

Abstract
A series of Cu(I)-amido complexes both lacking ancillary ligands and containing 1,10-phenanthroline (phen) as ligand have been prepared. These complexes react with iodoarenes to form arylamine products, and these data are consistent with the intermediacy of such complexes in catalytic Ullmann amination reactions. The stoichiometric reactions of the Cu(I)-amido complexes with iodoarenes are autocatalytic, with the free CuI generated during the reaction serving as the catalyst. Such autocatalytic behavior was not observed for reactions of iodoarenes with Cu(I)-amidates, -imidates and -phenoxides. The selectivity of these complexes for two sterically distinct aryl halides under various conditions imply that the autocatalytic reaction proceeds by forming highly-reactive [CuNPh2]n lacking phen. Reactions with radical probes imply that the reactions of phen-ligated Cu(I)-amido complexes with iodoarenes occur without the intermediacy of aryl radicals. DFT calculations of the oxidative addition of iodoarenes to Cu(I) species are consistent with faster reactions of iodoarenes with CuNPh2 species lacking phen in DMSO than with phen-ligated LCuNPh2. The free energy barrier computed for reaction of PhI with (DMSO)CuNPh2 was 21.8 kcal/mol, while the free energy barrier computed for reaction of PhI with (phen)CuNPh2 was 33.4 kcal/mol.
A century has passed since the discovery of Ullmann reactions in which arylamines couple with aryl halides mediated by copper.1 The synthetic scope of this process remained limited for many decades and often required stoichiometric amounts of copper and high reaction temperatures (typically 200 °C) to obtain satisfactory yields. More recently, copper catalysts containing ancillary ligands have been used, and these systems react with broader scope and at lower temperatures.2 In 1999, Goodbrand and Hu reported the coupling of arylamines with aryl iodides catalyzed by a combination of 1,10-phenanthroline (phen) and CuCl under mild conditions.3 Thereafter, a variety of ligands, such as 1,2-diamines,4 1,3-diketones,5 and others6 have been used with copper to promote the Ullmann amination reaction.
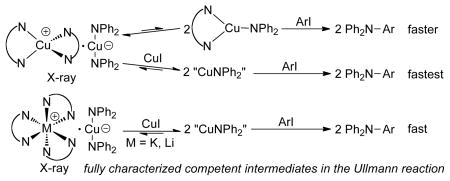
Despite significant improvements in the scope and conditions for the copper-catalyzed arylation of amines, little direct information is available on the mechanism of this reaction. Ligandless “CuNPh2” was proposed by Paine7 in 1987 to be an intermediate in Ullman reactions catalyzed by CuI,8 and tetramers of similar species [CuNR2]4 (R = alkyl) have been reported.9 Ligated Cu(I)-phenoxides, -amidates and -imidates have recently been characterized and shown to be likely intermediates in the Cu-catalyzed formation of biaryl ethers and N-aryl amides.10,11 However, Cu(I)-amido complexes (LCuNR2) containing a ligand that generates synthetically useful catalysts has not been characterized.12
Anionic copper(I) amides also could be intermediates in Ullman reactions. Organocuprates (MCuR2) are among the most reactive nucleophiles in organic chemistry,13 and similar cuprates have been proposed to form from the reaction of lithium amides with copper halides when the ratio of LiNR2 to CuX is greater than one14 (a condition that exists in the catalytic Ullmann reaction). However, amido cuprate complexes of the type MCu(NR2)2 have not been isolated, characterized or assessed as potential intermediates in the Ullmann aminations.
We report the synthesis and characterization of a series of Cu(I)-amido complexes, some lacking additional ligands and some ligated by phen. These complexes are chemically and kinetically competent to be intermediates in the Ullmann amination or lead to such intermediates. Under some conditions, these complexes react with iodoarenes by an autocatalytic process in which CuI serves as the catalyst. Our data imply that the autocatalysis occurs by generation of a highly-reactive [CuNPh2]n species lacking a phen ligand. Experimental and theoretical studies imply that these complexes react with iodoarenes without the intermediacy of aryl radicals and this result implies the involvement of Cu(III) intermediates.15,16
Our synthesis of Cu(I)-amido complexes is outlined in Scheme 1. Phen-ligated complex 1, and cuprates 2–4 in which phen and a crown ether ligate to K+ or Li+, were prepared by the reaction of CuOtBu with HNPh2 or a mixture of HNPh2 and either LiNPh2 or KNPh2 in the presence of phen or 18-crown-6 in THF. Cuprates 2 and 3 were also prepared from phen, LiNPh2 or KNPh2, and double salt 1 in THF. Finally, ligandless cuprate 5 was prepared from HNPh2, KNPh2 and CuOtBu in the absence of any dative ligand. These complexes were characterized by elemental analysis and NMR spectroscopy. Solid state structures of complexes 1 and 3 were determined by single crystal X-ray diffraction.
