gene in patients with chronic hepatitis C. J Hepatol. 2010;52:153–9. doi: 10.1016/j.jhep.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 111.Mamtani M, Rovin B, Brey R, et al. CCL3L1 gene-containing segmental duplications and polymorphisms in CCR5 affect risk of systemic lupus erythaematosus. Ann Rheum Dis. 2008;67:1076–83. doi: 10.1136/ard.2007.078048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mamtani M, Anaya JM, He W, Ahuja SK. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun. 2010;11:155–60. doi: 10.1038/gene.2009.71. [DOI] [PubMed] [Google Scholar]
- 113.Burns JC, Shimizu C, Gonzalez E, et al. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J Infect Dis. 2005;192:344–9. doi: 10.1086/430953. [DOI] [PMC free article] [PubMed] [Google Scholar]