

Abstract
Myocarditis is an inflammation of the myocardium which often follows virus infections. Coxsackievirus B3 (CVB3), as a marker of the enterovirus group, is one of the most important infectious agents of virus-induced myocarditis. Using a CVB3-induced myocarditis model, we show that injection α-galactosylceramide (α-GalCer), a ligand for invariant natural killer (NK) T (iNK T) cells, can protect the mice from viral myocarditis. After the systemic administration of α-GalCer in CVB3 infected mice, viral transcription and titres in mouse heart, sera and spleen were reduced, and the damage to the heart was ameliorated. This is accompanied by a better disease course with an improved weight loss profile. Compared with untreated mice, α-GalCer-treated mice showed high levels of interferon (IFN)-γ and interleukin (IL)-4, and reduced proinflammatory cytokines and chemokines in their cardiac tissue. Anti-viral immune response was up-regulated by α-GalCer. Three days after CVB3 infection, α-GalCer-administered mice had larger spleens. Besides NK T cells, more macrophages and CD8+ T cells were found in these spleens. Upon stimulation with phorbol myristate acetate plus ionomycin, splenocytes from α-GalCer-treated mice produced significantly more cytokines [including IFN-γ, tumour necrosis factor-α, IL-4 and IL-10] than those from untreated mice. These data suggest that administration of α-GalCer during acute CVB3 infection is able to protect the mice from lethal myocarditis by local changes in inflammatory cytokine patterns and enhancement of anti-viral immune response at the early stage. α-GalCer is a potential candidate for viral myocarditis treatment. Our work supports the use of anti-viral treatment early to reduce the incidence of virus-mediated heart damage.
Keywords: α-galactosylceramide, coxsackievirus, myocarditis, NK T cell
Introduction
Cardiovascular diseases are the major cause of human death and have been linked to many different factors. The group B Coxsackievirus type 3 (CVB3), a member of the Picornaviridae family, is considered to be a cause of acute and fulminant viral myocarditis and has been associated with dilated cardiomyopathy [1]. Despite improvements in studying viral pathology, structure and molecular biology, as well as improved disease diagnosis, there is still no virus-specific drug or vaccine in clinical use [1]. There are three distinct stages in virus-induced myocarditis: viraemic injury, immune infiltration and reclamation. Earlier studies have suggested that the mechanisms of viral myocarditis include direct myocyte injury by CVB3 and subsequent immune-mediated damage of the heart [2,3]. However, general immunosuppressive therapy did not benefit patients with myocarditis, raising the need for induction of anti-viral immunity in viral myocarditis therapy.
Natural killer T (NK T) cells are an innate subset of T cells. They were named originally because of co-expression of a T cell receptor (TCR) along with typical surface receptors for NK cells. NK T cells are part of the innate immune system. They effect rapid killing and cytokine responses without the need for extensive cell proliferation or differentiation. NK T cells have a semi-invariant Vα14-Jα18 TCR repertoire specific for lipid antigens that are presented on the major histocompatibility complex (MHC) class I-like CD1d molecule. NK T cells have been found to influence diverse immune responses, including the surveillance for tumours, the maintenance of self-tolerance and the clearance of microorganisms. It was shown that NK T cells participate in the response to various microbial pathogens including bacteria, fungi, parasites and viruses [4–7]. In terms of viral immunity, there is evidence that NK T cells are involved in immune surveillance or clearance of herpes simplex virus 1 and 2, respiratory syncitial virus, hepatitis B and C virus [8–12].
α-Galactosylceramide (α-GalCer) was discovered originally by the Pharmaceutical Division of the Kirin Brewery Company during a screen for reagents derived from the marine sponge Agelas mauritianus that prevent tumour metastasis in mice [13]. It is now also a well-known invariant NK T (iNK T) TCR ligand and it has been possible to target and track specifically iNK T cells. It has been revealed that α-GalCer can be used in various models of autoimmunity, such as experimental autoimmune encephalomyelitis (EAE), type 1 diabetes and experimental autoimmune uveitis (EAU) [14,15], but the role of α-GalCer in myocarditis therapy is not well understood. In the present study, we examine the role of α-GalCer on a CVB3-induced myocarditis model. Our data show that administration of α-GalCer during CVB3 infection is able to treat viral myocarditis by an up-regulated immune response at early stage of infection. Induction of efficient anti-viral immunity reduces the viraemia early in infection, thereby reducing the incidence of virus-mediated heart damage.
Materials and methods
Mice
Male BALB/c mice (6–8 weeks) were purchased from Shanghai Laboratory Animal Center, Chinese Academy of Sciences. All mice were maintained in the Shanghai Jiaotong University School of Medicine animal facilities under specific pathogen-free conditions. Each test group consisted of six or seven mice. All experiments were carried out more than three times. The animal protocol was approved by the review board of Shanghai Jiaotong University School of Medicine.
Virus, virus infection and virus titration
CVB3 (Nancy strain) was a gift from Professor Jingxing Liu (Department of Microbiology, Shanghai Second Medical University, Shanghai, China). Virus stocks were prepared in monolayers of HeLa cells using a multiplicity of infection of 0·1 plaque-forming units (pfu)/cell in Dulbecco's modified Eagle's medium (DMEM). Virus was collected by freezing and thawing and was stored at −80°C. Viral titres were determined on HeLa cell monolayers using a standard plaque assay technique. Mice were infected by intraperitoneal (i.p.) injection of 106 pfu of CBV3 in 0·2 ml of phosphate-buffered saline (PBS). α-GalCer (2 µg; Alexis Biochemicals, San Diego, CA, USA) was administered by i.p. injection at the same time of viral challenge or 3 days after virus infection. The vehicle that suspends α-GC (0·5% Tween-20/PBS) was used as control.
