

Abstract
Crohn's disease and ulcerative colitis are inflammatory bowel diseases (IBD) characterized by chronic relapsing mucosal inflammation. Tumour necrosis factor (TNF)-α, a known agonist of the mitogen-activated protein kinase (MAPK) pathway, is a key cytokine in this process. We aimed first to determine whether p38 MAPK is activated in IBD inflamed mucosa, and then studied the effect of four different p38α inhibitory compounds on MAPK phosphorylation and secretion of proinflammatory cytokines by IBD lamina propria mononuclear cells (LPMCs) and organ culture biopsies. In vivo phospho-p38α and p38α expression was evaluated by immunoblotting on intestinal biopsies from inflamed areas of patients affected by Crohn's disease and ulcerative colitis, and from normal mucosa of sex- and age-matched control subjects. Both mucosal biopsies and isolated LPMCs were incubated with four different p38α selective inhibitory drugs. TNF-α, interleukin (IL)-1β and IL-6 were measured in the organ and cell culture supernatants by enzyme-linked immunosorbent assay. We found higher levels of phospho-p38α in the inflamed mucosa of IBD patients in comparison to controls. All the p38α inhibitory drugs inhibited p38α phosphorylation and secretion of TNF-α, IL-1β and IL-6 from IBD LPMCs and biopsies. Activated p38α MAPK is up-regulated in the inflamed mucosa of patients with IBD. Additionally, all the p38α selective inhibitory drugs significantly down-regulated the activation of the MAPK pathway and the secretion of proinflammatory cytokines.
Keywords: Crohn's disease, lamina propria mononuclear cell, p38 inhibitor, TNF-α, ulcerative colitis
Introduction
Crohn's disease (CD) and ulcerative colitis (UC) represent the two main types of inflammatory bowel diseases (IBD). The aetiology of IBD is unknown, although there is now rather convincing evidence that changes in the way the host immune system deals with the normal microbial flora is involved. It is generally agreed, however, that IBD are characterized by chronic relapsing inflammation of the gastrointestinal tract that arises from inappropriate activation of the mucosal immune system. Both CD and UC are characterized by an imbalance between pro- and anti-inflammatory cytokines leading to destruction of normal tissue integrity [1].
Tumour necrosis factor (TNF)-α plays a central role in the initiation and amplification of the mucosal inflammation observed in IBD patients [2], and is also one of the best-characterized agonists of the mitogen-activated protein kinase (MAPK) pathway [3,4]. The latter has a role in controlling inflammation, cell differentiation, cell growth, cell death and malignancy [5,6]. The p38 MAPK is composed of four related Ser/Thr kinase isoforms that share 60–80% of amino-acid sequence, but differ in their tissue-specific expression and sensitivity to chemical inhibitors [6,7]. The most thoroughly studied isoform is p38α. It is expressed ubiquitously and has an important role in regulating gene expression in the gut. This kinase is a point of convergence for multiple signalling processes activated in inflammation, and thus it represents an attractive target for the identification of new therapeutic strategies in IBD [8]. p38α has also been reported to be the most important isoform in inflammatory cells involved in the mucosa of patients with IBD [9]. However, this is somewhat controversial, as other studies failed to find any evidence for p38 activation in IBD [10,11].
p38α inhibitors have been proposed as a new therapeutic approach to control inflammation in IBD. However, controversial results have been found regarding efficacy in reducing the inflammatory response and specificity of inhibition both in animal models of IBD and clinical trials. For example, in mouse colitis, p38 inhibition increases mucosal damage [12]. Most inhibitors have been dropped out in clinical trials for different reasons [10,13].
The aim of the present study was, first, to determine the activation status of p38 in order to establish if this pathway is activated in IBD. The second goal of the project was to study the effect of p38α inhibitory drugs on the p38 phosphorylation and secretion of proinflammatory cytokines in gut samples from patients with IBD.
Materials and methods
Patients and tissues
Twenty-five patients with active CD, 34 patients with active UC and 18 age- and sex-matched control subjects, who showed normal mucosa at histology, were included in the study. In IBD patients, perendoscopic mucosal biopsy specimens were taken from macroscopically inflamed colonic areas. In addition, surgical specimens were collected from inflamed colon of six CD patients and two UC patients. Clinical data from patients with CD and UC are, respectively, shown in Table 1 and Table 2. Colonic samples from four subjects undergoing surgery for colon carcinoma were included as control. Some biopsy samples were homogenized and used for immunoblotting analysis. Some other biopsies were used for lamina propria mononuclear cell (LPMC) isolation. Written informed consent was obtained in all cases and the project was approved by the local Ethics Committee.
Table 1.
