

Abstract
Activation of the alpha7 receptor (α7nAChR) has been shown to be important in inflammation and immune regulation, and is also essential in the neural cholinergic anti-inflammatory pathway. The aim of this study was to investigate the role of α7nAChR in the development of experimental arthritis and immune activation. Mice lacking the α7nAChR were immunized with collagen II and the development of arthritis was assessed. Another group of α7nAChR-deficient mice was immunized with ovalbumin, spleen and lymph node cells were isolated and the proliferative responses to restimulation with ovalbumin or concanavalin A were investigated. We could demonstrate significantly milder arthritis and less cartilage destruction, together with a decrease of T cell content in lymph nodes in mice lacking the α7nAChR compared to wild-type controls. In addition, mice lacking the α7nAChR had a deficient proliferative response to concanavalin A, whereas antigen presentation-dependent proliferation was not affected. These results indicate important roles for α7nAChR in arthritis development as well as in regulation of T cell-dependent immunological mechanisms. In addition, the data implicate α7nAChR as a therapeutic target for modulation of adaptive immune responses.
Keywords: acetylcholine, alpha 7 nicotinic acetylcholine receptor, arthritis, collagen II, proliferation
Introduction
Acetylcholine (Ach) is a classical neurotransmitter involved in both cerebral and autonomous nerve transmission. Moreover, downstream cholinergic action can control the immune system via the vagus nerve (reviewed in [1,2]). This central cholinergic anti-inflammatory pathway is dependent upon the α7 subunit of the nicotinic acetylcholine receptor (α7nAChR), which is known to mediate efficient inhibition of proinflammatory cytokine synthesis from macrophages [3]. In addition, there is accumulating evidence suggesting an additional important role for non-neuronal cholinergic mechanisms in immune regulation (reviewed in [4,5]). T cells, macrophages and other immune cells are capable of producing Ach and both muscarinic and nicotinic cholinergic receptors are expressed on their surfaces, including α7nAChR. Ach regulates the function of immune cells, including B and T cells, via different cholinergic receptors (reviewed in [5,6]). Concerning muscarinic receptors, m2 receptors have been demonstrated to have a role in immunoregulation [7].
Of the known nicotinic cholinergic receptors, α7nAChR is considered to be the most important for immune regulatory actions [3,5]. Mice lacking α7nAChR have been demonstrated to produce an excess of proinflammatory cytokines in response to lipopolysaccharide (LPS) stimulation [3]. Increased production of antigen-specific antibodies has also been reported in α7nAChR-deficient mice [8]. However, the effects of α7 gene depletion may show different patterns concerning innate and adaptive immunity, and recently a decreased severity of autoimmune encephalitis was reported in α7 gene knock-out mice [9]. α7nAChR expression is pronounced in the synovium of rheumatoid arthritis (RA) patients [10–12] and Ach-producing enzymes have been detected in the joint [13]. In vitro, cholinergic stimulation can decrease cytokine production from RA synovial fibroblasts [11]. In addition, vagus and parasympathetic activity is decreased in systemic inflammatory conditions such as rheumatoid arthritis (RA), also being correlated to systemic levels of proinflammatory cytokines [14]. Although Ach can mediate both central and peripheral cholinergic immune-regulating mechanisms, the role of Ach in chronic inflammation and RA has not been studied in detail. Herein we demonstrate the importance of α7nAChR for development of collagen-induced arthritis and immune activation.
Materials and methods
Animals
Mice deficient in the α7nAChR (chrna7tm1Bay, The Jackson Laboratory, Bar Harbor, ME, USA) (knock-out) and wild-type C57BL/6 mice were housed and bred in specific pathogen-free facilities. The mice were maintained under climate-controlled conditions with a 12-h light/dark cycle and fed standard rodent chow and water ad libitum. All experiments were approved by the Stockholm North Ethical Committee, Sweden.