To detect virus titres in sera or organs, organs were harvested aseptically, washed with sterile PBS and homogenized with serum-free cell culture medium (DMEM). Cell debris was removed by centrifugation and supernatants were subjected to 10-fold dilutions in DMEM. The virus titre was determined by tissue culture infectious dose 50 (TCID50) assays.
Histochemical staining
To assess inflammation and myocardial injury during CVB3 replication in the heart, 200 µl Evans Blue dye (10 mg/ml in PBS) was injected intravenously (i.v.) before harvesting hearts at day 7 after CVB3 infection. Hearts were fixed overnight in 4% buffered paraformaldehyde and embedded in paraffin. Tissue sections (5 µm) were stained with haematoxylin and eosin to assess inflammatory infiltrations. Sections were also observed under fluorescence microscopy to visualize intracellular uptake of Evans blue dye, an indicator of membrane permeability that reflects early injury in cardiomyocytes.
Serum biochemistry
Mouse blood samples were collected and centrifuged at 1800 g for 10 min. Serum samples were harvested and then stored at −20°C until analysis was performed. Serum creatine kinase (CK), CK-myocardial band (CK-MB), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) activities were determined on a spectrophotometer (7180 Clinical Analyzer, Hitachi High-Technologies, Japan) using commercial kits (Shanghai Kehua, China).
Flow cytometry
Cells were suspended in fluorescence activated cell sorter (FACS) buffer [PBS-1% bovine serum albumin (BSA)] for cell surface staining. After preincubation with anti-mouse CD16/32 monoclonal antibody (mAb) for 10 min, the cells were stained at 4°C for 20 min with fluorescence-labelled mAb [phycoerythrin (PE) anti-mouse pan NK, peridinin chlorophyll (PerCP) anti-CD3, fluorescein isothiocyanate (FITC) anti-mouse CD8, PE anti-mouse CD4, FITC anti-mouse CD11b or Allophycocyanin (APC) anti-mouse F4/80 and isotype control; eBioscience, San Diego, CA, USA]. Stained cells were washed twice and then analysed on a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) using CellQuest software (BD Biosciences).
ELISA
For cytokine measurements, splenocytes (1·5 × 106 per well) derived from CVB3-infected mice were cultured in triplicate in DMEM with 10% fetal calf serum (FCS). Cells were stimulated with 20 ng/ml phorbol myristate acetate (PMA) and 0·5 µM ionomycin (Sigma, Saint Louis, MO, USA) for 8 h. The supernatants were collected from the cell cultures for the measurement of interferon (IFN)-γ, tumour necrosis factor (TNF)-α, interleukin (IL)-4 and IL-10 by enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions. A standard curve was performed for each plate and used to calculate the absolute concentrations of the indicated cytokines. Cytokine levels are expressed as mean ± standard deviation (s.d.) of triplicate cell cultures.
Real-time polymerase chain reaction (PCR)
Total RNA was isolated from cell pellets using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and first-strand cDNA was synthesized subsequently using Sensiscript RT Kit (Qiagen), according to the manufacturer's instructions. mRNA expression of CVB3, cytokines or chemokines was determined by real-time PCR using SYBR Green master mix (Applied Biosystems, Carlsbad, CA, USA). Nucleotide sequences of specific primers are listed in Table 1. Thermocycle conditions comprised an initial holding at 50°C for 2 min and subsequently at 95°C for 10 min, which was followed by a two-step PCR programme consisting of 95°C for 15 s, and 60°C for 60 s for 40 cycles. Data were collected and analysed quantitatively on an ABI Prism 7900 sequence detection system (Applied Biosystems). Mouse β-actin gene was used as an endogenous control for sample normalization. Results were presented relative to the expression of β-actin.
Table 1.
Specific primers used in real-time polymerase chain reaction analysis.
| Name | Primer | Sequence (5′→ 3′) |
|---|---|---|
| β-actin | FW | TGTCCACCTTCCAGCAGATGT |
| RV | AGCTCAGTAACAGTCCGCCTAGA | |
| CVB3 | FW | ATCAAGTTGCGTGCTGTG |
| RV | TGCGAAATGAAAGGAGTGT | |
| IFN-γ | FW | TCAAGTGGCATAGATGTGGAAGAA |
| RV | TGGCTCTGCAGGATTTTCATG | |
| IL-4 | FW | ACAGGAGAAGGGACGCCAT |
| RV | GAAGCCCTACAGACGAGCTCA | |
| IL-6 | FW | TTCCATCCAGTTGCCTTCTTG |
| RV | TTGGGAGTGGTATCCTCTGTGA | |
| MCP-1 | FW | CGGAATTCGCCACCATGCAGGTCCCTGTCAT |
| RV | GCTCTAGACTAGTTCACTGTCACACT | |
| MIP-2 | FW | CACTTCAGCCTAGCGCCAT |
| RV | AGGTCAGTTAGCCTTGCCT | |
| TNF-α | FW | GACGTGGAACTGGCAGAAGAG |
| RV | GCCACAAGCAGGAATGAGAAG |
CVB3: Coxsackievirus B3; IFN: interferon; IL: interleukin; MCP: monocyte chemoattractant protein; MIP: macrophage inflammatory protein; TNF: tumour necrosis factor. FW: forward; RV: reverse.