Scheme 1.

The solid state structure of complex 1 (Figure 1) is an ion pair consisting of a tetrahedral, cationic copper ligated by phen and a linear, anionic copper ligated by two amides. In DMSO, the molar conductivity of a 1.0 mM solution of 1 was high (16.9 Ω−1 cm2 mol−1), relative to that of a 1.0 mM solution of neutral ferrocene (0.3 Ω−1 cm2 mol−1) and similar to that of a 1.0 mM solution of the ionic [Bu4N][BPh4] (23.5 Ω−1 cm2 mol−1). In THF, the conductivity of a 1.0 mM solution of 1 was 13.7 μΩ−1 cm−1 (13.7 Ω−1 cm2 mol−1), whereas the conductivity of a 65.5 mM solution of [(n-octyl) 4N][Br] was 65.1 μΩ−1 cm−1 (0.99 Ω−1 cm2 mol−1) and that of a 65.5 mM solution of ferrocene was 0.0 μΩ−1 cm−1 (0.0 Ω−1 cm2 mol−1). These data indicate that, unlike ligated Cu(I)-amidates, -imidates and -phenoxides which vary in structure with the polarity of the solvent,10 ligated Cu(I)-amido complexes are predominantly double salts in both polar and less polar solvents.
Figure 1.

(a) ORTEP drawings of 1 (a) and 3 (b) with 50% ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg) for 1 (a): Cu2-N5, 1.8659(15); N5-Cu2-N5, 180.0. Selected bond lengths (Å) and angles (deg) for 3 (b): Cu1-N1, 1.860(2); Cu1-N2, 1.879(2); N1-Cu1-N2, 176.45(10).
The solid state structure of complex 3 contains two potassium cations ligated by six phenanthrolines and two bis-amidocopper anions (Figure 1). Two of the six phen ligands in the dimer are shared by two potassium cations in which each nitrogen of the bridging ligand is coordinated to two potassium cations, such that each potassium cation is eight-coordinate.
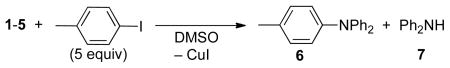
The reactions of these Cu(I)-amido complexes with iodoarenes are summarized in Table 1. Phen-ligated complex 1 reacted with p-iodotoluene in DMSO at 80 °C to afford triarylamine 6 in 95% yield after 1.5 h (Table 1). Cuprates 2 and 3, in which Li and K cations are ligated with phen, reacted similarly with p-iodotoluene in DMSO at 110 °C and 80 °C in 1 h and 3 h, respectively, to give one equivalent of 6 per copper in 84% and 90% yields. The second amido group in 2 and 3 forms diphenylamine, presumably by abstracting a proton from DMSO. Cuprates 4 and 5, which do not contain ancillary ligands to bind copper, did not react with p-iodotoluene when heated at 80 °C for 3 h; at 110 °C they formed 6 in modest yields (54% and 44%). The combination of phen and complex 4 containing the crown ether and complex 5 containing no added ligand reacted with p-iodotoluene at 80 °C to give 6 in >95% and 56% yields, respectively. These results indicate that dative ligands, such as phen, can convert less reactive cuprates to a more reactive species, possibly by substitution of the phen for one of the two amido ligands to form a ligated copper amide.
Table 1.
