

Abstract
Matrix metalloproteinase-9 (MMP-9) is a well known regulator and effecter of many cellular processes including wound healing. In the cornea, either too much or too little MMP-9 can be detrimental to overall wound repair. We investigated the secreted factors as well as the intracellular signaling pathways and the promoter sequences that mediate this regulation. Primary culture rabbit corneal epithelial cells were treated with various cytokines alone or in different combinations and MMP-9 induction was assessed by gel zymography. Pharmacological inhibitors were used to determine the intracellular signaling pathways induced by the cytokines tested and deletion promoter constructs were created to determine the regions of the MMP-9 promoter involved in the cytokine regulation, thereby assessing the exact transcription factors binding the MMP-9 promoter. We found that two cytokine families, TGF-β and IL-1, act additively in an isoform non-specific manner to induce MMP-9 in this cell type. Our data suggest TGF-β mediated MMP-9 induction may be regulated by the NF-kB, Smad3, and JNK pathways, whereas the IL-1β mediated induction may be regulated by the NF-kB and p38 pathways. Inhibition of the p38, NF-kB, or JNK pathways significantly reduced, but did not abrogate, basal MMP-9 levels. Inhibition of the ERK pathway did not have an effect on MMP-9 mediated expression in either the treated or untreated co-transfected cells.
Keywords: Corneal epithelial cells, MMP-9, signal transduction, TGF-β, IL-1
Introduction
Matrix metalloproteinases (MMPs) are a pleiotropic family of enzymes capable of regulating and affecting various systemic processes such as development, wound healing, and multiple disease states[1, 2]. One of the first MMPs discovered and best characterized member of the family is MMP-9, also known as 92 kDa type IV collagenase or gelatinase B[3]. This enzyme has been shown to affect several aspects of epithelial dynamics[4]. While MMP-9 is not normally present in the unwounded surface of the corneal epithelium, it appears at the migrating front of epithelium resurfacing a wound[4]. Following epithelial closure, MMP-9 expression spreads distally throughout the wound site correlating with remodeling in the basement membrane zone beneath the epithelium[4].
In MMP-9 KO mice, the rate of wound re-epithelialization is faster in both skin and cornea and is correlated with an increase in the proliferation rate of the epithelial cells and altered cytokine profiles[4]. The lack of MMP-9 also results in a reduced ability to remove the provisional fibrin matrix deposited in the wound bed resulting in opacity and diminished visual acuity. Therefore, while epithelial resurfacing in MMP-9 KO mice occurs faster than in wild-type control littermates, the quality of repair is reduced. Over-expression of MMP-9 is also detrimental. In models of chronic wound healing, failure to re-epithelialize is associated with increased expression of MMP-9 and is correlated with impaired restoration of the underlying basement membrane[5, 6]. Similarly, over-expression of MMP-9 also occurs in other cutaneous and ocular surface disorders including epidermolysis bullosa, cicatricial pemphigoid, and corneal epithelial erosions[7-10]. In a model of ocular surface dry eye disease, over-expression of MMP-9 has been linked to disruption of corneal epithelial barrier integrity; this symptom is alleviated in MMP-9 KO mice[11]. In summary, abnormal expression, high or low, of MMP-9 expressed by the surface epithelia can be detrimental to maintenance and repair of the tissue. Thus defining the external cues and the internal signaling pathways that control MMP-9 expression is very important as they may serve as targets for therapeutic interventions.
The production of MMP-9, like most other members of the MMP family, is regulated primarily at the transcriptional level. Most studies focus on the 500-600 base pairs directly upstream of the transcription start site, as this region of the MMP-9 gene is highly conserved among humans, mice, and rabbits[12]. Also, studies in transgenic mice have shown that this DNA fragment accurately promotes transcription of a reporter gene during embryonic development, angiogenesis, epithelial response to ultraviolet radiation, and wound healing[13].
There are many external cues which can induce various transcription factors (TFs) to act on the MMP-9 promoter, though evidence linking the extracellular cues to the intracellular pathways is scarce. In cell culture models, MMP-9 production has been shown to be regulated by external factors such as osmolarity shifts[14], high glucose[15], plating density[16], phorbol esters[17], and several growth factors and cytokines including interleukin 1β (IL-1β)[18], tumor necrosis factor-alpha (TNF-α)[18], transforming growth factor β (TGF-β)[18], basic fibroblast growth factor (FGF-2)[19], and platelet activating factor (PAF)[20]. Additionally, hyaluronan and several sulfated GAGs have been shown to stimulate MMP-9 in both dermal and corneal explants[21].
In order to control gene transcription, external signals must be transduced via intracellular signaling cascades. There exists a host of intracellular signaling cascades which can be activated depending on the stimulus and many of them can cross-talk directly or indirectly with one another to activate TFs and alter gene transcription. The large number of factors capable of inducing MMP-9 expression emphasizes all of the diverse processes in which MMP-9 is involved. The host of TFs involved must be in place in order to respond to any stimulus in a precise way so as not to allow over or under expression of this gene, as either of which has been shown to result in pathology.
Previous work has indicated that in corneal epithelial cells, MMP-9 expression was not regulated by cytokine treatment[16]. However, more recently it has been shown that MMP-9 can be regulated by cytokines in these cells under certain conditions[18]. The work here was meant to determine the conditions which make corneal epithelial cells responsive to cytokines and to define the downstream intracellular signaling pathways mediating MMP-9 gene expression.
Materials and Methods
Cell Culture
The freshly isolated corneal epithelial cells used in this study express a profile of gelatinolytic MMPs that recapitulates expression in wounded corneal epithelium in situ, with prominent levels of MMP-9 secreted into the medium constitutively, but only minimal amounts of MMP-2[22, 23]. With passing, this ratio changes, with MMP-2 becoming prominent while MMP-9 becomes essentially undetectable [23], similar to the profile produced by corneal fibroblasts[24].
Primary cultures of rabbit corneal epithelial cells were prepared as described previously[23, 25], with some slight modifications. Briefly, rabbit eyes (Pel-Freeze, Rogers, AR) were shipped overnight on ice in Hanks Balanced Salt Solution (HBSS) containing penicillin/streptomycin. Corneas were dissected from eyes by cutting just outside of the corneal/scleral interface, the endothelial tissue layer was removed from each by scraping with a scalpel and the resulting two-layered corneas were incubated in 0.25% Dispase II (Gibco, Carlsbad, CA) in basal Keratinocyte Media (KM) without calcium (Gibco, Carlsbad, CA) overnight at 4°C. The following day, the temperature was increased to 37°C for 45 minutes and the epithelium was then gently scraped from each of the corneal stromal layers with a scalpel. Epithelial sheet fragments were removed to a solution of 0.25% trypsin/EDTA (Gibco, Carlsbad, CA) and further incubated for 10 minutes at 37°C to separate individual cells. Finally, cells were counted on a hemocytometer and plated into 24-well culture dishes (Nunclon; Nalge Nunc International, Naperville, IL) for experiments.
The homogeneity of corneal epithelial cells prepared using this method was confirmed by indirect immunofluorescent staining using an antibody to the corneal epithelial cell-specific marker, keratin 3/12 (Chemicon, Temecula, CA).
Primary corneal epithelial cells were plated at a density of 3*106 cells per well in a 24-well culture dish and covered with 500μl of KM containing 5% FBS (Invitrogen, Carlsbad, CA), 50 U/mL penicillin (Cellgro, Herndon, VA), 50 μg/mL streptomycin (Cellgro, Herndon, VA), and 0.5 μg/mL amphotericin B (Gibco, Carlsbad, CA). Cells were incubated at 37°C in 5% CO2 overnight to allow them time to adhere and spread on the dish. The next day, cells were washed 3 times with PBS (Gibco, Carlsbad, CA) for 10 minutes each to remove FBS and then 350 μl of KM containing no additives, except the cytokines or inhibitors being assayed, was added and cells were again allowed to incubate at 37°C in 5% CO2 for 24 hours. Following 24 hours of treatment, media or cell lysates were harvested for analysis. Results were obtained from three independent experiments.