Splenic CD3+ cells and antigen-presenting cell (APC) purification and stimulation
CD3+ cells and APCs were selected from mouse splenocytes using CD3 MicroBeads and anti-MHC class II MicroBeads (Miltenyi Biotech, Auburn, CA, USA), respectively. For the α-GalCer stimulation assay, 2 × 105 splenic CD3+ cells were cultured in 96-well round-bottomed plates and stimulated with 20 ng/ml α-GalCer or with α-GalCer-pulsed APCs (2 × 105; the splenic APCs were pulsed with 20 ng/ml of α-GalCer for 3h, irradiated and washed extensively) for 72h. In the blocking experiments, anti-CD1d mAb 1B1 (BD Pharmingen, San Diego, CA, USA) was added at 10 µg/ml, 30 min before the addition of α-GalCer.
Statistics
Student's t-test was used to analyse the differences between the groups. One-way analysis of variance (anova) was performed initially to determine whether an overall statistically significant change existed before using the two-tailed paired or unpaired Student's t-test. A P-value of < 0·05 was considered statistically significant.
Results
Administration of α-GalCer results in less weight loss and reduced mortality in CVB3-infected mice
To test the effect of α-GalCer treatment during CVB3 infection, we challenged BALB/C mice with CVB3 and injected the mice with 2 µg α-GalCer (i.p.) or vehicle medium for α-GalCer (0·5%Tween-20, PBS). α-GalCer was administered simultaneously with i.p. CVB3 inoculation. Mice were monitored for survival and their body weight was determined over the following 2 weeks (Fig. 1). From 3 days post-infection, mice that received CVB3 virus alone began to demonstrate progressive weight loss. Infected mice that were administered α-GalCer showed comparatively lower weight loss, although their body weight did not increase as much as those mice without virus infection (Fig. 1a). Four days post-lethal virus challenge, mice began to die. As shown in Fig. 1b, more than 90% of α-GalCer untreated mice died during the first 2 weeks, but nearly all α-GalCer-treated mice survived without symptoms. These results are consistent with the body weight profile. We also administered the α-GalCer post-inoculation with virus. Three days after CVB3 infection when symptoms were shown in infected mice, α-GalCer were administered daily from day 3 to day 8. Consistently, α-GalCer showed a significant inhibitory effect on the severity of myocarditis compared with a vehicle control (Fig. 1c and d).
Fig. 1.
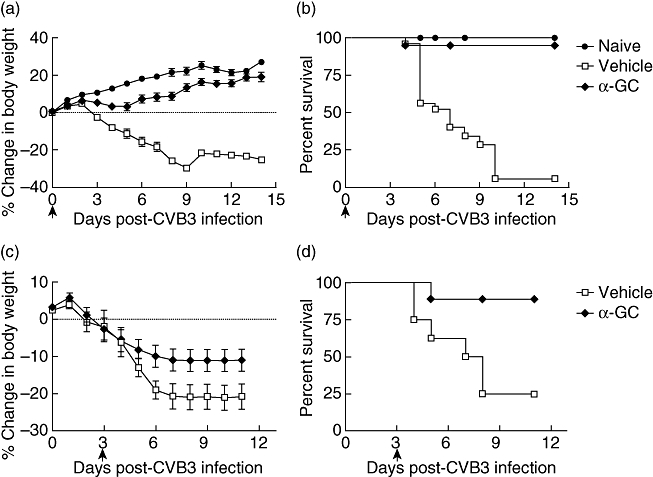
Survival and weight profile of Coxsackievirus B3 (CVB3)-infected mice treated with or without α-galactosylceramide (α-GalCer). BALB/c mice were infected with CVB3 to induce myocarditis as described in Materials and methods. α-GalCer or vehicle control were administered via intraperitoneal injection at the same time of viral challenge (a,b). In the treatment protocol, α-GalCer were administered daily from day 3 to day 8 (c,d). Mouse body weight (a,c) and survival (b,d) were scored and monitored daily. Each group consisted of five to seven mice. Data are representative of three independent experiments.
Tissue injuries and viral loads are reduced in α-GalCer-treated mice
Coxsackievirus B is the most common viral culprit of myocarditis. It induces myocyte damage and can cause myocarditis in the first week of infection. In CVB3-infected mice, severe heart injury was readily detectable by histochemistry. Histological analysis of cardiac tissue sections from CVB3-infected mice treated with α-GalCer exhibited markedly reduced inflammation. There are fewer inflammatory cell infiltrations in α-GalCer-treated mouse cardiac tissue (Fig. 2a and b). To recognize necrotic cells in mouse heart, Evans blue dye was injected as an indicator of cardiomyocyte plasma membrane integrity. More injuries were observed in the hearts of mice that did not receive α-GalCer (Fig. 2c and d). CKMB, CK, LDH and AST are cardiac enzymes. The blood level of these enzymes will increase in the damaged heart. Serum cardiac enzyme data from CVB3-infected mice are shown in Fig. 2e. The blood cardiac enzyme levels, include AST, CK and CKMB, were significantly lower in α-GalCer-treated mice than in non-treated mice, which indicated fewer heart injuries.
Fig. 2.