Reactivity of complexes 1–5 with p-iodotoluene.
 | ||||||
|---|---|---|---|---|---|---|
| entry | complex | additive | temp (°C) | time (h) | % yield 6a | 7/1–5b |
| 1 | 1 | none | 80 | 1.5 | 95 | - |
| 2 | 2 | none | 110 | 1 | 84 | 1.14c |
| 3 | 3 | none | 80 | 3 | 90 | 0.62 |
| 4 | 4 | none | 110 | 3 | 54 | 1.24c |
| 5 | 4 | phen (1 equiv) | 80 | 2.5 | >95 | 0.80 |
| 6 | 5 | none | 110 | 3 | 44 | 1.48c |
| 7 | 5 | phen (3 equiv) | 80 | 3 | 56 | 1.24c |
Yield by GC with an internal standard;
Molar ratio of amine 7 formed, relative to the starting complex 1–5. The balanced equation for reactions of 2–5 requires a proton from solvent or adventitious water;
Each of complexes 1–5 contains two amido groups. In the balance equation, one amido group forms the triarylamine and one forms diarylamine. Some diarylamine is apparently formed in competition with the process that forms triarylamine, leading to a ratio that is greater than one.
Although phen-ligated double salt 1 and cuprates 2 and 3 react with aryl halides to form triarylamines, monitoring of these reactions by 1H NMR spectroscopy shows that the events during this process are complex. Figure 2 shows the profile of the decay of 1 from its reaction with 4-iodotoluene. This sigmoidal behavior is characteristic of an autocatalytic process. The normalized concentration vs time plots overlap when the concentration of iodoarene is doubled, indicating that this process is first order in iodoarene.
Figure 2.

a. Decay of complex 1 from reaction with p-iodotoluene alone (red), with added CuI (blue) or with added [phenCuI]2 (green) at 60 °C. b. Decay of complex 1 from reaction with p-iodotoluene alone (red), with 0.010 M (blue) or 0.020 M (green) 1,10-phenanthroline at 80 °C.
Further experiments showed that the autocatalysis results from the generation of CuI. The reaction of 1 with p-iodotoluene conducted with 0.5 equiv of added CuI was faster and occurred to form triarylamine 6 with a first order decay of 1 (Figure 2a). In contrast, this was unaffected by added ligated copper iodide [phenCuI]2. In the presence of 1 equiv of CuI, the reaction occurred at 30 °C with a first order decay and a half-life (t1/2) of 37 min (kobs = 3.1 × 10−4 s−1); no reaction of complex 1 with p-iodotoluene occurred at this temperature in the absence of added CuI. The reaction of 1 with p-iodotoluene in the presence of CuI was first order in added CuI and in iodoarene.
The effect of added phen on the reaction of complex 1 with p-iodotoluene was not straightforward. The reaction of complex 1 with p-iodotoluene in the presence of added phen was slower than that in the absence of added phen, but the reactions with 0.010 M and 0.020 M added phen occurred with identical rates (Figure 2b). The reaction at 80 °C with 0.010 M added phen was first order in 1, with a half-life (t1/2) of 32 min (kobs = 3.6 × 10−4 s−1). Thus, the slower rate with added phen does not result from reversible dissociation of phen to generate the active species, as is often concluded when a reduction in rate is observed with added ligand. Instead, these data imply that the added phen consumes the free CuI that is generated in the absence of added phen and responsible for the autocatalysis and that the reaction occurs through the pathway lacking the autocatalysis in the presence of added phen. Reactions of 1 with varied concentrations of iodoarenes and added phen showed that the reaction was first order in this reagent.
Similar autocatalysis was observed for reactions of other copper amides. The reaction of cuprates 2 and 3 with p-iodotoluene occurred with sigmoidal reaction profiles (see Supporting Information), and the same reactions with added CuI were faster and occurred with an exponential decay. The reaction of 2 with 1 equiv of CuI occurred with t1/2 = 9.1 min (kobs = 1.3 × 10−3 s−1) at 100 °C, and the reaction of 3 with 2 equiv of CuI occurred with t1/2 = 20.6 min (kobs = 5.6 × 10−4 s−1) at 80 °C.
To assess the relationship between complexes 1–5 and intermediates in Ullmann reactions catalyzed by Cu(I)-phen species and to determine if the intermediates in the autocatalytic reactions are the same as those in reactions when free CuI is sequestered by added phen, we studied the selectivity of these complexes toward the sterically distinct o- and p-iodotoluenes. We then compared the distribution of products from these stoichiometric reactions in presence and absence of phen to those of catalytic reactions.