Except for the dose-dependence experiments, cytokines were used at a concentration of 10 ng/ml. The following cytokines were obtained from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO): TGF-β1, TGF-β2, IL-1α, IL-1β, EGF, HB-EGF, FGF-2, PDGF-BB. PMA (Sigma-Aldrich, St. Louis, MO) was used at a concentration of 1 μM as a positive control to induce MMP-9 expression. In some experiments, cells were treated with cholera toxin at a concentration of 30 ng/ml (Sigma-Aldrich, St. Louis, MO) or AG1478 at a concentration of 50 ng/ml (Sigma-Aldrich, St. Louis, MO).
For pathway inhibition experiments, various compounds were used to inhibit different intracellular signaling cascades. Cells were treated for 1 hour with compounds alone then media was removed and media containing the inhibitor and TGF-β and/or IL-1 was added and cells were further incubated for 24 hours. JSH-23 (Calbiochem, Darmstadt, Germany) was used at a concentration of 30 μM to inhibit NF-kB activity. SB202190 (Calbiochem, Darmstadt, Germany) was used at a concentration of 20 μM to inhibit p38 activity. PD98059 (Calbiochem, Darmstadt, Germany) was used at a concentration of 40 μM to inhibit ERK activity. SP600125 (Calbiochem, Darmstadt, Germany) was used at a concentration of 30 μM to inhibit JNK activity. SIS-3 (Calbiochem, Darmstadt, Germany) was used at a concentration of 10 μM to inhibit Smad3 activity.
Zymographic Analysis of MMP-9 Production
Zymographic analysis was used to analyze the relative amounts of MMP-9 secreted into the cell culture medium over a fixed period of time by CECs treated in various ways. The method was performed according to standard procedure in our lab as described previously[16, 26, 27]. Briefly, following 24 hours of experimental treatment as above, cell-conditioned media was harvested for zymography. Equal volumes (16μl) of media collected from each well were used for analysis. 4 μl loading buffer was added, and 15 μl of this mixture was loaded into an 8% PAGE gel containing 0.1% gelatin (Sigma-Aldrich, St. Louis, MO). Gels were run for 45 minutes at 200 volts. Gels were then incubated in 2.5% Triton-X (EMD Biosciences, Darmstadt, Germany) for 1 hour at room temperature on a rocker. Gels were thoroughly washed with distilled water and incubated overnight in renaturing buffer (10mM CaCl2, 50mM Tris pH7.5) at 37°C. The next morning, gels were briefly washed and then stained (25g Coomassie blue, 150μl Isopropanol, 50ml acetic acid, 300ml H2O) for 1 hour at room temperature on a rocker. Gels were then de-stained in distilled water until bands were clearly visible. Gels were then scanned for densitometry analysis by ImageJ software (Bio-Rad, Hercules, CA).
Analysis of Transcriptional promoter activity
The Pr22 construct was created as described previously[13] and contains A 541-base pair fragment extending from -522 to +19 of the rabbit MMP-9 gene driving expression of a β-Galactosidase (β-gal) reporter protein on a pNASSβ backbone (Promega, Madison, WI). As discussed above, this construct accurately drives expression of a reporter gene during embryonic development, angiogenesis, epithelial response to ultraviolet radiation and wound healing[13]. The Pr22 construct was used as a template for creating the deletion constructs: 440GelB, 330GelB, 90GelB, and empty vector (Figure 4A). The 440GelB, 330GelB, and 90GelB deletion constructs were amplified from the original Pr22 by polymerase chain reaction using 5′-CTTTCCAGGAGGGAGGACGTA-3′, 5′-CTTTCGCTGAGCTGACCACTTG-3′, and 5′-ACCGGCCCTGAGTCAGCACTTGCCTGCAG-3′ primers, respectively, with 5′-ACTGGCCGTCGTTTTACAACGTC-3′ being used as the reverse primer for all constructs. These primers amplified the MMP-9 promoter region from -440, -330, and -90 base pairs before the transcription start site respectively. The firefly luciferase construct, PGL-3 (Promega, Madison, WI), which is driven by the SV-40 promoter, was a generous gift from Dr. Wei Li.
Figure 4. Regions of MMP-9 promoter mediating TGF-β and IL-1 stimulation of MMP-9 in CECs.
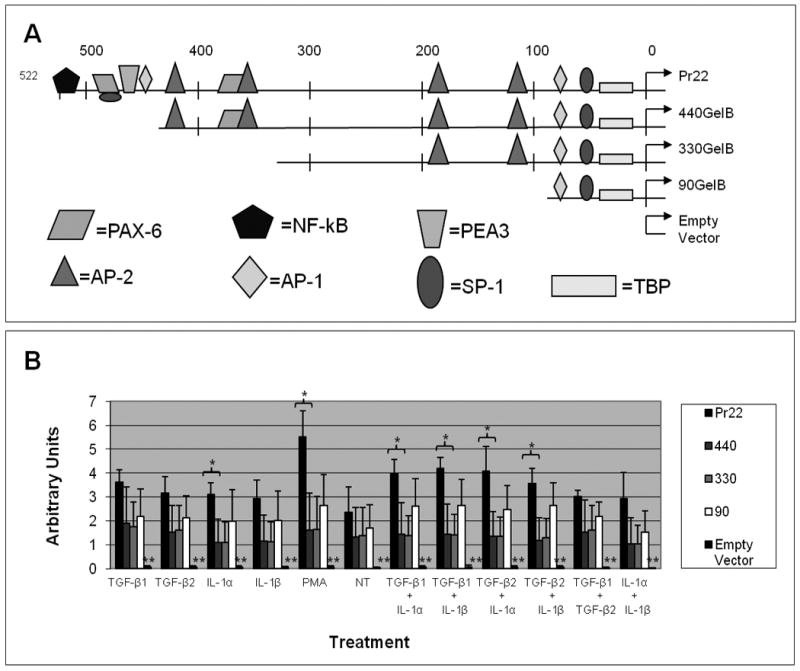
MMP-9 promoter activity in rabbit CECs stimulated by TGF-β1, TGF-β2, IL-1α, and IL-1β. (A) Schematics depicting the various 5′-deleted MMP-9 promoter constructs linked to a β–Gal reporter gene. (B) Cultures were co-transfected with a luciferase construct under the SV-40 promoter and one of the various 5′-deleted MMP-9 promoter constructs containing a β–Gal reporter gene. Transiently transfected cells were left untreated or were stimulated with 10 ng/ml of TGF-β1, TGF-β2, IL-1α, or IL-1β alone or in combinations for 24 hours and promoter activities were analyzed by dividing β–Gal activity by luciferase activity. Values are the average of results obtained from three independent experiments performed in duplicate. Error bars indicate standard deviation. * indicates a p-value of less than 0.05. ** indicates a p-value of less than 0.005 between the empty vector and any of the other constructs.
Cells were transfected using Genejammer (Stratagene, Cedar Creek, TX) according to the manufacturer's protocol. Cells were transfected with the β-gal construct (driven by the full length or deletion constructs of the MMP-9 promoter) and luciferase construct (driven by a SV-40 promoter) at a 4:1 ratio (1.6 μg of B-gal construct + 0.4 μg of luciferase construct per 3 μl genejammer) for 16 hours. Cells were then washed three times for 10 minutes each in PBS and treated with or without TGF-β (10ng/ml) or IL-1 (10ng/ml) alone or together for 24 hours at 37°C in a humidified incubator.
For reporter expression assay, cells were washed once with PBS and then lysed in 1X Reporter Lysis Buffer (promega, Madison, WI). Lysates were spun at top speed at 4°C for 10 minutes to pellet cell debris and the supernatant were transferred to fresh tubes and stored at -80°C. β-gal and Luciferase assays were run according to manufacturer's protocols (Promega, Madison, WI). β-gal activity was then divided by luciferase activity in order to normalize each value.