Clinical features of patients with Crohn's disease (n = 31).
| Characteristics and parameters | n | Median (range) |
|---|---|---|
| Age (years) | 33·4(17–64) | |
| First attack | 4 | |
| Intestinal location | ||
| Small bowel and colon | 24 | |
| Colon only | 7 | |
| Disease behaviour | ||
| Fistulizing | 4 | |
| Stricturing | 7 | |
| Luminal | 20 | |
| Duration of disease(months) | 69·9(2–218) | |
| Number of recurrences | 2·7(0–6) | |
| Crohn's Disease Activity Index [38] | 207(104–415) | |
| Treatment | ||
| Mesalazine | 13 | |
| Mesalazine + topical steroids | 4 | |
| Mesalazine + antibiotics | 8 | |
| Mesalazine + AZA/6-MP/MTX | 6 |
AZA: azathioprine; 6-MP: 6-mercaptopurine; MTX: methotrexate.
Table 2.
Clinical features of patients with ulcerative colitis (n = 36).
| Characteristics and parameters | n | Median (range) |
|---|---|---|
| Age (years) | 37·1(18–54) | |
| First attack | 4 | |
| Intestinal location | ||
| Pancolitis | 12 | |
| Left-sided colitis | 24 | |
| Duration of disease(months) | 59·1(3–195) | |
| Number of recurrences | 3·2(0–6) | |
| Clinical Activity Index [39] | 7·1(2–12) | |
| Treatment | ||
| Mesalazine | 8 | |
| Mesalazine + topical steroids | 13 | |
| Mesalazine + AZA/6-MP | 5 |
AZA: azathioprine; 6-MP: 6-mercaptopurine.
Cell isolation
LPMCs were isolated as described previously [14]. Briefly, surgical colonic specimens were collected in complete RPMI-1640 medium (Sigma-Aldrich, Poole, UK) supplemented with 10% fetal calf serum and antibiotics. After washing with Hanks' balanced salt solution (HBSS) Ca and Mg free (Sigma-Aldrich), the epithelial layer was removed with 1 mM ethylenediamine tetraacetic acid (EDTA)/HBSS (Sigma-Aldrich) for two 30-min periods of incubation at 37°C with shaking. After EDTA treatment, mucosal samples were denuded of epithelial cells, and were cultured subsequently at 37°C for 1 h with shaking in a humidified CO2 incubator in complete RPMI-1640 medium with 1 mg/ml collagenase type IA from Clostridium histolyticum (Sigma-Aldrich) and 10 U/ml DNase I (Roche Diagnostics, Burgess Hill, UK). The crude cell suspension was allowed to stand for 60 min for sedimentation of debris. Cells from the supernatant were washed twice, resuspended in complete RPMI-1640 medium containing 10% fetal calf serum, and kept on ice until used. Cells were not used if viability did not exceed 90%.
Cell culture
LPMCs (1 × 106/ml) were cultured in duplicate with recombinant human TNF-α (10 ng/ml; R&D Systems, Abingdon, UK), or with different concentrations (0·01–10 µM) of the well-characterized p38 inhibitor SB203580 [15] or three novel, selective p38α MAPK inhibitor compounds (designated 1, 2 and 3; GlaxoSmithKline, Stevenage, UK). The p38α inhibitor profile of these compounds is shown in Table 3. Compound 1 is a member of the pyrimidin class [16], and compounds 2 and 3 are nicotinamides [17]. All compounds act as p38 kinase inhibitors through the competition with adenosine triphosphate (ATP) for binding, and have comparable solubility and cell permeability. As drugs were diluted in dimethylsulphoxide (DMSO), 0·1% DMSO was used as control condition. After 30 min- (for immunoblotting determination of p38α) or 48 h-culture [for enzyme-linked immunosorbent assay (ELISA) determination of cytokines], cells were stored at –70°C.
Table 3.
p38 inhibitor profile of the three novel p38 inhibitor compounds used. All values are pIC50s in non-cellular assay systems.
| p38 inhibitor | p38α | p38β | p38γ/p38δ | Broader kinase selectivity |
|---|---|---|---|---|
| 1 | 7·6 | 7·4 | < 5 | No issues |
| 2 | 7·2 | 6·9 | < 5 | No issues |
| 3 | 7·2 | 6·9 | < 5 | No issues |
| SB203580 | 7·1 | 7·1 | < 5 | Some non-selectivity issues (JNK) |
JNK: Jun N-terminal kinase.
Organ culture
Biopsy specimens were placed on iron grids in the central well of an organ culture dish and the dishes placed in a tight chamber with 95%O2/5%CO2 at 37°C [14]. All the above-mentioned p38 MAPK inhibitors (0·01–10 µM) or recombinant human TNF-α (10 ng/ml; R&D Systems) were added to the medium containing RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% HL-1 (Cambrex Bio Science, Wokingham, UK), 100 U/ml penicillin and 100 µg/ml streptomycin. After 60 min- (for immunoblotting determination of p38) or 24 h-culture (for ELISA determination of cytokine production), biopsies were snap-frozen and stored at –70°C.