Results from these competitive reactions are provided in Table 2. The reaction of HNPh2 with o- and p-iodotoluenes catalyzed by a 1:1 ratio of CuI to phen in the presence of K3PO4 formed a 19:81 ratio of 6 to 8, and the reaction of KNPh2 with o- and p-iodotoluenes in presence of stoichiometric amounts of a 1:1 ratio of CuI and phen formed an indistinguishable 20:80 ratio of these products (eq 1 and 2). A similar selectivity (14:86) was also observed at the early stage of the reaction of complex 1 with o- and p-iodotoluenes (2 min, 11% yield) when the concentration of free CuI was low and the autocatalytic pathway was not yet dominant. In contrast, the reaction of 1 with o- and p-iodotoluenes in the presence of CuI formed nearly equal amounts of the two amine products (49:51). This selectivity is similar to that of the reaction of KNPh2 with stoichiometric CuI and no added phen (47:53).
Table 2.
Selectivity of complexes 1–5 for o- and p-iodotoluenes.
 | |||||
|---|---|---|---|---|---|
| entry | complex | additive | temp (°C) | yield (%)a | product ratio (8:6) |
| 1 | 1 | none | 80 °C | 85 | 34:66 |
| 2 | 1 | CuI (1 equiv) | 30 °C | 72 | 49:51 |
| 3 | 1 | phen (1 equiv) | 80 °C | 72b | 20:80 |
| 4 | 1 | none | 80 °C | 11c | 14:86c |
| 5 | 2 | none | 100 °C | 82 | 15:85 |
| 6 | 2 | CuI (4 equiv) | 80 °C | 77 | 43:57 |
| 7 | 3 | none | 80 °C | 92 | 10:90 |
| 8 | 3 | CuI (4 equiv) | 80 °C | 84 | 43:57 |
| 9 | 4 | CuI (1 equiv) | 80 °C | 89 | 47:53 |
| 10 | 5 | CuI (1 equiv) | 80 °C | 80 | 47:53 |
The ratio of products from reaction of complex 1 with o- and p-iodotoluenes and no added phen (34:66) was between that of the reaction of HNPh2 with o- and p-iodotoluenes catalyzed by a 1:1 ratio of CuI to phen in the presence of K3PO4 base and the reaction of 1 and added CuI with the iodoarenes. However, the ratio of products from the reaction of 1 with o- and p-iodotoluenes in the presence of 1 equiv of phen to sequester free CuI formed a ratio (20:80) of the two products that is similar to that of catalytic reactions with added phen and the reaction of complex 1 before autocatalysis. Thus, we conclude that the reaction of 1 in the presence of phen occurs through complex 1 or, more likely, the neutral form of 1 [Cu(phen)(NPh2)], but the portion of the reaction of 1 with aryl iodide without added phen that is autocatalytic in CuI likely proceeds by the transfer of an amido group from the Cu(NPh2)2 − unit to CuI to generate a [CuNPh2]n species lacking phen (Scheme 2).17 This species is likely less selective than 1 because it contains a more open coordination sphere.
Scheme 2.

 |
(1) |
 |
(2) |
The reactions of cuprates 2 and 3, which contain phen-ligated alkali metal cations, and the reactions of cuprates 4 and 5, which lack ancillary ligands capable of binding Cu(I), with o- and p-iodotoluenes in the presence of CuI were equally unselective. Thus, the reactions of these cuprates in the presence of CuI generate the same species that we propose to be [CuNPh2]n.18,19
To probe the potential intermediacy of free aryl radicals during these reactions, we have conducted the reaction of complex 1 with o-(allyloxy)iodobenzene 9 in the presence and absence of phen. The aryl radical from this iodoarene is known to undergo cyclization to form the methyl radical 11 with kobs = 9.6 × 109 s−1 in DMSO with subsequent formation of 12.20 Reaction of complex 1 with this arene formed amine 10 in 16% yield with added phen and 60% yield without added phen. No detectable amount of cyclized 12 was observed by GC-MS in either reaction mixture (eq 3). Because our detection limit for 12 is 0.1% of the amount of 10, recombination would need to be >103 faster than cyclization. Thus, any free aryl radical must react with the copper(II) amide21 to form the arylamine with a pseudo first-order rate constant greater than 1013 s−1,22 which is the lifetime of a transition state.23 Clearly, a bimolecular trapping of two reactive intermediates present in low concentrations is unlikely to occur on this timescale. Thus, the low selectivity from reaction without added phen and added CuI is unlikely to result from a pathway involving free aryl radicals and more likely from an open coordination sphere without a bidentate dative ligand.