Statistical Analysis
Data were analyzed with general linear models to account for effects of days of experiment and replicates. For both TGF-β and IL-1, the effects of inhibitors were assessed by tests of statistical interaction between the cytokine and the presence of each inhibitor. Significance was determined by a p value of less than 0.05.
Results
Effect of cytokines on MMP-9 expression in CECs
There was no significant difference in MMP-9 production as measured by gel zymography when freshly isolated cultures of corneal epithelial cells were stimulated by various cytokines individually (EGF, HB-EGF, FGF-2, IL-1β, TGF-β1, TGF-β2, or PDGF-BB) (Figure 1). Increasing cAMP levels by treating cells with cholera toxin also failed to alter MMP-9 expression (Figure 1). However when combined, TGF-β2, IL-1β, FGF-2, HB-EGF, and PDGF-BB acted to strongly up-regulate MMP-9 (Figure 2A). When individual cytokines were removed from this mixture one by one, there was no decrease in expression except when TGF-β or IL-1 was removed; this reduced the production of MMP-9 (Figure 2B). When cells were treated in pair-wise combinations, only the combination of TGF-β and IL-1 was sufficient to stimulate MMP-9 expression (Figure 2C). Furthermore, TGF-β and IL-1 stimulated MMP-9 in a dose-dependent manner; as little as 1 ng/ml of one cytokine combined with 10 ng/ml of the other was sufficient for stimulation of MMP-9 above basal levels (Figure 3A). This regulation was not isoform specific and any combination of IL-1α or IL-1β with either TGF-β1 or TGF-β2 was sufficient to strongly induce MMP-9, though IL-1α and IL-1β together or TGFβ-1 and TGFβ-2 together failed to stimulate MMP-9 discernibly (Figure 3B).
Figure 1. Cytokines do not significantly affect MMP-9 expression in CECs with or without cholera toxin.
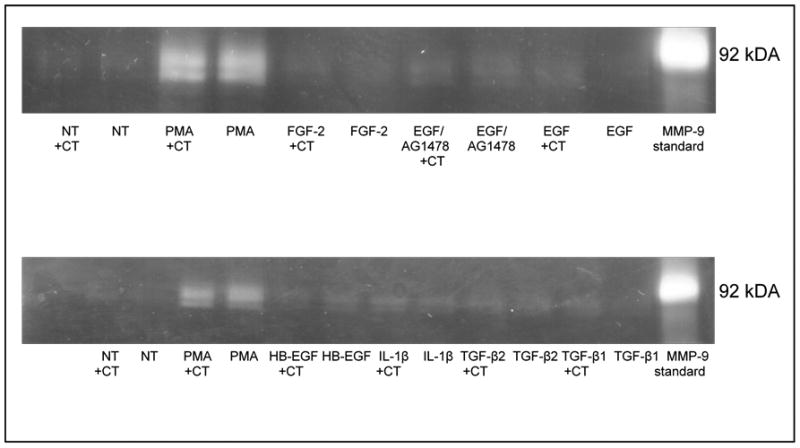
Zymographic analysis of MMP-9 secreted into the media by CECs following TGF-β1, TGF-β2, IL-1β, HB-EGF, EGF, and FGF-2 treatments (10 ng/mL) with or without cholera toxin (30 ng/mL), a known up-regulator of cAMP. Treatment with an EGFR inhibitor, AG1478 (50 ng/ml), had no effect on MMP-9 expression with or without EGF treatment. Similarly, cholera toxin failed to affect MMP-9 expression in conjunction with any of the various cytokines. PMA (1μM) was used as a positive control for MMP-9 stimulation. A human MMP-9 standard was run in parallel for sizing of the rabbit MMP-9 zymogram bands.
Figure 2. TGF-β and IL-1 families work together to induce MMP-9 in CECs.
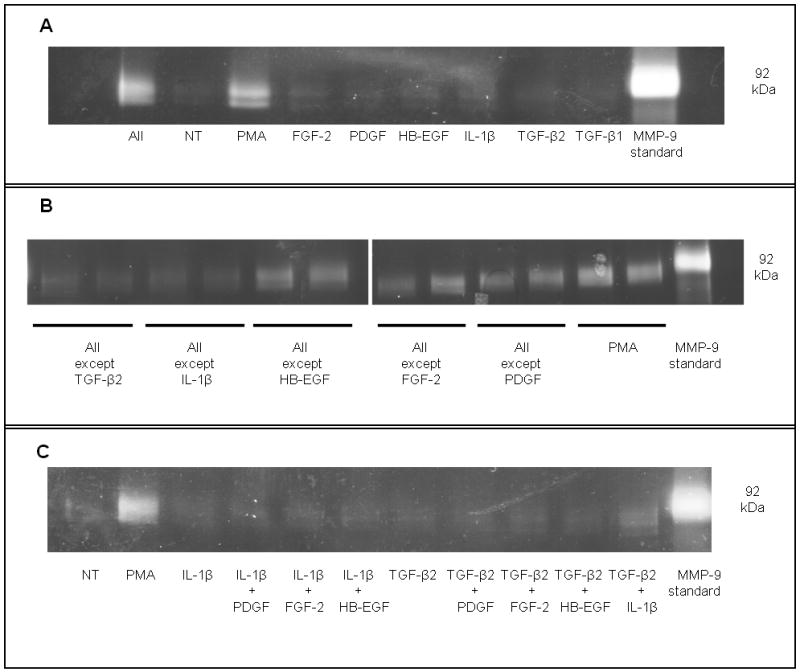
Zymographic analysis of MMP-9 secreted into the media by CECs showing that (A) a combination of cytokines (TGF-β1, TGF-β2, IL-1, HB-EGF, PDGF-BB, and FGF-2 at 10 ng/ml each) can act together to induce MMP-9 expression in CECs. (B) Only removal of either TGF-β or IL-1 from this cytokine cocktail resulted in a significant decrease in MMP-9 expression. (C) The combination of TGF-β2 and IL-1β but not any other combination of cytokines was sufficient to induce a significant up-regulation of MMP-9 in CECs. PMA (1μM) was used as a positive control for MMP-9 stimulation for all experiments. A human MMP-9 standard was run in parallel for sizing of the rabbit MMP-9 zymogram bands.
Figure 3. TGF-β and IL-1 stimulation of MMP-9 in CECs is dose-dependent but not isoform dependent.
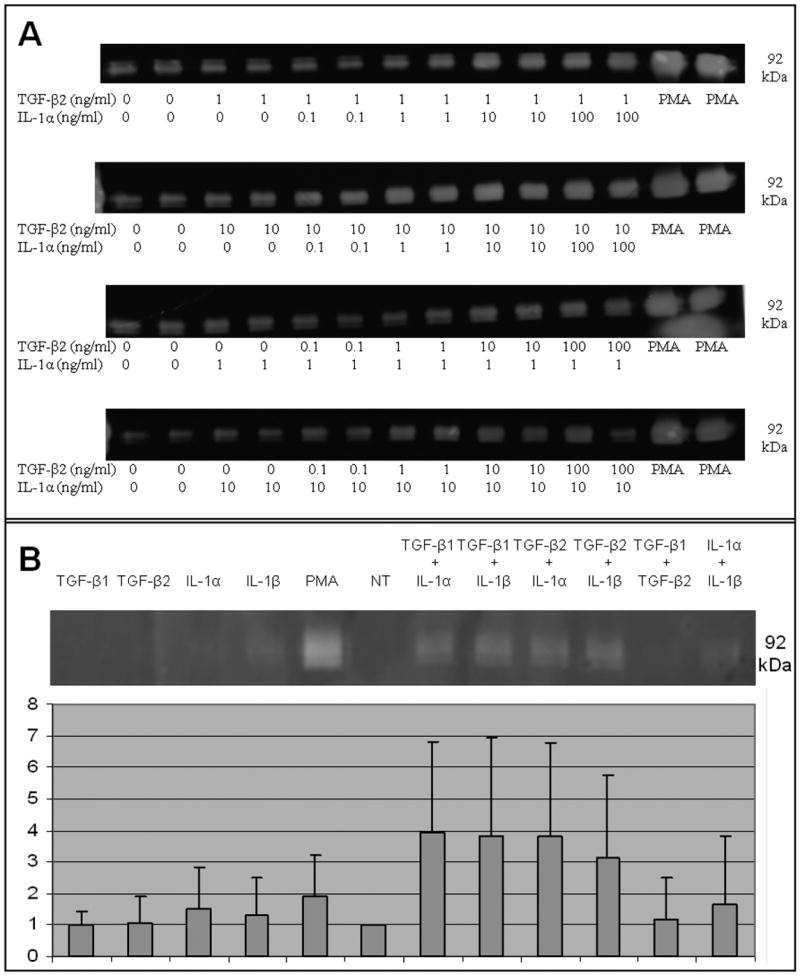
Zymographic analysis of MMP-9 secreted into the media by CECs following cytokine treatment showing (A) TGF-β2 and IL-1β act in a dose-dependent manner to induce MMP9 in CECs. As little as 1 ng of one cytokine combined with 10 ng of the other cytokine is sufficient for stimulation of MMP-9 above basal levels. (B) The stimulation of MMP-9 in CECs by the TGF-β and IL-1 families is not isoform specific; any combination of TGF-β1 or TGF-β2 with either IL-1α or IL-1β is sufficient to up-regulate MMP-9 expression in CECs. Densitometry was performed using the Bio-Rad software “ImageJ”, and MMP-9 stimulation was plotted on a bar graph. Each value is the mean of results obtained from three independent experiments. Error bars indicate standard deviation. ** indicates a p-value of less than 0.005.