Arthritis
Chicken collagen type II (CII) (Morwell Diagnostics GmbH, Zumikon, Switzerland) (2 mg/ml) was emulsified 1:1 with a mixture of equal volumes incomplete and complete Freund's adjuvant (Difco, Detroit, MI, USA) at 4°C. One hundred µl of the emulsion was immunized subcutaneously at the base of the tail under isoflurane anesthesia (Abbot Scandinavia AB, Solna, Sweden). After 28 days the mice were given a booster immunization with equal volumes of CII (2 mg/ml) and incomplete Freund's adjuvant in an emulsion. Arthritis score was assessed for every animal every day from onset and onwards and the cumulative score of individual animals was compared on a group basis. The intraphalangeal joints of digits, metacarpophalangeal joints and the wrist in the forepaw and ankle joint in the hindpaw were each considered as one category of joint. Each paw was given a score of 0–3, as follows: 0 = unaffected, 1 = one type of joint affected, 2 = two types of joint affected and 3 = the entire paw affected. Evaluation of arthritis was performed by staff members blinded to the identity of the animals. The individual mice were killed 14 days after onset of disease. In a previous experiment the day of maximal arthritis score had been determined as day 14. Blood and paws were collected and the paws were fixed in zamboni solution at 4°C overnight. After fixation the paws were decalcified as described previously [15] and 7-µm cryosections were cut and mounted on glass slides (Super Frost/Plus, Menzel-Glaser, Braunschweig, Germany).
Histological staining
Paw sections were stained with haematoxylin followed by Safranin O (0·1%). Cell infiltration, cartilage destruction evidenced as proteoglycan depletion and bone erosion were each graded on a scale of 0–3, where 0 corresponds to normal joint without cell infiltration, proteoglycan depletion or bone erosion and 3 corresponds to severe inflammation, completely destroyed or destained cartilage or fully eroded bone, including ankylosis [16].
Fluorescence activated cell sorter (FACS) staining of lymph node and spleen cells
Mice were immunized with an emulsion of CII and complete Freund's adjuvant (CFA) as described above. After 10 days the mice were killed and cells from the draining lymph node and spleen were purified. The cells were blocked with Fc block (BD Biosciences, San Jose, CA, USA) for 30 min and stained with the following fluorophore-conjugated antibodies in titres 1/200 Biolegend: phycoerythrin (PE)-conjugated CD4, CD25, immunoglobulin (Ig)D and anti-granulocytes (GR1), antigen presentation cell (APC)-conjugated CD3, IgM and F4/80, peridinin chlorophyll (PerCP)-conjugated CD4, CD8, CD19 and CD11b (all antibodies from BD Biosciences, San Jose, CA, USA). Analyses were performed on a FACSCalibur (BD Biosciences, San Jose, CA, USA) with CellQuest software.
Ovalbumin (OVA) stimulation
Mice (19 wild-type, 17 knock-out) were immunized subcutaneously at the base of the tail with 100 µl of an emulgate of 1:1 ovalbumin (50 µg/ml) (Sigma, St. Louis, MO, USA) and CFA. After 10 days mice were killed, blood collected, spleen and lymph node cells were purified and cultured in complete medium without stimulation or stimulated with ovalbumin (10 or 50 µg/ml) or concanavalin A (ConA) (5 µg/ml) (Sigma). After 72 h thymidine (Perkin Elmer, Boston, MA, USA) was added and the proliferation of lymph node cells measured using a 1450 Microbeta counter (Wallac, Turku, Finland) 16 h after the addition of thymidine.
Statistical analysis and ethical permission
Differences in arthritis score were analysed using repeated-measures analysis of variance (anova) and Bonferroni/Dunn's post-hoc test. Score differences individual days between wild-type and α7nAChR were analysed using the Mann–Whitney U-test. Comparisons of arthritis onset, histological score and analyses of immune cells in lymph nodes and spleen in wild-type and α7nAChR gene-deleted mice were assessed by the Mann–Whitney U-test. Stimulation index was calculated for each sample (OVA-stimulation/no stimulation) and group differences were analysed using the Mann–Whitney U-test.
P ≤ 0·05 was considered to be significant. The study was approved by the local ethics committee.
Results
Depletion of α7nAChR results in less severe arthritis score
We have previously reported expression of α7nAChR expression in synovial tissue of arthritis patients [10]. Herein we wanted to investigate the role of α7nAChR in the development of experimental arthritis. α7nAChR-deficient and wild-type mice were immunized with chick collagen type II and the development of arthritis was evaluated daily. The clinical scores were significantly lower in α7nAChR-deficient mice compared to wild-type mice throughout the course of the disease (Fig. 1a). The onset time for mice deficient in α7nAChR compared to wild-type mice was somewhat delayed (mean days of onset 31 and 24, respectively), but the difference did not reach statistical significance (P = 0·14).