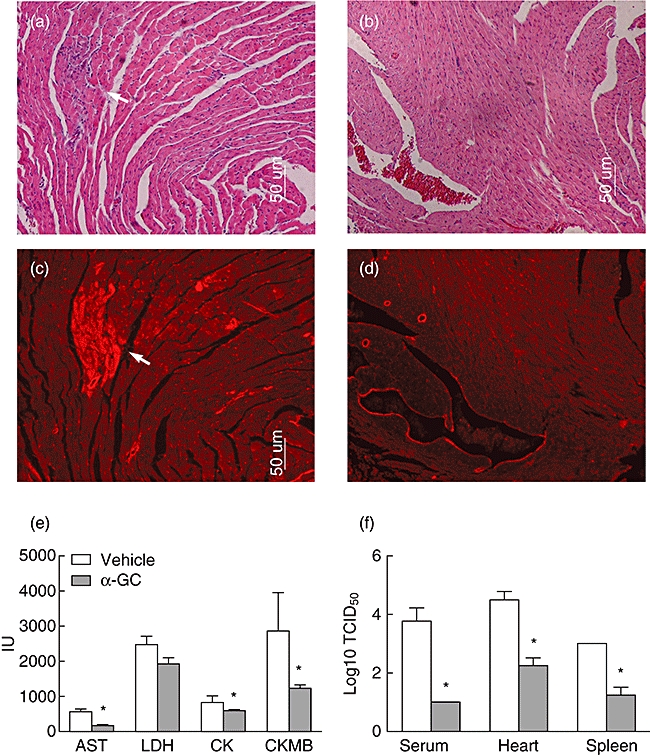
Tissue damage and viral loads of Coxsackievirus B3 (CVB3)-infected mice treated with or without α-galactosylceramide (α-GalCer). BALB/c mice were challenged with CVB3 and treated with α-GalCer (b,d) or vehicle alone (a,c). Samples were taken 7 days after CVB3 challenge. (a,b) Paraffin sections of heart tissue were stained with haematoxylin and eosin (×100). Arrow points to inflammatory cells infiltration. (c,d) Red fluorescence from the same sections and derived Evans blue dye that was taken up by injured cells (arrow) given 4 h before death. (e) Enzyme profiles of CVB3-infected mouse sera. Blood samples were collected at 3 days post-infection. Aspartane aminotransferase (AST), lactate dehydrogenase (LDH), creatine kinase (CK) and creatine kinase muscle band (CKMB) levels were determined as described in Materials and methods. (f) The amount of infectious virus particles were analysed in spleen, heart or serum samples of all animals. Experimental groups consisted of three mice, and experiments were repeated three times. The mean ± standard deviation is shown. Asterisks represent statistical differences between groups. (P < 0·05 as determined by Student's t-test).
To test the viral loads in CVB3-infected mice, 3 days post-infection mice were killed and the amount of infectious virus particles was analysed in serum, heart and spleen by TCID50titrations. As shown clearly in Fig. 2f, due to the α-GalCer injection the viral titres in the tissue and sera were greatly reduced.
α-GalCer changes local inflammatory cytokine patterns and enhances immune response at early stage of CVB3 infection
One of the most prominent characteristics of Coxsackievirus infection of the heart is the marked cellular inflammation after infection. The immune response can be beneficial by limiting viral replication and can also be detrimental if directed against heart antigens or by killing potentially viable cells that are in close proximity to infected cells. We analysed viral RNA and inflammatory cytokine and chemokine expression levels in the heart by real-time PCR. Seven days after CVB3 infection, fewer proinflammatory cytokines (TNF-α, IL-6) and chemokines [major chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-2] were found in α-GalCer-treated mouse tissue, which was consistent with decreased inflammatory cell infiltration in α-GalCer-treated mouse tissue (Fig. 3). However, in the early stage, 3 days after virus infection, more IFN-γ and IL-4 were found in treated mouse heart tissues (Fig. 3).
Fig. 3.
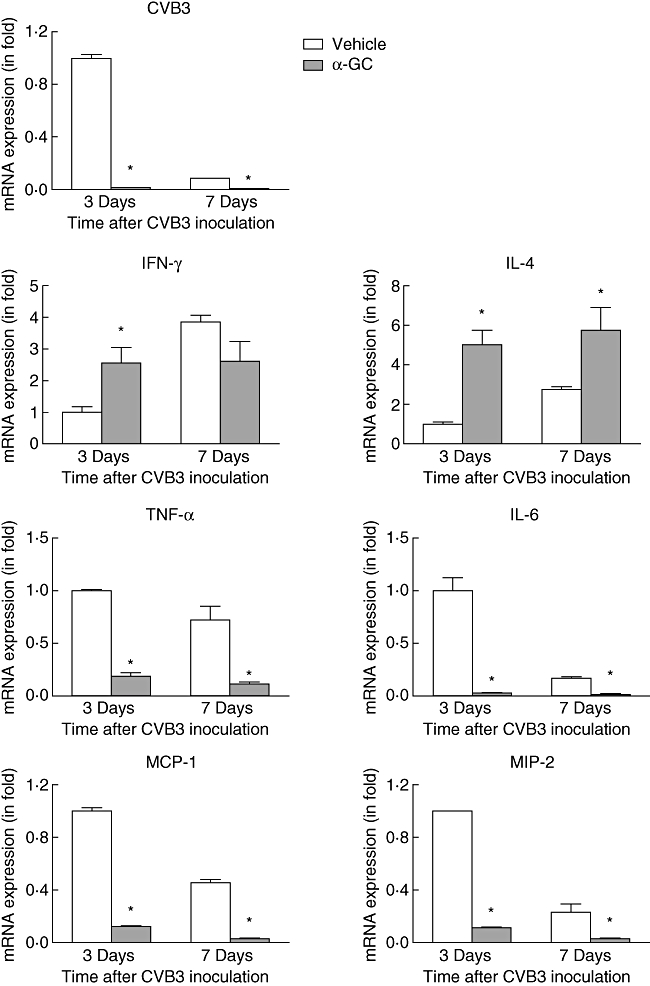
Viral RNA copies and transcriptional expression of cytokines and chemokines in Coxsackievirus B3 (CVB3)-infected mouse heart. RNA was prepared from hearts of CVB3-infected mice treated with or without α-galactosylceramide (α-GalCer) and subjected to real-time polymerase chain reaction. The values from the mice without treatment were set to 1 and the relative mRNA units represent the fold induction over untreated mice. Data are presented as means ± standard error. Asterisks represent statistical differences between groups.