These results indicate that stimulation of MMP-9 production in freshly isolated CECs requires dual treatment with both IL-1 and TGF-β.
Transcriptional Promoter
Transcriptional promoter analysis was used to determine the promoter elements needed to stimulate MMP-9 transcription by IL-1 and TGF-β. The 541 base pair DNA fragment of the rabbit MMP-9 gene used for these experiments contains many TF binding elements[13] (Figure 4b). These elements can be grouped into three regions: proximal, distal, and intermediate[12]. The proximal region contains one AP-1 binding element located at bases (-82 to -76), the SP-1 site at (-49 to -56), and the TATA box at (-28 to -23). When transfected into rabbit corneal epithelial cells, the proximal region (-92 to 19) alone drives expression of a linked reported gene by over two fold as compared to a full length promoter construct (-519 to 19) when stimulated by PMA[12]. The distal region contains another AP-1 site at bases (-457 to -451) adjacent to the PEA3 element at bases (-465 to -459) and another SP-1 element at bases (-479 to -486) as well as two PAX-6 binding elements at (-467 to -492) and (-359 to -385) and an NF-κB binding element at bases (-522 to -514). Removal of the distal region from the full length promoter region results in reduced promoter activity. The intermediate region, between the proximal and distal regions, consists of four AP-2 binding elements. This intermediate region seems to act both as an inhibitory element and as a promoter element depending on the cellular environment. For example, a promoter construct containing both the intermediate and proximal regions shows a marked reduction in activity compared to the proximal region alone when stimulated by PMA showing that this region can inhibit PMA stimulated expression of MMP-9[28]. However, recently, it has been shown that Pax6 can work synergistically with AP-2 in the intermediate region to stimulate MMP-9 promoter activity in CECs[29]. Additionally, the ETS family of TFs has been implicated in MMP-9 promoter activity[30], though DNA binding studies have yet to be performed for this TF.
The full length -522 reporter construct recapitulated the effects of TGF-β, IL-1, and PMA on MMP-9 protein production. Reporter gene expression was increased by ∼2-4-fold above basal levels following combinatorial treatment with TGF-β and IL-1 (Figure 4). Individually, these cytokines stimulated only about half this activity. PMA stimulated expression of the reporter gene to a much higher level than the cytokines – by more than 5 fold. Deletion of the distal 80 base pairs of the MMP-9 promoter to -440 resulted in a significant decrease in promoter activity for all combinatorial treatments and PMA treatment, and also decreased basal MMP-9 promoter activity. While treatment with either TGF-β isoform still did result in a very slight stimulation of promoter activity above basal levels, it was considerably less than with the full length construct. Stimulation above basal levels with either IL-1 isoform was abolished completely by this deletion. Deletion of the distal 192 base pairs completely abolished all response to TGF-β and IL-1 treatments, though PMA stimulated expression and basal expression were still present at lower levels than the full length construct. Deletion of all but the proximal 90 base pairs allows for a higher basal expression level than either the -440 or the -330, but not the full length construct suggesting an inhibitory element in the MMP-9 promoter. Interestingly, having only the proximal region present also seems to partially restore responsiveness to both TGF-β/IL-1, though at very low levels. This same effect was observed for PMA responsiveness, as previously demonstrated.
These results indicate that the combination of TGF-β and IL1 stimulates the activity of the MMP-9 transcription promoter using the same regions of the transcriptional promoter as PMA.
Signal Transduction Pathways
We chose to examine the role of the Smad and NF-kB signaling pathways as they are the classical signaling pathways for TGF-β and IL-1, respectively. Additionally, we examined the role of all three branches of the MAPK pathway: p38, JNK, and ERK as they are potent signaling pathways that can also be activated by TGF-β and IL-1. Activation of these pathways following TGF-β and IL-1 treatment as well as abrogation of signaling following treatment with their specific inhibitor was shown by western blotting and immunocytochemistry (Supplementary figures 1 and 2). Blocking the p38, NF-kB, and JNK pathways individually using SB202190, JSH-23, and SP600125, respectively, significantly reduced but did not abrogate basal MMP-9 levels (Figure 5). Inhibition of the NF-kB, Smad3, or JNK pathways significantly reduced the TGF-β mediated stimulation of MMP-9 (p=0.002, 0.001, 0.009), whereas inhibition of the p38 pathway was less significant (p=0.083). IL-1β mediated stimulation of MMP-9 was significantly reduced following NF-kB or p38 inhibition (p=0.014 or 0.005 respectively). Inhibition of the ERK pathway did not have an effect on MMP-9 mediated expression in either treated or untreated cells.
Figure 5. Signaling Pathways Mediating TGF-β and IL-1 stimulation of MMP-9 in CECs.
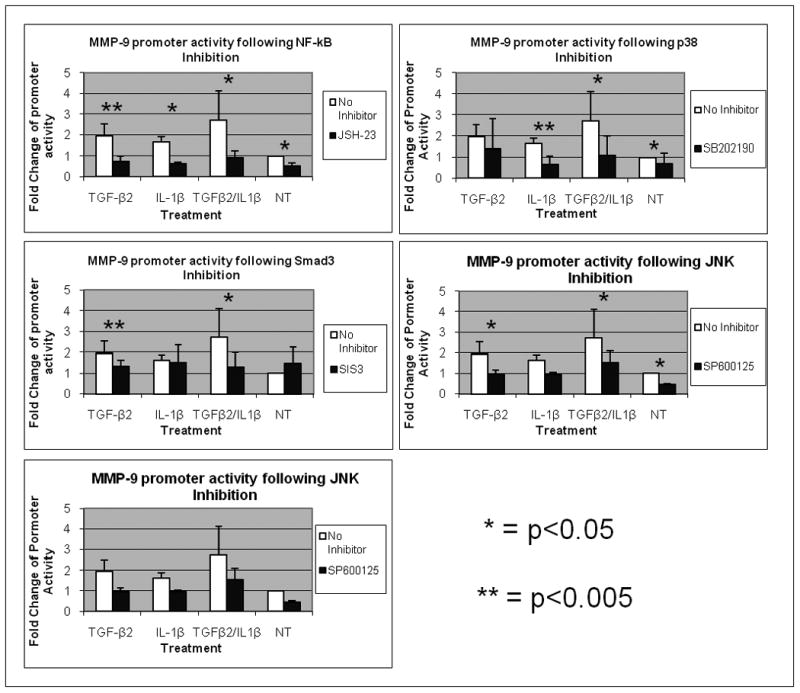
MMP-9 promoter activity in was measured in rabbit CEC cultures. Cultures were co-transfected with a luciferase construct under the SV-40 promoter and Pr22. Transiently transfected cells were left untreated or were stimulated with 10 ng/ml of TGF-β2 or IL-1β alone or in combinations with or without various intracellular signaling pathway inhibitors for 24 hours. Promoter activities were analyzed by dividing β–Gal activity by luciferase activity. Intracellular signaling pathway inhibitors (A) SB202190(20μM), (B) JSH-23 (30μM), (C) SP600125(30μM), (D) SIS-3(10μM), and (E) PD98059(40μM) were used. Each value is the mean of results obtained from three independent experiments performed in triplicate. Error bars indicate standard deviation. * indicates a p-value of less than 0.05. ** indicates a p-value of less than 0.005.