Fig. 1.

Less severe arthritis in mice deficient in alpha7 receptor (α7nAChR). Arthritis score in α7nAChR-deficient (knock-out) and wild-type mice immunized with collagen type II (CII), from day of onset until 14 days after onset. (a) Cumulative score. Values represent mean ± standard error of the mean (s.e.m.); (b) score each day. Values represent mean ± s.e.m., n wild-type = 6, knock-out = 5.
Suppression of synovial inflammation in α7nAChR-deficient mice
Histological staining of arthritic paws revealed milder disease in the α7nAChR-deficient mice compared to wild-type mice with significantly reduced bone erosion and less cartilage destruction, evidenced by decreased proteoglycan content in the cartilage of arthritic joints. In addition, a tendency (P = 0·07) for milder inflammation with fewer infiltrating inflammatory cells in the joints (Fig. 2a and b) was also evident in the α7nAChR-deficient mice.
Fig. 2.

Suppression of synovial inflammation in alpha7 receptor (α7nAChR)-deficient mice. Histological evaluation of α7nAChR-deficient and wild-type mice with collagen type II (CII)-induced arthritis. Tissue from paw was stained with haematoxylin followed by Safranin O (0·1%). (a) Cartilage destruction (CD), cell infiltration (Infilt) and bone erosion (BE) were each graded on a scale of 0–3. The histological score from both hind- and forepaws were investigated and added together, resulting in a maximum score of 12. Values represent mean ± standard error of the mean (s.e.m.); *P < 0·05. (b) Pictures showing a typical joint in mice with CII-induced arthritis (original magnification ×125). The lower pictures are higher magnifications of the upper showing details of synovial proliferation (original magnification ×250). The arrows depict proliferating synovial membrane and pannus formation in wild-type, and less cartilage destruction in α7nAChR-deficient mice synovium. (c) Distribution of immune cells in draining lymph nodes 10 days after CII immunization were analysed with fluorescence activated cell sorter using fluorophore-conjugated surface antibodies. Values represent mean ± s.e.m., n wild-type = 11, knock-out = 9; *P < 0·05.
Fewer T cells in α7nAChR-deficient lymphnodes
Next we investigated whether changes in the cellular distribution of immune cells or functional changes could explain the decreased arthritis in α7nAChR-deficient mice. Cells from draining lymph nodes and spleen were analysed 10 days after CII immunization. The following markers were analysed with FACS: CD3, CD4, CD8, CD25, CD19, IgD, IgM, GR1, F4/80 and CD11b. α7nAChR-deficient mice had fewer CD3+ cells in their draining lymph nodes compared to wild-type mice (Fig. 2c). There were no differences between the wild-type and α7nAChR-deficient mice for the other cells markers investigated, neither in lymph nodes nor spleen cells (data not included).
Depressed proliferative response in α7nAChR-deficient mice
In order to investigate possible effects of α7nAChR depletion on immunity, spleen and lymph node cells from α7nAChR-deficient mice and wild-type mice preimmunized with OVA were isolated and restimulated with OVA at two different concentrations (10 or 50 µg/ml), or the lymphocyte mitogen ConA. We could detect decreased mitogen-induced T cell proliferation in α7nAChR-deficient mice compared to wild-type (Fig. 3a and b), whereas there was no difference in the proliferation after OVA restimulation.
Fig. 3.
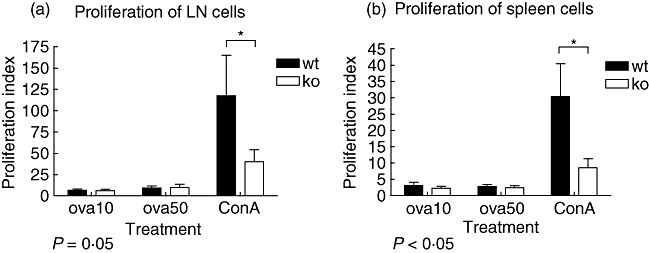
Alpha7 receptor (α7nAChR) affects the proliferation of immune cells. Mice were preimmunized with ovalbumin and after 10 days inguinal lymph nodes were collected. Purified lymph node cells were stimulated with concanavalin A or restimulated with ovalbumin (10 or 50 µg/µl), and 72 h later labelled thymidine was added to the cultures and proliferation was measured after another 16 h. Values are expressed as proliferation index, a ratio between unstimulated proliferation and stimulated (as indicated) proliferation. (a) Proliferation of lymph node cells from α7nAChR-deficient and wild-type mice. Values represent mean ± standard error of the mean (s.e.m.), n wild-type = 19, knock-out = 17. (b) Proliferation of spleen cells from α7nAChR-deficient and wild-type mice. Values represent mean ± s.e.m., n wild-type = 19, knock-out = 17.