Western blotting
Tissue and cell samples were lysed in 250 µl of ice-cold lysate buffer (10 mM EDTA, 50 mM pH 7·4 Tris-HCl, 150 mM sodium chloride, 1% Triton X100, 2 mM phenylmethylsulphonyl fluoride, 2 mM sodium orthovanadate, 10 mg/ml leupeptin and 2 mg/ml aprotinin). The amount of protein was determined by the Bio-Rad Protein assay (Bio-Rad Laboratories, Hemel Hempstead, UK). Protein samples were separated electrophoretically using a discontinuous sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) system at neutral pH (NUPAGE Bis-Tris Electrophoresis System; Invitrogen, Leicestershire, UK). Samples were boiled for 5 min in sample buffer containing 10% β-mercaptoethanol and then run under reduced conditions on 10% pre-cast gels following manufacturer's instructions. Total protein load was 40–65 µg per lane. After electrophoresis, proteins were electrotransferred immediately onto a nitrocellulose membrane (Bio-Rad Laboratories) using a semi-dry blotter (XCell II Blot Module; Invitrogen). Blots were probed with a rabbit anti-human phospho-p38α antibody (dilution 1:1000; R&D Systems) followed by the appropriate horseradish peroxidase-conjugated secondary antibody. The protein bands were visualized by enhanced chemiluminescence (ECL Plus; GE Healthcare, Bucks, UK) according to the manufacturer's instructions. The bands were scanned using an LKB Ultrascan XL Laser Densitometer (Kodak Ltd, Hemel Hempstead, UK) and quantified using the Adobe Photoshop CS software (version 8.0.1). Blots were stripped and analysed for total p38α as an internal loading control, using a mouse anti-human p38α antibody (dilution 1:500; R&D Systems). Normalization between blots was conducted by comparing the intensity of the band of the phospho-p38αversus the band intensity of the total p38α. We used the same exposure times in different blots for the total p38α or phospho-p38α. We performed single measurements for each patient and based the mean and standard deviation (s.d.) on this single measurement in a number of patients. A synthetic blocker phospho-peptide corresponding to residues surrounding Thr180 and Tyr182 (Cell Signaling, Hertfordshire, UK) was used to specifically block the phospho-p38α Western blot reactivity. Alkaline phosphatase (CIP; New England BioLabs, Hertfordshire, UK) was used to release phosphate groups from phosphorylated tyrosine, serine and threonine residues in proteins to show that the antibodies identified only phosphorylated proteins.
ELISA
TNF-α or interleukin (IL)-1β were measured by standard sandwich ELISA: human TNF-α/TNFSF1A DuoSet ELISA kit and human IL-1β (IL-1F2) DuoSet ELISA kit (both from R&D Systems). IL-6 was analysed by the electrochemiluminescence multiplex system Sector 2400 imager from Meso Scale Discovery (Gaithersburg, MD, USA), according to the manufacturer's instructions.
Statistical analysis
Data are displayed as means ± s.d. Statistical significance was determined by the non-parametric Mann–Whitney U-test to compare independent samples using the GraphPad Prism statistical PC program. A P-value of less than 0·05 was considered statistically significant. Experiments with each p38 inhibitor were conducted independently at least three times per patient.
Results
In vivo activation status of p38 MAPK
First we analysed by immunoblotting the activation of the p38 MAPK in the inflamed mucosa of CD and UC patients (Fig. 1). We used specific antibodies for the total p38α isoform (a monoclonal antibody against human recombinant p38α) or for the dual-phosphorylated form of this kinase (a polyclonal antiserum raised against the phospho-peptide). Mucosal biopsies from inflamed areas of UC and CD patients showed significantly (P < 0·0005 and P < 0·005, respectively) higher levels of the phospho-activated p38α isoform with a mean increase of 5·4- and 4·7-fold compared to controls, respectively. No significant difference was found between CD and UC mucosal specimens. Treating the blotted samples with alkaline phosphatase removed the immunoreactivity of the anti-phospho p38α antibody and, similarly, blocking peptide removed immunoreactivity for p38α (data not shown).
Fig. 1.
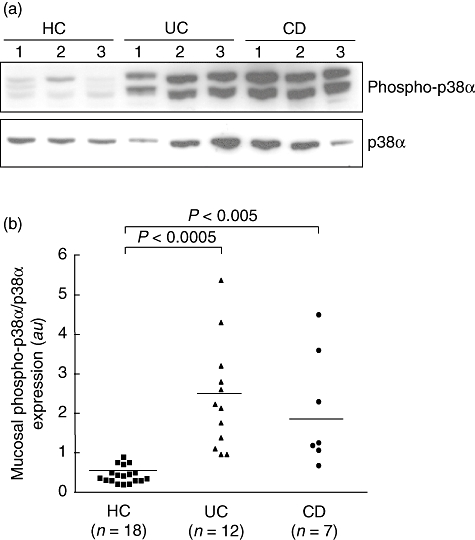
(a) Detection of the phosphorylated form of p38α mitogen-activated protein kinase (MAPK) (phospho-p38α) by immunoblotting in the inflamed mucosa from three patients with ulcerative colitis (UC) and three patients with Crohn's disease (CD), and in the normal mucosa of three healthy controls (HC). Blots were stripped and analysed for p38α. Each example is representative of experiments performed in 12 UC patients, seven CD patients and 18 control subjects. (b) Densitometry of Western blots. Phospho-p38α expression is normalized for p38α. Horizontal bars are mean; a.u.: arbitrary units.