These results indicate that TGF-β and IL-1 utilize an overlapping combination of signaling pathways to stimulate activity of the MMP-9 promoter. IL-1 uses p38 and NF-kB. TGF-β uses NF-kB, JNK, and Smad.
Discussion
Previously, our lab showed that media containing cytokines secreted by CECs was not sufficient to induce MMP-9 expression in other CECs in vitro, indicating CECs were relatively refractory to cytokine and growth factor stimulation[16]. Instead, it was shown that plating density and cell-cell interactions seemed to regulate MMP-9 expression because CECs plated at low density seemed to secret the same amount of MMP-9 into the media as CECs plated at a four-fold higher density[16]. However, more recent reports from other labs using different cell culture models have shown that there may be physiological relevant conditions where CECs can express MMP-9 in response to some secreted factors[18]. One difference that might account for the conflicting results is the type of media the CECs cells were treated with. While we used a basic media containing only the desired cytokine to be assayed, other labs used a media containing additives to stimulate cell growth such as cholera toxin and EGF which may have played a cooperative role in inducing MMP-9. Indeed, cAMP has been shown to be a co-activator of MMP-9 expression in uterine and cervical tissue in pregnant rats[31] and cholera toxin is a known inducer of cAMP. This led us to re-evaluate the effect of secreted factors on MMP-9 expression in CECs, and led to the hypothesis that cholera toxin, a known stimulator of the second messenger cAMP, was acting synergistically with the various cytokines to elicit MMP-9 expression.
We found that while MMP-9 in CECs can be regulated by cytokines present in the tear fluid or secreted from the CECs themselves, cAMP activation induced by cholera toxin did not seem to play a significant role in affecting this regulation. Instead, we have found that two of these cytokine families, TGF-β and IL-1, can act cooperatively in this cell type as opposed to some other cell types where these two families are antagonistic towards MMP-9 expression[32]. Furthermore, we have shown that these cytokines act via the NF-kB, Smad, p38, and JNK signaling pathways mainly on a specific region of the MMP-9 promoter located 440 to 522 base pairs upstream of the transcription start site.
TGF-β has three isoforms with the first two being well documented in the corneal epithelium. TGFβ-1 and -2 are present in the unwounded epithelium with TGF-β2 being the predominant isoform and TGF-β1 being the predominant isoform in the tear fluid; both these isoforms, especially, TGF-β2, are up-regulated following wounding [33]. The receptors for TGF-β, TGF-βRI and RII, are both present in the unwounded cornea at basal levels throughout the corneal epithelium. Following wounding, however, the receptors take on individual expression patterns. TGF-βRI is up-regulated throughout the epithelium while TGF-βRII is up-regulated specifically in the epithelial cells migrating to cover the wound [34]. These receptors work together to activate downstream signaling, so the areas of overlap are those where TGF-β signaling can occur.
IL-1α and -1β are also both expressed by corneal epithelial cells. Similar to TGF-β, IL-1 is up-regulated following wounding; however unlike TGF-β, IL-1α expression has been inversely correlated with cell-cell contact with the greatest expression being in cells with less contact[35]. Thus the enhanced expression of TGF-β and IL-1 as well as their cognate receptors correlates well with the localized induction of MMP-9 at the leading edge of the migrating corneal epithelium following wounding.
Signaling pathways mediating MMP-9 promoter stimulation
The results from the promoter deletion experiments have shown that both TGF-β and IL-1 require a small region of the promoter (-440 through -522) to significantly affect MMP-9 expression. Spotlighting the region of the promoter transducing the effect of TGF-β and IL-1 has trimmed the possible pathways mediating this effect; however, there are still several possibilities for direct and indirect regulation as this region contains NF-kB, AP-1, PEA-3, and SP-1 transcription factor binding elements. Interestingly, the effect of certain TFs on the MMP-9 promoter can vary depending on the cell type being assayed. For example, NF-kB has been shown to be an inducer of MMP-9 in rat brain astrocytes[36] as well as an inhibitor of MMP-9 in both monocytes and macrophages[37]. Similarly, SP-1 has been shown to activate the MMP-9 promoter in a transformed cell line[38] while it represses the promoter in corneal epithelial cells[39]. Thus, the promoter is a composite of many TF binding elements and depending on the stimulus and the cell type, these TFs can associate in different combinations in order to exquisitely control the expression of MMP-9.
The TGF-β receptor can couple directly to several intracellular signaling cascades including the Smad and all three branches of the MAPK pathway: p38, JNK, and ERK[40-42] and indeed has been shown to up-regulate all of these signaling pathways in corneal explants[43]. This receptor has also been shown to activate many other TF signaling pathways such as NF-kB[44, 45] and PI3K[46, 47] though evidence of this occurring in CECs is still lacking. We have found that the Smad3, JNK, and NF-kB pathways are the major mediators of TGF-β induced up-regulation of MMP-9 while NF-kB and p38 are the major mediators for IL-1, but how exactly do these mediators work?
Due to the many signaling pathways induced by TGF-β and IL-1 which mediate MMP-9 expression, there are many possible permutations of cross-talk (Figure 6). However, because TGF-β and IL-1β have a statistically additive rather than synergistic effect on MMP-9 stimulation it can be implied that if there is cross-talk between the signaling pathways, it occurs downstream of one or the other cytokine, but not between the two. Also, inhibiting various pathways individually was unable to abrogate MMP-9 expression, indicating that these pathways are not all directly acting on each other though we can't exclude the possibility that two or three of these pathways may be working in conjunction to induce a third.
Figure 6. Possible signaling pathways mediating the effects of TGF-β and IL-1 on the MMP-9 promoter.
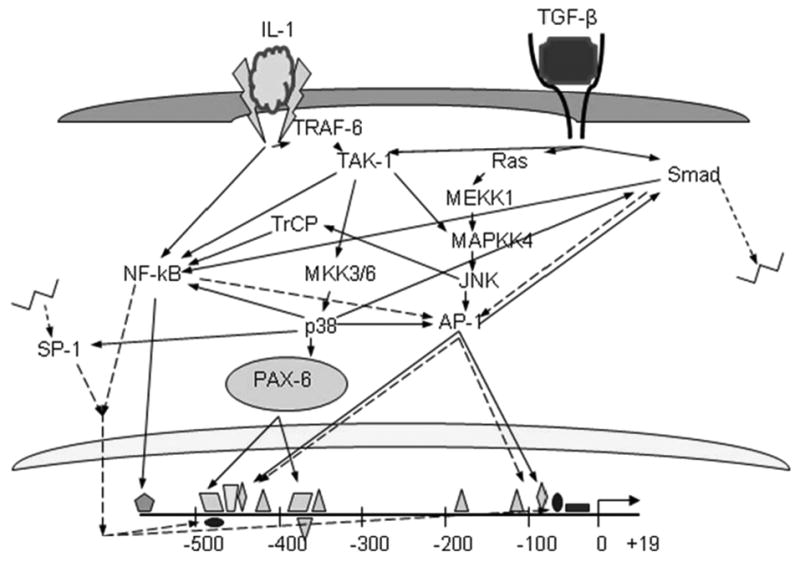
Direct phosphorylation of a downstream target is shown in solid arrow lines, indirect mechanisms such as synergism or down-stream gene upregulation is depicted with dotted arrow lines.