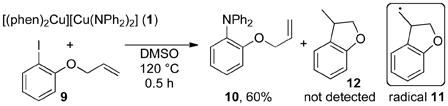 |
(3) |
Finally, to assess whether a copper amide lacking the phen ligand would be expected to add iodoarenes faster or slower than [(phen)Cu(I)(NPh2)], we calculated with DFT the barriers for oxidative addition of iodobenzene to [(phen)Cu(I)(NPh2)] and [(DMSO)Cu(I)NPh2]. 24,25 The formation of the Cu(III) species [(DMSO)Cu(NPh2)(Ph)(I)] lacking a dative ligand is thermodynamically uphill of the combination of (DMSO)Cu(I)(NPh2) and PhI by 17.1 kcal/mol and is formed with a ΔG‡ of 21.8 kcal/mol at 25 °C.26 This barrier is much lower than that for formation of [(phen)Cu(III)(NPh2)(Ar)(I)], which was computed to be 33.4 kcal/mol (see Supporting Information for details). These calculated barriers for oxidative addition are consistent with our proposal that the CuNPh2 species lacking phen in DMSO is more reactive than the phen-ligated LCuNPh2 species and that the species lacking phen reacts with aryl halides when CuI is added.
Some authors have proposed that the mechanism of Cu(I)-catalyzed C–N and C-O coupling reactions of haloarenes occur by radical pathways7,27 and others have proposed that they occur by non-radical pathways28 involving oxidative addition to form a Cu(III) intermediate (Scheme 3).25,29 The radical mechanism is proposed to be triggered by electron transfer from copper to the haloarene to form a haloarene radical anion, which eliminates the halide to form a neutral aryl radical. The aryl radical would then combine with Cu(II)-amide21 to afford the arylated amine and regenerate the Cu(I) catalyst.
Scheme 3.

Previous theoretical studies and reactions of radical probes with isolated Cu(I)-amidate, -imidate and -phenoxide complexes provided evidence against the involvement of aryl radicals in Ullmann-type reactions.10 This work provided some evidence for arylcopper(III) intermediates, and arylcopper(III) species have now been isolated.15 However, predicted barriers from Marcus theory using energies of reactants and products calculated by DFT led to the conclusion that the copper-catalyzed coupling of iodobenzene with methylamine, as a representative alkylamine, occurs by a radical path22 and that the Cu(III) species [LCu(III)(NHMe)(Ph)(I)] lies at too high an energy to be an intermediate. Our results from the reactions of isolated amidocopper(I) intermediates with the radical probe 9 suggest that the Cu(I)-catalyzed Ullmann reactions of diarylamines with iodoarenes do not proceed through free radial intermediates.30
We propose that the Ullmann reaction catalyzed by phenligated Cu(I) occurs by the pathway in Scheme 4. By this mechanism, the ligated CuI 13 is converted to the alkali metal cuprate 2 or 3 in the presence of excess amine and base. These cuprates then equilibrate with the neutral, monomeric form of 1, which undergoes oxidative addition of the iodoarene to form Cu(III) species 14. This Cu(III) species then reductively eliminates the amine product and regenerates 13. The autocatalysis by free CuI observed in the reactions of complexes 1–3 with iodoarenes would not be expected to occur as part of this cycle because the excess amine and base would convert free CuI to the lithium and potassium amido cuprates 2 and 3.
Scheme 4.