In vitro effect of MAPK inhibitors on p38 MAPK activity
Control LPMCs were cultured with TNF-α and p38α inhibitor compounds (1, 2, 3 and SB203580) for 30 min. As shown in Fig. 2, the TNF-α-induced phosphorylation of p38α was neutralized by compounds 2, 3 and SB203580, while compound 1 showed a lower efficacy. Then, we studied the effect of the p38α inhibitor compounds on unstimulated LPMCs isolated from inflamed areas of patients with IBD (Fig. 3). After 30 min culture, there was a dramatic decrease in the level of p38 phosphorylation, with compound 3 showing the strongest effect. We also demonstrated inhibition of the p38α pathway on IBD mucosal biopsies cultured ex vivo by all the compounds, with the exception of compound 2 that decreased, but not significantly, the level of phosphorylation of p38α (Fig. 4).
Fig. 2.
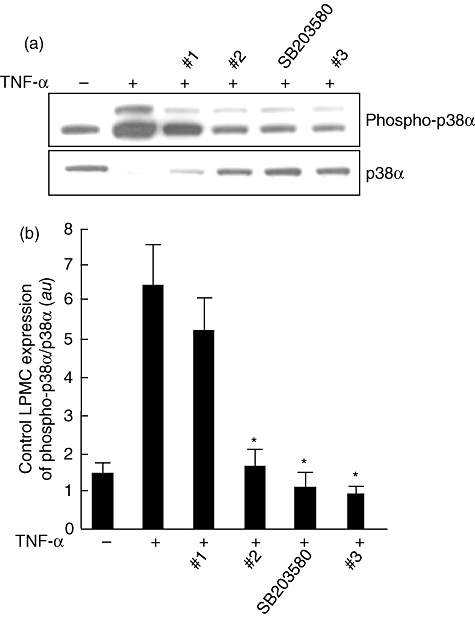
(a) Detection of the phosphorylated form of p38α (phospho-p38α) by immunoblotting in control lamina propria mononuclear cells (LPMCs) cultured with the four p38α inhibitor compounds. LPMCs (1 × 106/ml), isolated from colonic surgical specimens of four control subjects, were incubated for 30 min with or without 10 ng/ml recombinant human tumour necrosis factor (TNF)-α plus p38α inhibitor compounds 1, 2, 3 or SB203580, all at a final concentration of 10 µM. Blots were stripped and analysed for p38α. (b) Densitometry of Western blots. Phospho-p38α expression is normalized for p38α. Results are mean (standard deviation); a.u.: arbitrary units (*P < 0·05 versus cells treated with TNF-α only).
Fig. 3.
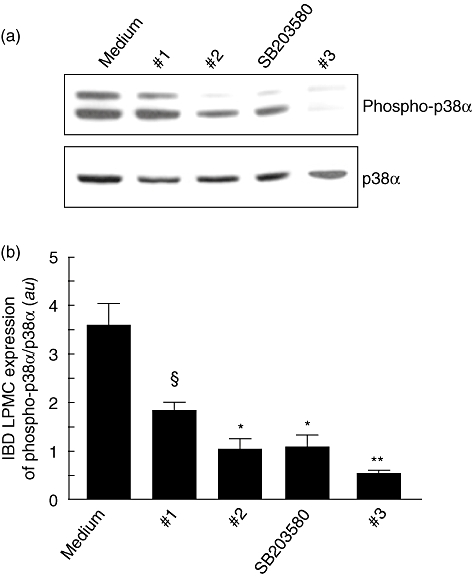
(a) Detection of the phosphorylated form of p38α (phospho-p38α) by immunoblotting in inflammatory bowel disease (IBD) lamina propria mononuclear cells (LPMCs) cultured with the four p38α inhibitor compounds. LPMCs (1 × 106/ml), isolated from inflamed areas of six Crohn's disease patients and two ulcerative colitis patients, were incubated for 30 min with p38α inhibitor compounds 1, 2, 3 or SB203580, all at a final concentration of 10 µM. Blots were stripped and analysed for p38α. (b) Densitometry of Western blots. Phospho-p38α expression is normalized for p38α. Results are mean (standard deviation); a.u.: arbitrary units (§P < 0·05, *P < 0·01 and **P < 0·001 versus cells treated with tumour necrosis factor-α only).
Fig. 4.