TGF-β and IL-1 Signaling via NF-kB
While the NF-kB pathway is the canonical IL-1/IL-1R signaling pathway, it can also be the downstream target of the TGF-β-activated-kinase (TAK-1) and Smad3 signaling pathways following TGF-β stimulation[48, 49]. NF-kB is a potent signaling molecule that can act directly by binding the NF-kB element in the MMP-9 promoter or indirectly via activation of other transcription factors or by up-regulation of gene expression of other transcription factors. A direct effect would be the simplest scenario and would correlate well with the promoter deletion results showing deletion of the promoter region containing the NF-kB binding element significantly reduces TGF-β induced MMP-9 expression (Figure 2.4). A second possibility is that NF-kB could be acting indirectly to activate or up-regulate the other TFs that can then activate MMP-9 gene expression. For example, NF-kB has been shown to up-regulate gene expression of both components of the AP-1 complex: c-jun [50] and c-fos [51]. A third possibility is that NF-kB could be acting cooperatively with other TFs to potentiate or enhance their effect, as NF-kB has been shown to cooperate with SP-1[52] or AP-1[53]. Finally, of course, there may be a combination of all three possibilities acting at once.
TGF-β signaling via JNK
NF-kB and JNK signaling share many similarities in the ways that they can affect MMP-9 promoter activity. While JNK is not the classical TGF-β signaling pathway, it is not a completely novel target. As TAK-1 can activate NF-kB in some circumstances, in others it can phosphorylate MKK4/7 which can then phosphorylate JNK and in turn activate c-jun and AP-1. Alternatively, TGF-β can activate JNK via the RAS/MEKK1/MKK4 pathway. AP-1, like NF-kB, can be acting in a number of ways though like NF-kB, it is most likely acting directly on the MMP-9 promoter. Indeed, several site-directed mutagenesis assays have implicated AP-1 as a critical component for MMP-9 gene expression, though these studies found the proximal rather than distal AP-1 site to be the more critical element([12, 54, 55]. A second possibility is that JNK or AP-1 is activating other TFs, as JNK has been shown to phosphorylate Smad3 and enhance nuclear translocation [41]. Also, JNK has been shown to activate the NF-kB pathway by activation of β-TrCP [56]. As mentioned above, AP-1 can also cooperate with other TFs such as Smad3 and PEA-3 rather than activate them to induce gene expression [41, 57, 58]. Finally of course, AP-1, like NF-kB, is a potent stress response TF and can induce the de novo expression of many of TFs or adaptor proteins which can aid in the activity of the MMP-9 promoter.
TGF-β signaling via Smad
Smad signaling is the canonical TGF-β signaling cascade where binding of TGF-β to its receptor induces R-Smad phosphorylation leading to binding by co-Smads and translocation to the nucleus, complexing with other co-activators, DNA binding, and promoter activation. While this pathway is simple enough, the mechanism by which the Smad pathway is mediating TGF-β induced up-regulation of MMP-9 may be more complex as there are no classical Smad binding elements in the MMP-9 promoter. This does not discount the possibility that the Smad TFs may be binding an unidentified promoter element or that they may be forming a complex and binding the DNA indirectly through another co-activator. For example, Smad3/4 can form a complex with AP-1 and this complex has been shown to be critical for TGF-β mediated AP-1 site activation independent of Smad3 DNA binding[59]. This complex also requires a SIM-containing subunit to bind a CCAG motif downstream of the AP-1 binding element, which the MMP-9 promoter has, further supporting this theory[59]. Alternatively, Smad3 has been shown to interact with SP-1 following nuclear translocation in order to induce target genes[60]. Further experiments are needed to determine if Smad3 is actually bound in a complex to the MMP-9 promoter and if so, in which complex. The Smad pathway may also be acting indirectly to induce MMP-9 by activating the NF-kB pathway as previously mentioned. Or, again like NF-kB, Smad may be inducing gene expression of other effecter TFs.
Given all myriad possibilities for cross-talk between signaling pathways only mediating the response to TGF-β, and not even considering the possibilities added by IL-1, the complexity of MMP-9 regulation can be staggering. It would be intriguing, however, if there was a simple elegant solution. For example, all pathways mentioned above seem to hinge on JNK activity. It is possible, therefore, that JNK is activating both NF-kB and AP-1 as well as Smad3 which can then possibly bind SP-1 or AP-1 along with NF-kB and PEA-3 and enhance promoter activity. This would explain why inhibition of these three pathways can inhibit TGF-β mediated MMP-9 expression and why inhibition of only one pathway is insufficient to completely block this expression. Alternatively, these many pathways may be in place as a failsafe, so if one pathway fails, the signal can be redirected through another pathway. While the efficiency of promoter stimulation may be lower this could account for the inability of one pathway to completely inhibit expression.
IL-1 signaling via p38
So far, we have discussed how TGF-β may be stimulating MMP-9 by the NF-kB, JNK, and Smad pathways as well as how IL-1 might be acting via NF-kB. However, we have also shown that the p38 signaling pathway mediates IL-1 induction of MMP-9. Aside from the canonical NF-kB pathway activated by IL-1R activation, this receptor can also phosphorylate TRAF6 which can then activate p38 via TAK1[61]. Contrary to inhibiting NF-kB which can block the signaling pathway at the most basic level (DNA binding) but not its upstream targets, understanding the mechanism by which the p38 pathway mediates the IL-1 stimulation of MMP-9 can be a bit more complex due to the fact that there is no p38 consensus binding element in the MMP-9 promoter, but this protein still has several possible downstream targets which can be activated and bind the DNA such as SP-1 or AP-1[62, 63] which are also in the distal 80 base pairs of the MMP-9 promoter construct which accounts for the response element to the IL-1 as well as TGF-β protein families. The p38 protein can also signal through a Smad-dependent or a Smad-independent pathway to induce NF-kB [45, 64]. Alternatively, p38 may be signaling through a completely separate pathway to activate ATF-2 which can heterodimerize with AP-1 and bind the AP-1 consensus sequence which is also in both the distal and proximal MMP-9 promoter response elements[65].
Thus, while we now have an idea of which signaling pathways are involved in regulating MMP-9 expression in CECs, we still have little idea of how these pathways are interacting and what transcription factors are actually binding the MMP-9 promoter region. Further investigation using gel-shift or mutation constructs are still needed to determine the exact mechanism(s) by which p38, JNK, Smad, and NF-kB are mediating the stimulation of MMP-9 in CECs by the TGF-β and IL-1 families.
While understanding the intracellular pathways mediating cells' responses to stimuli is undeniably important, it is equally important to link these intracellular pathways with extracellular stimuli which can then be linked to various disease states for possible treatments. Only recently have investigators began tying extracellular signaling cues with intracellular signaling pathways that control MMP-9 expression in the corneal epithelium. One such study found that SP-1 acts to repress MMP-9 expression via platelet activating protein (PAF) mediated release of this repressor from the promoter[39]. Our work adds to this base several novel possibilities by which the TGF-β and IL-1 families can be inducing MMP-9 in CECs.
It is widely accepted that TGF-β and IL-1 are powerful signaling molecules involved in many cellular processes and their altered expression profiles have shown to be associated with various pathologies displaying an over-expression of MMP-9. Over-expression of IL-1 as well as MMP-9, for example, has been associated with dry eye[66]. Common treatments for dry eye in the clinic involve treatment with anti-inflammatory agents which block IL-1 activity in a global manner. However, this also reduces the host's ability to ward off infection which is severely compromised in dry eye due to disruption of corneal epithelial integrity caused by an over-expression of MMP-9. Defining the pathways mediating the various downstream effects from TGF-β and IL-1 makes possible the separation of these processes, opening a window of opportunity for blocking the direct downstream up-regulation of MMP-9 by IL-1 without affecting the overall innate immune response to infection.