α-GalCer enhances immune response at early stage of CVB3 infection
Besides the immune response in the heart, we also assessed the systematic immune response of α-GalCer-treated and untreated mice. The glycosphingolipid α-GalCer is a potent agonist for iNK T cells. It is the only known antigen presented by CD1d and recognized by iNK T cells. Consistent with this theory, after α-GalCer treatment the NK T cell numbers in CVB3-infected mice were increased in the spleen (Fig. 4a). At the same time, α-GalCer treatment changed mouse systematic immune-response significantly. Three days after CVB3 infection, α-GalCer-treated mice showed larger spleens and more splenocytes than untreated mice (Fig. 4b). The larger spleens were caused by an increased in numbers of both macrophages (CD11b+F4/80+) and CD8+ T cells (Fig. 4c). These splenocytes are activated easily, as upon stimulation with PMA and ionomycin, α-GalCer-treated mouse splenocytes produced significantly more cytokines (IFN-γ, TNF-a, IL-4, IL-10) than those splenocytes from untreated mice. However, at 7 days after infection, no significant differences were found between α-GalCer-treated and untreated mouse splenoctyes (Fig. 5).
Fig. 4.
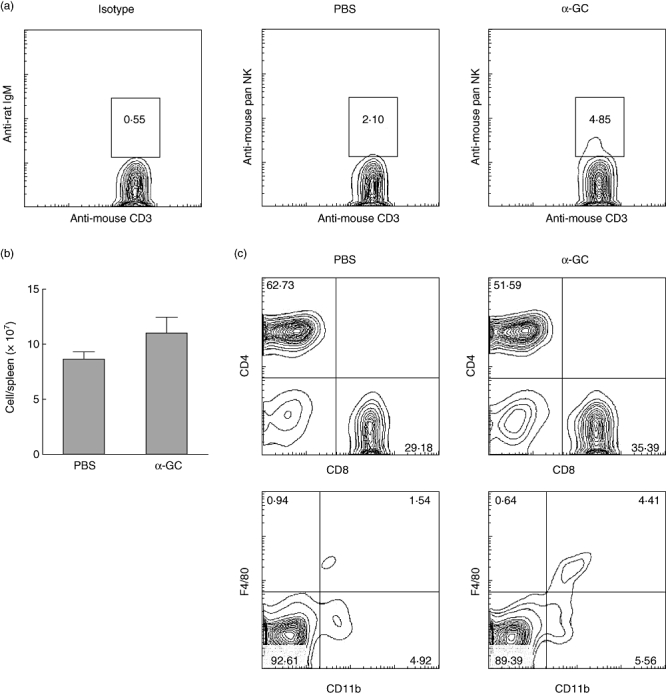
Natural killer (NK) T cells, T cell and macrophage frequency in Coxsackievirus B3 (CVB3)-infected mouse spleen are induced by α-galactosylceramide (α-GalCer). BALB/c mice were challenged with CVB3 and treated with α-GalCer or vehicle alone. Three days later, splenocytes were analysed by flow cytometry. (a) NK T cell frequency in CD3+ cells. (b) Splenocyte count. (c) CD4+CD8+ cell frequency in CD3+ cells, CD11b+ F4/80+ cell frequency in spleen cells.
Fig. 5.
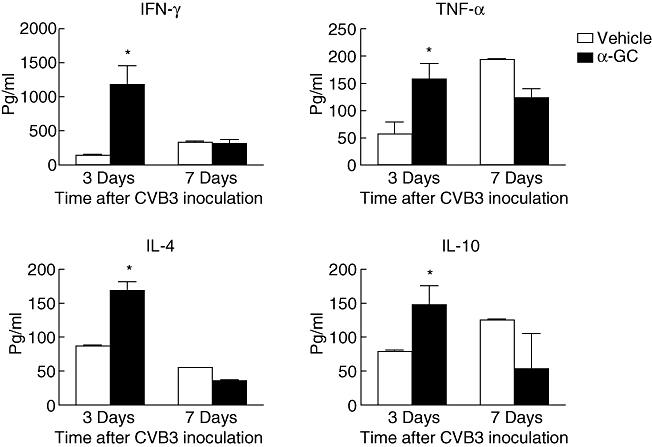
Ex vivo analysis of cytokine production profile in response to phorbol mysristate acetate (PMA) and ionomycin in Coxsackievirus B3 (CVB3)-infected mice treated with α-galactosylceramide (α-GalCer) or vehicle. BALB/c mice were challenged with CVB3 and treated with α-GalCer or vehicle alone. Splenocytes were isolated from mice at days 3 or 7 after infection. Cells were stimulated with 20 ng/ml PMA and 0·5 µM ionomycin for 8 h. Culture supernatants were assessed for the concentration of the indicated cytokines using enzyme-linked immunosorbent assay. Values represent mean concentration (pg/ml ± standard deviation) of triplicates. Results were reproduced in three independent experiments. Asterisks represent statistical differences between groups.
The above results indicate that α-GalCer treatment was associated with an enhanced immune response at early stage of CVB3 infection. Increased levels of cytokines, especially IFN-γ, were found in α-GalCer-treated mice. We further detected whether these increased cytokine levels come from α-GalCer-activated CD1d+CD3+ NK T cells. We purified cells from the splenocytes of naive mice and measured IFN-γ expression in response to α-GalCer in vitro. As shown in Fig. 6a, we found that the expression of IFN-γ is greatly enhanced in response to α-GalCer in CD3+ cells. To determine whether CD1d was involved in the responses to α-GalCer, we conducted antibody-blocking studies. IFN-γ secretion was abrogated by the simultaneous addition of α-GalCer and anti-CD1d mAb to CD3+ cells. Because α-GalCer can promote APC to express IL-12 which can, in turn, stimulate IFN-γ secretion, we further detected cytokine production profiles in response to α-GalCer-pulsed APC in CD3+ cells. Our results indicated that those cells responded to α-GalCer-pulsed APC to produce IFN-γ and IL-4, and these phenomena can also be blocked by anti-CD1d mAb (Fig. 6b).