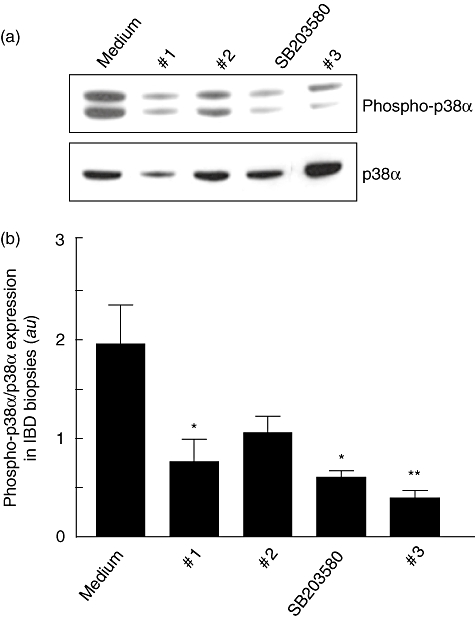
(a) Detection of the phosphorylated form of p38α (phospho-p38α) by immunoblotting in biopsy specimens collected from inflamed areas of 14 inflammatory bowel disease (IBD) (seven with Crohn's disease and seven with ulcerative colitis) and cultured for 60 min with p38α inhibitor compounds, namely 1, 2, 3 or SB203580, all at a final concentration of 10 µM. Blots were stripped and analysed for p38α. (b) Densitometry of Western blots. Phospho-p38α expression is normalized for p38α. Results are mean (standard deviation); a.u.: arbitrary units (*P < 0·05 and **P < 0·005 versus cells treated with tumour necrosis factor-α only).
In vitro and ex vivo effect of p38 inhibitors on proinflammatory cytokine production
As shown in Fig. 5a, the basal production of TNF-α by IBD LPMCs (mean 45·8 ± 10·7 pg/ml) was reduced significantly in a dose-dependent manner by compound 1 (at 0·1 µM: 18·9 ± 5·9 pg/ml, P < 0·05; at 1 µM: 11·3 ± 1·8 pg/ml, P < 0·001; at 10 µM: 5·1 ± 1·1 pg/ml, P < 0·0005), compound 2 (at 0·1 µM: 18·0 ± 5·8 pg/ml, P < 0·05; at 1 µM: 12·2 ± 2·0 pg/ml, P < 0·001; at 10 µM: 5·2 ± 1·9 pg/ml, P < 0·0005), SB203580 (at 0·1 µM: 30·1 ± 8·8 pg/ml; at 1 µM: 24·8 ± 4·9 pg/ml, P < 0·05; at 10 µM: 18·1 ± 2·1 pg/ml, P < 0·001) and compound 3 (at 0·1 µM: 21·2 ± 4·9 pg/ml, P < 0·05; at 1 µM: 13·6 ± 4·7 pg/ml, P < 0·001; at 10 µM: 6·4 ± 1·5 pg/ml, P < 0·0005). Figure 5b shows that the basal production of IL-1β (mean 660·2 ± 114·1 pg/ml) was reduced significantly by 10 µM compound 1 (513·9 ± 83·4 pg/ml, P < 0·05), 10 µM compound 2 (482·4 ± 83·9 pg/ml, P < 0·05), 10 µM SB203580 (549·1 ± 92·0 pg/ml, P < 0·05) and 10 µM compound 3 (523·0 ± 53·4 pg/ml, P < 0·05). As shown in Fig. 5c, the basal production of IL-6 (mean 1648·3 ± 271·0 pg/ml) was reduced significantly by 10 µM compound 1 (513·9 ± 83·4 pg/ml, P < 0·05), 10 µM compound 2 (1165·7 ± 152·9 pg/ml, P < 0·05) and 10 µM compound 3 (1001·4 ± 151·5 pg/ml, P < 0·001), but not 10 µM SB203580 (1405·2 ± 151·9 pg/ml).
Fig. 5.
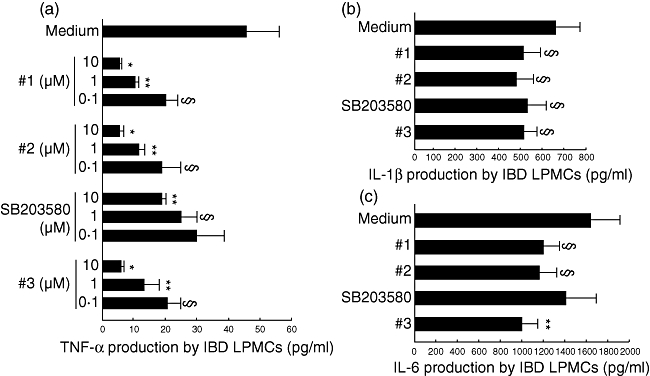
In vitro effect of the four p38 inhibitor compounds on tumour necrosis factor (TNF)-α (a), interleukin (IL)-1β (b) and IL-6 (c) production by inflammatory bowel disease (IBD) lamina propria mononuclear cells (LPMCs). TNF-α concentration (pg/ml) was detected by enzyme-linked immunosorbent assay (ELISA) in the supernatants of LPMCs isolated from inflamed areas of seven Crohn's disease patients and 12 ulcerative colitis patients, and cultured at the concentration of 1 × 106/ml for 48 h in the presence or absence of increasing concentrations (0·1, 1 and 10 µM) of p38α inhibitor compounds 1, 2, 3 or SB203580. IL-1β and IL-6 levels (pg/ml) were measured by ELISA in the supernatants of mucosal biopsies collected from inflamed areas of seven Crohn's disease patients and seven ulcerative colitis patients, and cultured in the presence or absence of p38α inhibitor compounds 1, 2, 3 or SB203580, all at a final concentration of 10 µM. Results are mean (standard deviation) (*P < 0·0005 versus medium only, **P < 0·001 and §P < 0·05 versus medium only).