Discussion
To our knowledge, this is the first report of the role of α7nAChR for development of chronic peripheral inflammation, with convincing evidence of decreased arthritis development in α7nAChR gene-deleted mice. Moreover, decreased T cell proliferation is a plausible mechanism for the protective effects of α7nAChR depletion on arthritis development.
The arthritis development and histological pattern in the wild-type paws were similar to the destructive arthritis induced in other mouse strains. Fourteen days after onset of arthritis the individual animals were killed, giving comparable data concerning histological joint inflammation. Histological staining of the inflammation in the joint confirmed the decreased arthritis development in α7nAChR-deficient mice, with significantly less cartilage destruction and bone erosion and a tendency for decreased infiltration of inflammatory cells in the synovium.
The decreased arthritis severity indicates that α7nAChR mediates important immune effects, probably through both the actions of neural stimulation [3,17,18] and direct effects on immune cells [11]. To date there has been only one previous study investigating the specific role of α7nAChR in experimental arthritis. Van Maanen and collaborators determined that both nicotine and a specific α7 agonist decreased collagen-induced arthritis score and synovial inflammation [19]. Notably, the down-regulating effects on arthritis were more pronounced with the specific α7nAChR agonist treatment than with the non-specific agonist nicotine, indicating a specific role for α7nAChR in arthritis propagation.
In order to investigate the basic functions of α7nAChR in arthritis development we adopted a different approach, in this context using gene-deleted mice. Furthermore, our purpose was to investigate if the cholinergic neural pathway, mediated specifically by α7nAChR, is of major importance as a constant immune regulating function or comes into action only in the event of excessive inflammatory conditions. Interestingly, instead of continually counteracting the arthritis-provoking mechanisms in the collagen-induced arthritis (CIA) model, the lack of α7nAChR resulted in impaired arthritis development and decreased synovial inflammation.
Extensive earlier investigations have described the α7nAChR receptor as being essential for neural immune regulation in the cholinergic pathway [3], and the importance of these mechanisms in peripheral and joint inflammation has been indicated by several recent findings. Thus spinal inhibition of map-kinases (MAPK; p38) effectively suppresses vagal outflow, resulting in decreased peripheral inflammation [20] and the same spinal p38 inhibition also attenuated experimental arthritis [21]. Recently, vagus suspension, utilized in order to obtain continuous vagal stimulation as an intervention after onset of experimental arthritis, was reported to suppress arthritis scores and synovial inflammation effectively [22]. Whether these mechanisms were active through peripheral immune suppression in lymph nodes and joints or systemic (i.e. splenic) down-regulating effects on immune cells is not known. However, this provides intriguing and complex questions, as no vagal innervations of joints have been affirmed.
In the present study it was not possible to separate basic α7nAChR neural and non-neural effects, which would demand organ-specific knock-out mice. However, based upon our data we may hypothesize that the neuroimmune cholinergic pathway is not exerting constant regulation of immunity. If that were the case, depletion of α7nAChR would probably have resulted in a deficient cholinergic anti-inflammatory control, resulting in a deterioration of the disease. Instead, we could detect a marked suppression of arthritis development which was accompanied by reduction in synovial inflammation and cartilage destruction. This notion is also corroborated by earlier investigations; for example, depletion of cholinergic neuroimmune interaction through vagotomy did not result in clear exacerbation of arthritis, but instead the effects were mild and non-significant [19]. Moreover, in an epidemiological study our own data also revealed that vagotomy was not associated with an increased risk for development of RA [23]. Finally, in concordance with our study, depletion of α7nAChR results in reduced severity of experimental autoimmune encephalomyelitis [9], again indicative of the importance of regulating cholinergic mechanisms during immune reactions.