Fig. 6.
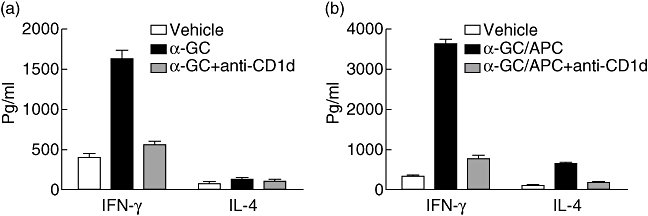
Activation of natural killer (NK) T cells by α-galactosylceramide (α-GalCer) in vitro. CD3+ cells were prepared and cultured either with 20 ng/ml α-GalCer (a) or with α-GalCer-pulsed antigen-presenting cells (APCs) (b). In blocking assay, CD1d monoclonal antibody 1B1 was added at 10 µg/ml, 30 min before the addition of α-GalCer; 72 h after stimulation, supernatants were collected and measured for the concentration interleukin-4 or interferon-γ using enzyme-linked immunosorbent assay. Values represent mean concentration (pg/ml ± standard deviation) of triplicates and are representative of three individual experiments.
Discussion
During the last 20 years, it has been demonstrated that Coxsackieviruses (especially serotype CVB3) are one of the major causes of acute and chronic myocarditis in humans [1]. While the molecular structure of virus itself and the genomic organization of viral RNA are well characterized, the prevention and therapy of CVB3-induced myocarditis are still not satisfactory. Immunosuppressive agents, anti-viral agents and natural medicines have been used in the treatment of patients with myocarditis. However, the efficacy of these agents has not been well established [16]. In this study, we provide compelling evidence that as an activator of iNK T, α-GalCer can protect susceptible mice from lethal CVB3 infection and subsequent myocarditis. The mechanisms of Coxsackievirus B-induced damage to cardiomyocytes are complex. The mechanisms involve mainly immune-mediated destruction of the myocardium, autoimmune-mediated destruction of cardiac cells and direct virus-induced cardiomyocyte injury. There are now several proposed treatment strategies that target specific points in myocarditis. The use of immunosuppressive agents (azathioprine, prednisone and cyclosporine) for the treatment of myocarditis seems logical, as one of the mechanisms thought to contribute to myocarditis is autoimmune destruction. Several groups have studied the efficacy of immunosuppressive therapy in clinical patients [17–19]. Some studies have shown positive results, in that patients receiving immunosuppressive therapy had haemodynamic and clinical improvement [18,19]. However, most studies indicated that there is not enough evidence to recommend immunosuppressive therapy routinely in patients with acute myocarditis; in fact, such treatment may be detrimental [20]. As direct viral cytotoxicity is one possible cause of myocardial damage, inducing anti-viral immunity efficaciously may halt the cascade of myocyte destruction. In this study we use α-GalCer, an iNK T cell activator, to treat viral myocarditis. After the systemic administration of α-GalCer, almost all mice survived from lethal CVB3 infection. Viral titres in mouse heart, sera and spleen were reduced, and the heart damage was ameliorated. This is accompanied by a better disease course with improved weight loss profile. These data indicated that use of anti-viral agents early in viral infection may improve disease outcomes. Instead of immunosuppressive agents, using agents to increase anti-viral immunity should be considered, especially with early treatment.
In this study, we used α-GalCer to improve anti-viral immunity in CVB3-infected mice. As a potent NK T cell agonist, α-GalCer induced significant iNK T cell activation in the mice. INK T cells have been implicated in numerous immune-regulatory and immune-protective roles. They perform diverse functions, from preventing tumour metastases, development of autoimmune diseases such as type I diabetes mellitus and experimental allergic encephalitis [21,22], to conferring protection against viruses, bacteria and parasites [23–25]. Numerous investigators have shown that iNK T cells have anti-viral activity [7,23]. Our study has focused specifically upon the possibility of anti-viral activity in CVB3 infection. Using a mouse model of CVB3 infection, we conclude that the anti-viral effect of α-GalCer induced iNK T activation in vivo. Wilson and Crowe have reported that iNK T proliferate rapidly and expand clonally 10–15-fold in the spleen and two- to threefold in the liver at approximately day 3 after α-GalCer injection [26–28]. In this study, we detected an iNK T cell population in the mouse spleens treated with or without α-GalCer. Three days after CVB3 infection, α-GalCer induced two- to threefold iNK T cells in the spleen.
α-GalCer-activated iNK T cells produce multiple cytokines over a period of several days [26,27,29]. Parekh and colleagues reported that the initial response has been shown to be dominated by IL-4 production and then to shift towards dominant IFN-γ production at ∼ 24 h, with little cytokine secretion after day 3 [30]. Consistent with their results, in our experiment, 3 days after CVB3 infection, more IFN-γ and IL-4, less TNF-α and IL-6 were detected in the virus target tissue of α-GalCer-treated mice. When we checked the systemic cytokine expression, all cytokines including IFN-γ, TNF-α, IL-4 and IL-10 were up-regulated after α-GalCer treatment. Because spleens were enlarged and more macrophages and CD8 T cells were also seen in α-GalCer-treated mouse spleens, it was concluded that administration of α-GalCer can up-regulate the immune response in the early course of viral infection.