In conclusion, MMP-9 has been shown to be stimulated in a wide array of tissues by a variety of stimuli. We have shown that in the corneal epithelium, two cytokine families, TGF-β and IL-1, co-operate to induce many intracellular signaling pathways which can stimulate multiple TFs to bind and activate the MMP-9 gene. This complex interplay of TFs also presents numerous points for dysregulation. A complete understanding of the signaling that goes on in the wound scenario will help in understanding various pathologies associated with this dysregulation and in designing rational therapeutics.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 EY12651 and P30 EY14801, and an unrestricted grant from Research to Prevent Blindness (RPB). MEF was an RPB Senior Scientific Investigator and held the Walter G. Ross Chair in Ophthalmic Research.
Contract grant Sponsor: NIH, Research to Prevent Blindness (RPB)
Contract grant number: R01 EY12651, P30 EY14801, unrestricted grant from RPB
References
- 1.Gijbels K, et al. Gelatinase B is present in the cerebrospinal fluid during experimental autoimmune encephalomyelitis and cleaves myelin basic protein. J Neurosci Res. 1993;36(4):432–40. doi: 10.1002/jnr.490360409. [DOI] [PubMed] [Google Scholar]
- 2.Gijbels K, et al. Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol. 1992;41(1):29–34. doi: 10.1016/0165-5728(92)90192-n. [DOI] [PubMed] [Google Scholar]
- 3.Somerville RP, Oblander SA, Apte SS. Matrix metalloproteinases: old dogs with new tricks. Genome Biol. 2003;4(6):216. doi: 10.1186/gb-2003-4-6-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohan R, et al. Matrix metalloproteinase gelatinase B (MMP-9) coordinates and effects epithelial regeneration. J Biol Chem. 2002;277(3):2065–72. doi: 10.1074/jbc.M107611200. [DOI] [PubMed] [Google Scholar]
- 5.Rayment EA, Upton Z, Shooter GK. Increased matrix metalloproteinase-9 (MMP-9) activity observed in chronic wound fluid is related to the clinical severity of the ulcer. Br J Dermatol. 2008;158(5):951–61. doi: 10.1111/j.1365-2133.2008.08462.x. [DOI] [PubMed] [Google Scholar]
- 6.Yager DR, Nwomeh BC. The proteolytic environment of chronic wounds. Wound Repair Regen. 1999;7(6):433–41. doi: 10.1046/j.1524-475x.1999.00433.x. [DOI] [PubMed] [Google Scholar]
- 7.Shimanovich I, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol. 2004;204(5):519–27. doi: 10.1002/path.1674. [DOI] [PubMed] [Google Scholar]
- 8.Lo Russo L, et al. Diagnostic pathways and clinical significance of desquamative gingivitis. J Periodontol. 2008;79(1):4–24. doi: 10.1902/jop.2008.070231. [DOI] [PubMed] [Google Scholar]
- 9.Afonso AA, et al. Tear fluid gelatinase B activity correlates with IL-1alpha concentration and fluorescein clearance in ocular rosacea. Invest Ophthalmol Vis Sci. 1999;40(11):2506–12. [PubMed] [Google Scholar]
- 10.Garrana RM, et al. Matrix metalloproteinases in epithelia from human recurrent corneal erosion. Invest Ophthalmol Vis Sci. 1999;40(6):1266–70. [PubMed] [Google Scholar]
- 11.Pflugfelder SC, et al. Matrix metalloproteinase-9 knockout confers resistance to corneal epithelial barrier disruption in experimental dry eye. Am J Pathol. 2005;166(1):61–71. doi: 10.1016/S0002-9440(10)62232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fini ME, et al. The rabbit gene for 92-kDa matrix metalloproteinase. Role of AP1 and AP2 in cell type-specific transcription. J Biol Chem. 1994;269(46):28620–8. [PubMed] [Google Scholar]
- 13.Mohan R, et al. Gelatinase B/lacZ transgenic mice, a model for mapping gelatinase B expression during developmental and injury-related tissue remodeling. J Biol Chem. 1998;273(40):25903–14. doi: 10.1074/jbc.273.40.25903. [DOI] [PubMed] [Google Scholar]
- 14.Li DQ, et al. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45(12):4302–11. doi: 10.1167/iovs.04-0299. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi H, et al. Matrix metalloproteinase activity is enhanced during corneal wound repair in high glucose condition. Curr Eye Res. 2000;21(2):608–15. [PubMed] [Google Scholar]
- 16.Bargagna-Mohan P, Strissel KJ, Fini ME. Regulation of gelatinase B production in corneal cells is independent of autocrine IL-1alpha. Invest Ophthalmol Vis Sci. 1999;40(3):784–9. [PubMed] [Google Scholar]
- 17.Fini ME, et al. Unique regulation of the matrix metalloproteinase, gelatinase B. Invest Ophthalmol Vis Sci. 1995;36(3):622–33. [PubMed] [Google Scholar]
- 18.Li DQ, et al. Regulation of MMP-9 production by human corneal epithelial cells. Exp Eye Res. 2001;73(4):449–59. doi: 10.1006/exer.2001.1054. [DOI] [PubMed] [Google Scholar]
- 19.Mohan R, et al. Curcuminoids inhibit the angiogenic response stimulated by fibroblast growth factor-2, including expression of matrix metalloproteinase gelatinase B. J Biol Chem. 2000;275(14):10405–12. doi: 10.1074/jbc.275.14.10405. [DOI] [PubMed] [Google Scholar]
- 20.Tao Y, Bazan HE, Bazan NG. Platelet-activating factor induces the expression of metalloproteinases-1 and -9, but not -2 or -3, in the corneal epithelium. Invest Ophthalmol Vis Sci. 1995;36(2):345–54. [PubMed] [Google Scholar]
- 21.Isnard N, Robert L, Renard G. Effect of sulfated GAGs on the expression and activation of MMP-2 and MMP-9 in corneal and dermal explant cultures. Cell Biol Int. 2003;27(9):779–84. doi: 10.1016/s1065-6995(03)00167-7. [DOI] [PubMed] [Google Scholar]
- 22.Azar DT, et al. Gelatinase B and A expression after laser in situ keratomileusis and photorefractive keratectomy. Arch Ophthalmol. 1998;116(9):1206–8. doi: 10.1001/archopht.116.9.1206. [DOI] [PubMed] [Google Scholar]
- 23.Fini ME, Girard MT. Expression of collagenolytic/gelatinolytic metalloproteinases by normal cornea. Invest Ophthalmol Vis Sci. 1990;31(9):1779–88. [PubMed] [Google Scholar]
- 24.Fini ME, Girard MT. The pattern of metalloproteinase expression by corneal fibroblasts is altered by passage in cell culture. J Cell Sci. 1990;97(Pt 2):373–83. doi: 10.1242/jcs.97.2.373. [DOI] [PubMed] [Google Scholar]
- 25.Strissel KJ, Rinehart WB, Fini ME. A corneal epithelial inhibitor of stromal cell collagenase synthesis identified as TGF-beta 2. Invest Ophthalmol Vis Sci. 1995;36(1):151–62. [PubMed] [Google Scholar]
- 26.Matsubara M, Zieske JD, Fini ME. Mechanism of basement membrane dissolution preceding corneal ulceration. Invest Ophthalmol Vis Sci. 1991;32(13):3221–37. [PubMed] [Google Scholar]
- 27.Matsubara M, et al. Differential roles for two gelatinolytic enzymes of the matrix metalloproteinase family in the remodelling cornea. Dev Biol. 1991;147(2):425–39. doi: 10.1016/0012-1606(91)90300-r. [DOI] [PubMed] [Google Scholar]
- 28.Sivak JM, et al. Pax-6 expression and activity are induced in the reepithelializing cornea and control activity of the transcriptional promoter for matrix metalloproteinase gelatinase B. Dev Biol. 2000;222(1):41–54. doi: 10.1006/dbio.2000.9694. [DOI] [PubMed] [Google Scholar]
- 29.Sivak JM, et al. Transcription Factors Pax6 and AP-2alpha Interact To Coordinate Corneal Epithelial Repair by Controlling Expression of Matrix Metalloproteinase Gelatinase B. Mol Cell Biol. 2004;24(1):245–57. doi: 10.1128/MCB.24.1.245-257.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura Y, et al. Ets-1 regulates TNF-alpha-induced matrix metalloproteinase-9 and tenascin expression in primary bronchial fibroblasts. J Immunol. 2004;172(3):1945–52. doi: 10.4049/jimmunol.172.3.1945. [DOI] [PubMed] [Google Scholar]