We also assessed the ex vivo effect of the four compounds in IBD organ culture biopsies (Fig. 6). The basal production of IL-1β (mean 657 ± 244 pg/ml) was reduced significantly by compound 1 (237 ± 89 pg/ml, P < 0·01), compound 2 (221 ± 72 pg/ml, P < 0·01), SB203580 (304 ± 116 pg/ml, P < 0·05) and compound 3 (324 ± 69 pg/ml, P < 0·05). The basal production of IL-6 (mean 4836 ± 1083 pg/ml) was reduced significantly by compound 1 (1232 ± 361 pg/ml, P < 0·005), compound 2 (985 ± 227 pg/ml, P < 0·005), SB203580 (2304 ± 486 pg/ml, P < 0·05) and compound 3 (1198 ± 507 pg/ml, P < 0·005). The basal production of TNF-α (mean 140·7 ± 49·1 pg/ml) was reduced significantly by compound 1 (107·3 ± 8·4 pg/ml, P < 0·05), compound 2 (99·4 ± 11·5 pg/ml, P < 0·05) and compound 3 (96·4 ± 5·6 pg/ml, P < 0·05), but not SB203580 (123·9 ± 15·8 pg/ml).
Fig. 6.
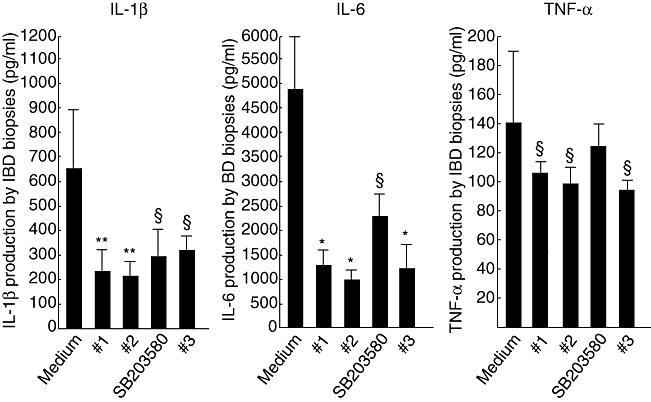
Ex vivo effect of the four p38 inhibitor compounds on interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α production by inflammatory bowel disease (IBD) biopsies. Cytokine levels (pg/ml) were measured by enzyme-linked immunosorbent assay in the supernatants of mucosal biopsies collected from inflamed areas of seven Crohn's disease patients and seven ulcerative colitis patients, and cultured in the presence or absence of p38α inhibitor compounds 1, 2, 3 or SB203580, all at a final concentration of 10 µM. Results are mean (standard deviation) (*P < 0·005, **P < 0·01 and §P < 0·05 versus medium only).
Discussion
In the present study we show that the p38α MAPK pathway is activated in IBD. Additionally, we demonstrate that four different p38α inhibitory compounds are effective in inhibiting the p38 pathway and inhibition of proinflammatory cytokine secretion.
So far, it has been controversial whether chronic gut inflammation involves the activation of p38 MAPK pathway. p38 MAPK and Jun N-terminal kinase (JNK) are both activator and receptor pathway targets of the proinflammatory cytokine TNF-α. Hommes et al. [10], using a semi-quantitative immunohistochemical method, showed that phospho-JNK is up-regulated in CD mucosal samples. We also found that phospho-JNK is up-regulated in IBD specimens (data not shown). In vivo and in vitro modulation using a guanylhydrazone inhibitor, CNI-1493, reduced the number of lamina propria TNF-α-positive cells. However, the analysis of p38 MAPK yielded inconsistent results [18]. Malamut et al. [11] found no increase in p38 and JNK in colonic specimens from patients with IBD. We have difficulty in explaining these somewhat counterintuitive negative results in light of our present findings.
A myriad cytokines are found at high levels in IBD and they have different roles in the initiation, regulation and perpetuation of the intestinal inflammation. Studies in animal models and patients revealed that IL-12 was increased in CD, but not UC, suggesting that CD is a T helper cell type 1 (Th1)-mediated inflammatory disease [18,19]. Conversely, there is considerable evidence that UC involves an atypical Th2-mediated disease. IL-5 and IL-13 are implicated in inflammation in humans and mouse models of UC [20–22]. Further cytokines that are up-regulated in IBD and link innate with adaptive immunity are IL-15, IL-16, IL-17, IL-18, IL-21, IL-27 and IL-32 [23–27]. Additionally, proinflammatory cytokines, such as IL-1, IL-6 and IL-8, are over-produced and help to sustain the ongoing inflammation [28,29]. In light of the critical role of Th1 CD4+ lymphocytes in IBD, the IL-23-dependent Th17 cells are highly pathogenic and elicit inflammation in inflammatory experimental models and IBD [30,31]. Accordingly, we found increased levels of TNF-α, IL-1β and IL-6 in biopsies and LPMCs isolated from IBD patients.