Cytokines are normally involved in the control of CVB3 replication and pathogenesis. Among them, IFN-γ is pluripotent in its ability to against CVB3 [1]. In our study, α-GalCer induced IFN-γ production in CVB3 infected mouse heart and spleen. α-GalCer can enhance IFN-γ secretion directly and indirectly in NK T cells. The mechanism by which IFN-γ abates CVB3 infection is most probably complex. Besides the direct interference of virus replication, IFN-γ also induces anti-viral cellular immunity. In our study, more macrophages in α-GalCer-treated mouse spleen may associate with IFN-γ produced by iNK Ts cell at the early stage. Indeed, IFN-γ is an important activator of macrophages [31]. Activated macrophages have multiple mechanisms with which to defend against virus infection. For example, nitric oxide production is enhanced in IFN-γ-activated macrophages. Nitric oxide has been linked to the anti-viral mechanism, as inhibition of inducible nitric oxide resulted in increased viral replication and mortality. Also, nitric oxide can act to specifically inactivate the P3C protease of CVB3, an enzyme necessary for CVB3 replication [32]. Moreover, Yaun et al. have reported recently on IFN-γ stimulation; cardiomyocytes can express CXCL10, which inhibits viral replication through recruitment of NK cells in CVB3-induced myocarditis [33].
Administration of α-GalCer during acute CVB3 infection is able to protect the mice from myocarditis. The mechanisms involve iNK T activation, changes in inflammatory cytokine patterns and enhancement of the immune response. In CVB3-induced myocarditis, the immune response can be beneficial by limiting viral replication and can also be detrimental if autoimmunity is induced. α-GalCer increased anti-virus immunity, perhaps also raising the risk of autoimmunity in myocarditis. In this study, α-GalCer was administered in CVB3 infection. At the early stage (3 days after infection), an up-regulated immune response was observed in α-GalCer-treated mice. Seven days after infection, α-GalCer-treated mice did not show a significantly increased immune response.This is due probably to increased activation-induced cell death (AICD) of iNK T cells shortly after α-GalCer administration to mice. After 3 days of marked population expansion, most iNK T cells died [34,35]. It is perhaps not surprising that after 7 days administration of α-GalCer mice did not show a strong immune response. In conclusion, our work has demonstrated that administration of α-GalCer during acute CVB3 infection is able to protect the mice from lethal myocarditis because, until now, there has been no virus-specific preventive or therapeutic procedure in clinical use. α-GalCer is a potential candidate for viral myocarditis treatment. Our work also supports the use of anti-viral treatment early in infection to reduce the incidence of virus-mediated heart damage.
Acknowledgments
The work was supported by grants from the National Natural Science Foundation of China (NSF-30600517, NSF-30972743), the Shanghai Commission of Science and Technology (08QA14043), Specialized Research Fund for the Doctoral Program of Higher Education (200802481081). Derek Johnson (Department of Pathology and Laboratory Science, University of Pennsylvania School of Medicine) read the paper.
Disclosure
None.
References
- 1.Henke A, Jarasch N, Wutzler P. Coxsackievirus B3 vaccines: use as an expression vector for prevention of myocarditis. Exp Rev Vaccines. 2008;7:1557–67. doi: 10.1586/14760584.7.10.1557. [DOI] [PubMed] [Google Scholar]
- 2.Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091–100. doi: 10.1161/01.cir.99.8.1091. [DOI] [PubMed] [Google Scholar]
- 3.Whitton JL, Feuer R. Myocarditis, microbes and autoimmunity. Autoimmunity. 2004;37:375–86. doi: 10.1080/08916930410001713089. [DOI] [PubMed] [Google Scholar]
- 4.Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 2008;4:e1000239. doi: 10.1371/journal.ppat.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinjo T, Fujita J, Kawakami K. Cooperative regulation of the host defense to cryptococcal infection by innate immune lymphocytes. Nippon Ishinkin Gakkai Zasshi. 2006;47:201–7. doi: 10.3314/jjmm.47.201. [DOI] [PubMed] [Google Scholar]
- 6.Stanley AC, Zhou Y, Amante FH, et al. Activation of invariant NKT cells exacerbates experimental visceral leishmaniasis. PLoS Pathog. 2008;4:e1000028. doi: 10.1371/journal.ppat.1000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galli G, Pittoni P, Tonti E, et al. Invariant NKT cells sustain specific B cell responses and memory. Proc Natl Acad Sci USA. 2007;104:3984–9. doi: 10.1073/pnas.0700191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujita K, Sandford AP, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Role of natural killer T (NKT) cells lacking interleukin (IL)-4 producing abilities on the CC-chemokine ligand 2-associated herpes simplex virus type 1 infection in human severe combined immunodeficiency (SCID) mouse chimeras. Burns. 2005;31:145–52. doi: 10.1016/j.burns.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Long BR, Erickson AE, Chapman JM, et al. Increased number and function of natural killer cells in human immunodeficiency virus 1-positive subjects co-infected with herpes simplex virus 2. Immunology. 2009;129:186–96. doi: 10.1111/j.1365-2567.2009.03170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larranaga CL, Ampuero SL, Luchsinger VF, et al. Impaired immune response in severe human lower tract respiratory infection by respiratory syncytial virus. Pediatr Infect Dis J. 2009;28:867–73. doi: 10.1097/INF.0b013e3181a3ea71. [DOI] [PubMed] [Google Scholar]