- 31.Lyons CA, et al. Regulation of matrix metalloproteinases (type IV collagenases) and their inhibitors in the virgin, timed pregnant, and postpartum rat uterus and cervix by prostaglandin E(2)-cyclic adenosine monophosphate. Am J Obstet Gynecol. 2002;187(1):202–8. doi: 10.1067/mob.2002.123543. [DOI] [PubMed] [Google Scholar]
- 32.Kitamura M, et al. Transfer of a mutated gene encoding active transforming growth factor-beta 1 suppresses mitogenesis and IL-1 response in the glomerulus. Kidney Int. 1995;48(6):1747–57. doi: 10.1038/ki.1995.473. [DOI] [PubMed] [Google Scholar]
- 33.LaGier AJ, et al. Inhibition of human corneal epithelial production of fibrotic mediator TGF-beta2 by basement membrane-like extracellular matrix. Invest Ophthalmol Vis Sci. 2007;48(3):1061–71. doi: 10.1167/iovs.06-0772. [DOI] [PubMed] [Google Scholar]
- 34.Zieske JD, et al. TGF-beta receptor types I and II are differentially expressed during corneal epithelial wound repair. Invest Ophthalmol Vis Sci. 2001;42(7):1465–71. [PubMed] [Google Scholar]
- 35.Strissel KJ, Rinehart WB, Fini ME. Regulation of paracrine cytokine balance controlling collagenase synthesis by corneal cells. Invest Ophthalmol Vis Sci. 1997;38(2):546–52. [PubMed] [Google Scholar]
- 36.Wu CY, et al. Involvement of p42/p44 MAPK, p38 MAPK, JNK and nuclear factor-kappa B in interleukin-1beta-induced matrix metalloproteinase-9 expression in rat brain astrocytes. J Neurochem. 2004;90(6):1477–88. doi: 10.1111/j.1471-4159.2004.02682.x. [DOI] [PubMed] [Google Scholar]
- 37.Ogawa K, et al. Suppression of matrix metalloproteinase-9 transcription by transforming growth factor-beta is mediated by a nuclear factor-kappaB site. Biochem J. 2004;381(Pt 2):413–22. doi: 10.1042/BJ20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshizaki T, et al. The expression of matrix metalloproteinase 9 is enhanced by Epstein-Barr virus latent membrane protein 1. Proc Natl Acad Sci U S A. 1998;95(7):3621–6. doi: 10.1073/pnas.95.7.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taheri F, Bazan HE. Platelet-activating factor overturns the transcriptional repressor disposition of Sp1 in the expression of MMP-9 in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2007;48(5):1931–41. doi: 10.1167/iovs.06-1008. [DOI] [PubMed] [Google Scholar]
- 40.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. Embo J. 2002;21(14):3749–59. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engel ME, et al. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274(52):37413–20. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 42.Lee MK, et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. Embo J. 2007;26(17):3957–67. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HS, et al. Doxycycline inhibits TGF-beta1-induced MMP-9 via Smad and MAPK pathways in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2005;46(3):840–8. doi: 10.1167/iovs.04-0929. [DOI] [PubMed] [Google Scholar]
- 44.Lu T, et al. Secreted transforming growth factor beta2 activates NF-kappaB, blocks apoptosis, and is essential for the survival of some tumor cells. Proc Natl Acad Sci U S A. 2004;101(18):7112–7. doi: 10.1073/pnas.0402048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gingery A, et al. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp Cell Res. 2008 doi: 10.1016/j.yexcr.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakin AV, et al. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275(47):36803–10. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 47.Piek E, et al. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112(Pt 24):4557–68. doi: 10.1242/jcs.112.24.4557. [DOI] [PubMed] [Google Scholar]
- 48.Ishinaga H, et al. TGF-beta induces p65 acetylation to enhance bacteria-induced NF-kappaB activation. Embo J. 2007;26(4):1150–62. doi: 10.1038/sj.emboj.7601546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakurai H, et al. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. J Biol Chem. 1999;274(15):10641–8. doi: 10.1074/jbc.274.15.10641. [DOI] [PubMed] [Google Scholar]
- 50.Brown RT, Ades IZ, Nordan RP. An acute phase response factor/NF-kappa B site downstream of the junB gene that mediates responsiveness to interleukin-6 in a murine plasmacytoma. J Biol Chem. 1995;270(52):31129–35. doi: 10.1074/jbc.270.52.31129. [DOI] [PubMed] [Google Scholar]
- 51.Fujioka S, et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol. 2004;24(17):7806–19. doi: 10.1128/MCB.24.17.7806-7819.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perkins ND, et al. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. Embo J. 1993;12(9):3551–8. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stein B, et al. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. Embo J. 1993;12(10):3879–91. doi: 10.1002/j.1460-2075.1993.tb06066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowe DL, Tsang KJ, Shemirani B. Jun N-terminal kinase 1 mediates transcriptional induction of matrix metalloproteinase 9 expression. Neoplasia. 2001;3(1):27–32. doi: 10.1038/sj.neo.7900135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon C, et al. The p38 SAPK pathway regulates the expression of the MMP-9 collagenase via AP-1-dependent promoter activation. Exp Cell Res. 2001;271(2):344–55. doi: 10.1006/excr.2001.5374. [DOI] [PubMed] [Google Scholar]
- 56.Spiegelman VS, et al. Induction of beta-transducin repeat-containing protein by JNK signaling and its role in the activation of NF-kappaB. J Biol Chem. 2001;276(29):27152–8. doi: 10.1074/jbc.M100031200. [DOI] [PubMed] [Google Scholar]
- 57.Hesselbrock DR, et al. PEA3, AP-1, and a unique repetitive sequence all are involved in transcriptional regulation of the breast cancer-associated gene, mammaglobin. Breast Cancer Res Treat. 2005;89(3):289–96. doi: 10.1007/s10549-004-2622-z. [DOI] [PubMed] [Google Scholar]
- 58.D'Orazio D, et al. Cooperation of two PEA3/AP1 sites in uPA gene induction by TPA and FGF-2. Gene. 1997;201(1-2):179–87. doi: 10.1016/s0378-1119(97)00445-9. [DOI] [PubMed] [Google Scholar]
- 59.Liberati NT, et al. Smads bind directly to the Jun family of AP-1 transcription factors. Proc Natl Acad Sci U S A. 1999;96(9):4844–9. doi: 10.1073/pnas.96.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jungert K, et al. Smad-Sp1 complexes mediate TGFbeta-induced early transcription of oncogenic Smad7 in pancreatic cancer cells. Carcinogenesis. 2006;27(12):2392–401. doi: 10.1093/carcin/bgl078. [DOI] [PubMed] [Google Scholar]
- 61.Yamaguchi K, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270(5244):2008–11. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 62.D'Addario M, Arora PD, McCulloch CA. Role of p38 in stress activation of Sp1. Gene. 2006;379:51–61. doi: 10.1016/j.gene.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 63.Pramanik R, et al. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 2003;278(7):4831–9. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- 64.Calleros L, et al. Low cell cholesterol levels increase NFkappaB activity through a p38 MAPK-dependent mechanism. Cell Signal. 2006;18(12):2292–301. doi: 10.1016/j.cellsig.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 66.Luo L, et al. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Invest Ophthalmol Vis Sci. 2004;45(12):4293–301. doi: 10.1167/iovs.03-1145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.