TNF-α is a key cytokine in inflammatory reactions, and the use of anti-TNF-based therapy has proved to be effective in IBD patients [2]. However, targeting the p38α MAPK pathway is an alternative and potentially more favourable target, as it is amenable to orally administered small molecules and is downstream of TNF-α. Thus, the use of p38α inhibitors might inhibit the effects of TNF-α and other p38-activating cytokines in a variety of cell types, such as fibroblasts, to inhibit MMP production; endothelial cells, to inhibit adhesion molecule expression; and epithelial cells, to inhibit TNF-α induced apoptosis and increased permeability. However, despite the evidence supporting the importance of p38 in inflammation, there is no conclusive information about the therapeutic potential of MAPK inhibition in IBD. One of the most common p38α inhibitors used is SB203580, included in our study. It has also been used in many in vivo studies, including dextran sulphate sodium (DSS)- and trinitrobenzene sulphonic acid (TNBS)-colitis [12,32]. Paradoxically, however, in a TNBS-colitis model, SB203580 treatment increases TNF-α secretion and exacerbates gut damage [12]. Badger et al. [33] showed that inflammation could be controlled in mouse colitis by targeting p38α. Hommes et al. [10] used a JNK/p38 inhibitor, CNI-1493, in patients with CD in the only effective therapeutic trial in humans. However, these investigators could not demonstrate either an increase in the level of activated-p38α or a modulation of the p38 pathway with this drug. Waetzig et al. [34] studied the role of p38α in the TNF-α signalling pathway by using the classical p38αβ inhibitor SB203580. In keeping with our data, they first found a marked activation of p38α in IBD. However, inhibition of p38α in cultured biopsies only decreased TNF-α secretion in patients with low or moderate inflammation. Waetzig et al. [35] next investigated the role of p38α in TNF-α signalling in patients responder and non-responder to infliximab. In all patients, a peak of p38α activation was detected 24 h after the infliximab infusion, which is consistent with our previous studies showing that outside–in signalling via infliximab binding to membrane TNF-α activates p38 [14]. Although apoptosis was induced by infliximab in all patients, only responder patients showed increased levels of activated ATF-2, a transcription factor downstream of p38α, inhibitable in vitro by 10 µM SB203580. Surprisingly, apoptosis was not affected by the inhibitor. This may suggest that infliximab induces apoptosis only in responder patients through activation of caspase-3 independent of p38 activation.
BIRB796, a selective p38 inhibitor, has also been studied in human endotoxaemia and IBD [13]. Although a strong inhibition of proinflammatory cytokine production and p38 phosphorylation was achieved, the drug was not useful in IBD because of liver toxicity side effects and no evidence of clinical efficacy [36]. Interestingly, it has been shown recently that p38 is activated in intestinal microvascular endothelial cells and fibroblasts from IBD patients, thus promoting T cell adhesion and transmigration. Blockade of p38 phosphorylation with SB203580 decreased the expression of adhesion molecules and secretion of chemokines [37].
In conclusion, we found high levels of phospho-p38α in both CD and UC mucosal samples. Interfering with the activation of this MAPK pathway might be effective in IBD patients. The challenge is to identify selective non-toxic compounds with in vivo efficacy.
Acknowledgments
We gratefully acknowledge the contribution made by GlaxoSmithKline (Stevenage, UK) that funded this study and provided all the compounds tested.
Disclosure
None of the authors have any conflict of interest.
References
- 1.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–98. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 2.van Deventer SJ. Review article: targeting TNF alpha as a key cytokine in the inflammatory processes of Crohn's disease-the mechanisms of action of infliximab. Aliment Pharmacol Ther. 1999;13:3–8. doi: 10.1046/j.1365-2036.1999.00024.x. [DOI] [PubMed] [Google Scholar]
- 3.Minden A, Lin A, McMahon M, et al. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–23. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 4.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 5.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 6.Cook R, Wu CC, Kang YJ, Han J. The role of the p38 pathway in adaptive immunity. Cell Mol Immunol. 2007;4:253–9. [PubMed] [Google Scholar]
- 7.Wang XS, Diener K, Manthey CL, et al. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:23668–74. doi: 10.1074/jbc.272.38.23668. [DOI] [PubMed] [Google Scholar]
- 8.Ulfgren AK, Andersson U, Engström M, et al. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum. 2000;43:2391–6. doi: 10.1002/1529-0131(200011)43:11<2391::AID-ANR3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 9.Waetzig GH, Seegert D, Rosenstiel P, et al. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–51. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 10.Hommes D, van den Blink B, Plasse T, et al. Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn's disease. Gastroenterology. 2002;122:7–14. doi: 10.1053/gast.2002.30770. [DOI] [PubMed] [Google Scholar]
- 11.Malamut G, Cabane C, Dubuquoy L, et al. No evidence for an involvement of the p38 and JNK mitogen-activated protein in inflammatory bowel diseases. Dig Dis Sci. 2006;51:1443–53. doi: 10.1007/s10620-006-9116-2. [DOI] [PubMed] [Google Scholar]