Although there have been several reports characterizing the cholinergic non-neuronal system in general and the α7nAChR receptor expression specifically, little is known about the physiological function of cholinergic receptors in immune cells [4]. Moreover, the importance of cholinergic mechanisms in antigen presentation and adaptive immunity has not been elucidated. The data from the present study indicate a major role for α7nAChR in immune modulation and chronic inflammation such as arthritis, and it is plausible that the decrease in arthritis propagation of α7nAChR-depleted mice could be due at least partly to more general immune-modifying mechanisms. We thus investigated adaptive immune mechanisms both in the experimental CIA model and using OVA immunization. We could demonstrate a significant reduction on CD3+ T cells in draining lymph nodes from α7nAChR-deficient mice. In addition, restimulation of spleen and lymph node cells with OVA in vitro did not result in altered T cell proliferation, suggesting that antigen presentation is not dependent upon α7nAChR. However, incubation with ConA resulted in significantly decreased T cell proliferation in α7nAChR-deficient mice, both in spleen and lymph node cells. Taken together, these results suggest that α7nAChR affects basal T cell activity and possibly also development of CD3+ T cells.
The decreased proliferative response in α7nAChR-deficient mice is in accordance with earlier reports of α7nAChR influence on the development of some T and B cell subsets [4,6]. Moreover, lymphocyte ACh production in spontaneously hypertensive rats (SHR) has been suggested to be related to T cell dysfunction, such as decreased T cell proliferation in response to ConA [24].
We cannot explain fully the discrepancy between APC-dependent and ConA-induced T cell proliferation, but a plausible explanation could be related to different macrophage influence on T cell function in α7nAChR-deficient versus wild-type mice. Macrophage production of proinflammatory cytokines such as tumour necrosis factor (TNF) [3] can result in inhibition of T cell function [25], and conversely, TNF inhibition has been associated with T cell expansion [26]. A general increase in TNF and other macrophage-derived cytokines has been detected in α7nAChR-deficient mice injected with LPS [3]. This hyperactivity of myeloid cells may thus hamper ConA-induced T cell function in these mice through excessive production of TNF and other proinflammatory cytokines.
Taken together, although specific cholinergic stimulation has proved efficient for anti-inflammatory action on innate immune responses, the effects on adaptive immunity are more complex. We provide evidence that depletion of α7 results in a decrease of experimental arthritis and synovial inflammation, and is associated with a decreased proliferative immune response. These data demonstrate the importance of the α7 receptor in regulation of T cell-dependent immunological mechanisms and provide implications for α7 as a therapeutic target for modulation of adaptive immunity.
Acknowledgments
This study was supported by grants from the Gustav V 80 Year Foundation, The Swedish Rheumatism Association, the Åke Wibergs Foundation, the Magnus Bergvalls Foundation, the Swedish Research Council and the Nanna Svartz Foundation. We thank Dr R. A. Harris for linguistic advice and Marianne Engström for technical assistance.
Disclosure
None.
References
- 1.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–96. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosas-Ballina M, Tracey KJ. Cholinergic control of inflammation. J Intern Med. 2009;265:663–79. doi: 10.1111/j.1365-2796.2009.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 4.Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 2004;9:2063–85. doi: 10.2741/1390. [DOI] [PubMed] [Google Scholar]
- 5.Fujii T, Takada-Takatori Y, Kawashima K. Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J Pharmacol Sci. 2008;106:186–92. doi: 10.1254/jphs.fm0070109. [DOI] [PubMed] [Google Scholar]
- 6.Kawashima K, Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci. 2003;74:675–96. doi: 10.1016/j.lfs.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 7.Jönsson M, Norrgård O, Forsgren S. Presence of a marked nonneuronal cholinergic system in human colon: study of normal colon and colon in ulcerative colitis. Inflamm Bowel Dis. 2007;13:1347–56. doi: 10.1002/ibd.20224. [DOI] [PubMed] [Google Scholar]