- 11.Ito H, Ando K, Ishikawa T, et al. Role of Valpha14 + NKT cells in the development of hepatitis B virus-specific CTL: activation of Valpha14 + NKT cells promotes the breakage of CTL tolerance. Int Immunol. 2008;20:869–79. doi: 10.1093/intimm/dxn046. [DOI] [PubMed] [Google Scholar]
- 12.Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut. 2006;55:869–77. doi: 10.1136/gut.2005.076463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi E, Motoki K, Natori T, Uchida T, Fukushima H, Koezuka Y. Enhancing effects of agelasphin-11 on natural killer cell activities of normal and tumor-bearing mice. Biol Pharm Bull. 1996;19:350–3. doi: 10.1248/bpb.19.350. [DOI] [PubMed] [Google Scholar]
- 14.Miyake S, Yamamura T. Therapeutic potential of glycolipid ligands for natural killer (NK) T cells in the suppression of autoimmune diseases. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:315–22. doi: 10.2174/1568008054863772. [DOI] [PubMed] [Google Scholar]
- 15.Grajewski RS, Hansen AM, Agarwal RK, et al. Activation of invariant NKT cells ameliorates experimental ocular autoimmunity by a mechanism involving innate IFN-gamma production and dampening of the adaptive Th1 and Th17 responses. J Immunol. 2008;181:4791–7. doi: 10.4049/jimmunol.181.7.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunetti L, DeSantis ER. Treatment of viral myocarditis caused by coxsackievirus B. Am J Health Syst Pharm. 2008;65:132–7. doi: 10.2146/ajhp060586. [DOI] [PubMed] [Google Scholar]
- 17.Mason JW, O'Connell JB, Herskowitz A, et al. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med. 1995;333:269–75. doi: 10.1056/NEJM199508033330501. [DOI] [PubMed] [Google Scholar]
- 18.Camargo PR, Snitcowsky R, da Luz PL, et al. Favorable effects of immunosuppressive therapy in children with dilated cardiomyopathy and active myocarditis. Pediatr Cardiol. 1995;16:61–8. doi: 10.1007/BF00796819. [DOI] [PubMed] [Google Scholar]
- 19.Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation. 2001;104:39–45. doi: 10.1161/01.cir.104.1.39. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Liu J, Yang M. Corticosteroids for viral myocarditis. Cochrane Database Syst Rev. 2006;4 doi: 10.1002/14651858.CD004471.pub2. CD004471. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Ji F, Zhang Y, et al. Cooperation of invariant NKT cells and CD4 + CD25 + T regulatory cells in prevention of autoimmune diabetes in non-obese diabetic mice treated with alpha-galactosylceramide. Acta Biochim Biophys Sin. 2008;40:381–90. doi: 10.1111/j.1745-7270.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 22.Ouyang Q, Chen K, Wang X, et al. [The study of changes on NKT cells of experimental autoimmune encephalomyelitis (EAE) mice] Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2009;25:894–6. [PubMed] [Google Scholar]
- 23.Ho LP, Denney L, Luhn K, Teoh D, Clelland C, McMichael AJ. Activation of invariant NKT cells enhances the innate immune response and improves the disease course in influenza A virus infection. Eur J Immunol. 2008;38:1913–22. doi: 10.1002/eji.200738017. [DOI] [PubMed] [Google Scholar]
- 24.Ranson T, Bregenholt S, Lehuen A, et al. Invariant V alpha 14 + NKT cells participate in the early response to enteric Listeria monocytogenes infection. J Immunol. 2005;175:1137–44. doi: 10.4049/jimmunol.175.2.1137. [DOI] [PubMed] [Google Scholar]
- 25.Ronet C, Darche S, Leite de Moraes M, et al. NKT cells are critical for the initiation of an inflammatory bowel response against Toxoplasma gondii. J Immunol. 2005;175:899–908. doi: 10.4049/jimmunol.175.2.899. [DOI] [PubMed] [Google Scholar]
- 26.Wilson MT, Johansson C, Olivares-Villagomez D, et al. The response of natural killer T cells to glycolipid antigens is characterized by surface receptor down-modulation and expansion. Proc Natl Acad Sci USA. 2003;100:10913–18. doi: 10.1073/pnas.1833166100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crowe NY, Uldrich AP, Kyparissoudis K, et al. Glycolipid antigen drives rapid expansion and sustained cytokine production by NK T cells. J Immunol. 2003;171:4020–7. doi: 10.4049/jimmunol.171.8.4020. [DOI] [PubMed] [Google Scholar]
- 28.Parekh VV, Lalani S, Van Kaer L. The in vivo response of invariant natural killer T cells to glycolipid antigens. Int Rev Immunol. 2007;26:31–48. doi: 10.1080/08830180601070179. [DOI] [PubMed] [Google Scholar]
- 29.Parekh VV, Wilson MT, Olivares-Villagomez D, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115:2572–83. doi: 10.1172/JCI24762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parekh VV, Singh AK, Wilson MT, et al. Quantitative and qualitative differences in the in vivo response of NKT cells to distinct alpha- and beta-anomeric glycolipids. J Immunol. 2004;173:3693–706. doi: 10.4049/jimmunol.173.6.3693. [DOI] [PubMed] [Google Scholar]
- 31.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saura M, Zaragoza C, McMillan A, et al. An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity. 1999;10:21–8. doi: 10.1016/S1074-7613(00)80003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan J, Liu Z, Lim T, et al. CXCL10 inhibits viral replication through recruitment of natural killer cells in coxsackievirus B3-induced myocarditis. Circ Res. 2009;104:628–38. doi: 10.1161/CIRCRESAHA.108.192179. [DOI] [PubMed] [Google Scholar]
- 34.Eberl G, MacDonald HR. Rapid death and regeneration of NKT cells in anti-CD3epsilon- or IL-12-treated mice: a major role for bone marrow in NKT cell homeostasis. Immunity. 1998;9:345–53. doi: 10.1016/s1074-7613(00)80617-2. [DOI] [PubMed] [Google Scholar]
- 35.Eberl G, MacDonald HR. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol. 2000;30:985–92. doi: 10.1002/(SICI)1521-4141(200004)30:4<985::AID-IMMU985>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]