- 12.ten Hove T, van den Blink B, Pronk I, et al. Dichotomal role of inhibition of p38 MAPK with SB 203580 in experimental colitis. Gut. 2002;50:507–12. doi: 10.1136/gut.50.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Branger J, van den Blink B, Weijer S, et al. Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J Immunol. 2002;168:4070–7. doi: 10.4049/jimmunol.168.8.4070. [DOI] [PubMed] [Google Scholar]
- 14.Di Sabatino A, Pender SL, Jackson CL, et al. Functional modulation of Crohn's disease myofibroblasts by anti-tumor necrosis factor antibodies. Gastroenterology. 2007;133:137–49. doi: 10.1053/j.gastro.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 15.Cuenda A, Rouse J, Doza Y, et al. SB 203580 is a specific inhibitor of a MAP kinase homolog which is stimulated by cellular stresses and interleukin-1. FEBS Letters. 1995;364:229–33. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 16.Boehm JC, Widdowson K, Callahan J, Wan Z. Preparation of 2,4,8-trisubstituted-8H-pyrido[2,3-d]pyrimidin-7-ones as CSBP/p38 kinase inhibitors. PCT Int Appl. 2003;68:WO 2003088972. [Google Scholar]
- 17.Aston N, Bamborough P, Walker A. Preparation of nicotinamides as p38 inhibitors. PCT Int Appl. 2003;75:WO 2003068747. [Google Scholar]
- 18.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–90. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monteleone G, Biancone L, Marasco R, et al. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–78. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 20.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–70. [PubMed] [Google Scholar]
- 21.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–64. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Heller F, Fuss IJ, Nieuwenhuis EE, et al. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–38. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 23.Seegert D, Rosenstiel P, Pfahler H, et al. Increased expression of IL-16 in inflammatory bowel disease. Gut. 2001;48:326–32. doi: 10.1136/gut.48.3.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maerten P, Shen C, Colpaert S, et al. Involvement of interleukin 18 in Crohn's disease: evidence from in vitro analysis of human gut inflammatory cells and from experimental colitis models. Clin Exp Immunol. 2004;135:310–17. doi: 10.1111/j.1365-2249.2004.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishiwaki T, Ina K, Goto H, et al. Possible involvement of the interleukin-15 and interleukin-15 receptor system in a heightened state of lamina propria B cell activation and differentiation in patients with inflammatory bowel disease. J Gastroenterol. 2005;40:128–36. doi: 10.1007/s00535-004-1510-y. [DOI] [PubMed] [Google Scholar]
- 26.Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis. 2006;65:61–4. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seiderer J, Elben I, Diegelmann J, et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn's disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 2008;14:437–45. doi: 10.1002/ibd.20339. [DOI] [PubMed] [Google Scholar]
- 28.Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6 and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–81. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brandt E, Colombel JF, Ectors N, et al. Enhanced production of IL-8 in chronic but not in early ileal lesions of Crohn's disease (CD) Clin Exp Immunol. 2000;122:180–5. doi: 10.1046/j.1365-2249.2000.01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker C, Wirtz S, Blessing M, et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J Clin Invest. 2003;112:693–706. doi: 10.1172/JCI17464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollenbach E, Neumann M, Vieth M, et al. Inhibition of p38 MAP kinase- and RICK/NF-kappaB-signaling suppresses inflammatory bowel disease. FASEB J. 2004;18:1550–2. doi: 10.1096/fj.04-1642fje. [DOI] [PubMed] [Google Scholar]
- 33.Badger AM, Bradbeer JN, Votta B, et al. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 1996;279:1453–61. [PubMed] [Google Scholar]
- 34.Waetzig GH, Seegert D, Rosenstiel P, et al. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–51. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 35.Waetzig GH, Rosenstiel P, Nikolaus S, et al. Differential p38 mitogen-activated protein kinase target phosphorylation in responders and nonresponders to infliximab. Gastroenterology. 2003;125:633–4. doi: 10.1016/s0016-5085(03)00979-x. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber S, Feagan B, D'Haens G, et al. BIRB796 Study Group. Oral p38 mitogen-activated protein kinase inhibition with BIRB 796 for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4:325–34. doi: 10.1016/j.cgh.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 37.Scaldaferri F, Sans M, Vetrano S, et al. The role of MAPK in governing lymphocyte adhesion to and migration across the microvasculature in inflammatory bowel disease. Eur J Immunol. 2009;39:290–300. doi: 10.1002/eji.200838316. [DOI] [PubMed] [Google Scholar]
- 38.Best WR, Becktel JM, Singleton JW, Kern F., Jr Development of a Crohn's disease activity index: National Cooperative Crohn's Disease Study. Gastroenterology. 1976;70:439–44. [PubMed] [Google Scholar]
- 39.Rachmilewitz D. Coated mesalazine (5-aminosalicylic acid) versus sulphasalazine in the treatment of active ulcerative colitis: a randomized study. BMJ. 1989;298:82–6. doi: 10.1136/bmj.298.6666.82. [DOI] [PMC free article] [PubMed] [Google Scholar]