- 8.Fujii YX, Fujigaya H, Moriwaki Y, et al. Enhanced serum antigen-specific IgG1 and proinflammatory cytokine production in nicotinic acetylcholine receptor alpha7 subunit gene knockout mice. J Neuroimmunol. 2007;189:69–74. doi: 10.1016/j.jneuroim.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol. 2009;183:6681–8. doi: 10.4049/jimmunol.0902212. [DOI] [PubMed] [Google Scholar]
- 10.Westman M, Engstrom M, Catrina AI, Lampa J. Cell specific synovial expression of nicotinic alpha 7 acetylcholine receptor in rheumatoid arthritis and psoriatic arthritis. Scand J Immunol. 2009;70:136–40. doi: 10.1111/j.1365-3083.2009.02266.x. [DOI] [PubMed] [Google Scholar]
- 11.Waldburger JM, Boyle DL, Pavlov VA, Tracey KJ, Firestein GS. Acetylcholine regulation of synoviocyte cytokine expression by the alpha7 nicotinic receptor. Arthritis Rheum. 2008;58:3439–49. doi: 10.1002/art.23987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maanen MA, Stoof SP, Zanden EP, et al. The alpha7 nicotinic acetylcholine receptor on fibroblast-like synoviocytes and in synovial tissue from rheumatoid arthritis patients: a possible role for a key neurotransmitter in synovial inflammation. Arthritis Rheum. 2009;60:1272–81. doi: 10.1002/art.24470. [DOI] [PubMed] [Google Scholar]
- 13.Grimsholm O, Rantapaa-Dahlqvist S, Dalen T, Forsgren S. Unexpected finding of a marked non-neuronal cholinergic system in human knee joint synovial tissue. Neurosci Lett. 2008;442:128–33. doi: 10.1016/j.neulet.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein RS, Bruchfeld A, Yang L, et al. Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med. 2007;13:210–15. doi: 10.2119/2006-00108.Goldstein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed M, Bjurholm A, Schultzberg M, Theodorsson E, Kreicbergs A. Increased levels of substance P and calcitonin gene-related peptide in rat adjuvant arthritis. A combined immunohistochemical and radioimmunoassay analysis. Arthritis Rheum. 1995;38:699–709. doi: 10.1002/art.1780380519. [DOI] [PubMed] [Google Scholar]
- 16.Ostberg T, Wahamaa H, Palmblad K, et al. Oxaliplatin retains HMGB1 intranuclearly and ameliorates collagen type II-induced arthritis. Arthritis Res Ther. 2008;10:R1. doi: 10.1186/ar2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saeed RW, Varma S, Peng-Nemeroff T, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201:1113–23. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallowitsch-Puerta M, Tracey KJ. Immunologic role of the cholinergic anti-inflammatory pathway and the nicotinic acetylcholine alpha 7 receptor. Ann NY Acad Sci. 2005;1062:209–19. doi: 10.1196/annals.1358.024. [DOI] [PubMed] [Google Scholar]
- 19.van Maanen MA, Lebre MC, van der Poll T, et al. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009;60:114–22. doi: 10.1002/art.24177. [DOI] [PubMed] [Google Scholar]
- 20.Waldburger JM, Boyle DL, Edgar M, et al. Spinal p38 MAP kinase regulates peripheral cholinergic outflow. Arthritis Rheum. 2008;58:2919–21. doi: 10.1002/art.23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyle DL, Jones TL, Hammaker D, et al. Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med. 2006;3:e338. doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang P, Han D, Tang T, Zhang X, Dai K. Inhibition of the development of collagen-induced arthritis in Wistar rats through vagus nerve suspension: a 3-month observation. Inflamm Res. 2008;57:322–8. doi: 10.1007/s00011-008-8070-1. [DOI] [PubMed] [Google Scholar]
- 23.Carlens C, Brandt L, Klareskog L, Lampa J, Askling J. The inflammatory reflex and risk for rheumatoid arthritis: a case–control study of human vagotomy. Ann Rheum Dis. 2007;66:414–16. doi: 10.1136/ard.2006.055285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimoto K, Matsui M, Fujii T, Kawashima K. Decreased acetylcholine content and choline acetyltransferase mRNA expression in circulating mononuclear leukocytes and lymphoid organs of the spontaneously hypertensive rat. Life Sci. 2001;69:1629–38. doi: 10.1016/s0024-3205(01)01237-1. [DOI] [PubMed] [Google Scholar]
- 25.Cope AP, Liblau RS, Yang XD, et al. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med. 1997;185:1573–84. doi: 10.1084/jem.185.9.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Notley CA, Inglis JJ, Alzabin S, McCann FE, McNamee KE, Williams RO. Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J Exp Med. 2008;205:2491–7. doi: 10.1084/jem.20072707. [DOI] [PMC free article] [PubMed] [Google Scholar]