Proposed catalytic cycle without autocatalysis
In summary, we have prepared and characterized a series of amido cuprates, some having phen-ligated Cu(I) countercations, some with phen-ligated alkali metal cations, and some with alkali metal cations lacking bound phen. Free CuI accelerates the reactions of each of these complexes. Double salt 1 was more reactive with iodoarenes than were the alkali metal cuprates 2–5. The relative reactivities in the presence of added CuI were 1 ≫ 3 > 2. These rates relate to reactions of amines with aryl halides, base and stoichiometric copper during which CuI would be generated. In the absence of added CuI, the relative rates for reactions of the cuprates with iodotoluene were complicated by autocatalysis but followed the rough trend 1 > 3 > 1 + phen > 2 > 4 ~ 5. The low reactivity of cuprates 4 and 5, which lack phen, implies that the reactions of 2 and 3 occur through a phen-ligated Cu(I) species, such as [Cu(phen)-NPh2]. The inherent reactivity of [Cu(phen)NPh2] with aryl iodides was assessed by conducting the reactions in the presence of added phen to sequester CuI, and these reactions occurred in high yields at 80 °C. The reaction of 1 with a radical probe imply the reactions occur without Ph• intermediates, most likely through Cu(III) intermediates from oxidative addition of the iodoarene. DFT calculations further support the mechanism proceeding through Cu(III) intermediates since the formation of such Cu(III) species is predicted to occur with a low energy barrier.
Supplementary Material
Acknowledgments
We thank the NIH NIGMS (GM-55382) for support of this work.
Footnotes
Supporting Information Available: Experimental procedures, computational details and characterization of complexes. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Ullmann F. Ber Dtsch Chem Ges. 1903;36:2382. [Google Scholar]
- 2.(a) Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]; (b) Ley SV, Thomas AW. Angew Chem Intl Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 3.Goodbrand HB, Hu NX. J Org Chem. 1998;64:670. [Google Scholar]
- 4.Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 5.(a) de Lange B, Lambers-Verstappen MH, Schmieder-van de Vondervoort L, Sereinig N, de Rijk R, de Vries AHM, de Vries JG. Synlett. 2006:3105. [Google Scholar]; (b) Shafir A, Buchwald SL. J Am Chem Soc. 2006;128:8742. doi: 10.1021/ja063063b. [DOI] [PubMed] [Google Scholar]
- 6.Kwong FY, Buchwald SL. Org Lett. 2003;5:793. doi: 10.1021/ol0273396. [DOI] [PubMed] [Google Scholar]; Kwong FY, Klapars A, Buchwald SL. Org Lett. 2002;4:581. doi: 10.1021/ol0171867. [DOI] [PubMed] [Google Scholar]
- 7.Paine AJ. J Am Chem Soc. 1987;109:1496. [Google Scholar]
- 8.(a) Manifar T, Rohani S, Bender TP, Goodbrand HB, Gaynor R, Saban M. Ind Eng Chem Res. 2005;44:789. [Google Scholar]; (b) Bethell DJ, IL, Quan PM. J Chem Soc, Perkin Trans 2. 1985:1789. [Google Scholar]
- 9.(a) Gambarotta SBM, Floriani C, Chiesi-Villa A, Guastini C. J Chem Soc, Dalton Trans. 1987:1883. [Google Scholar]; (b) Tsuda T, Watanabe K, Miyata K, Yamamoto H, Saegusa T. Inorg Chem. 1981;20:2728. [Google Scholar]; (c) Hope H, Power PP. Inorg Chem. 1984;23:936. [Google Scholar]
- 10.(a) Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2008;130:9971. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tye Jesse W, Weng Z, Giri R, Hartwig John F. Angew Chem Intl Ed. 2010;49:2185. doi: 10.1002/anie.200902245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Strieter ER, Bhayana B, Buchwald SL. J Am Chem Soc. 2008;131:78. doi: 10.1021/ja0781893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Strieter ER, Blackmond DG, Buchwald SL. J Am Chem Soc. 2005;127:4120. doi: 10.1021/ja050120c. [DOI] [PubMed] [Google Scholar]
- 12.Blue ED, Davis A, Conner D, Gunnoe TB, Boyle PD, White PS. J Am Chem Soc. 2003;125:9435. doi: 10.1021/ja0353659. [DOI] [PubMed] [Google Scholar]
- 13.Krause N, Gerold A. Angew Chem Intl Ed. 1997;36:186. [Google Scholar]
- 14.Tsuda T, Miwa M, Saegusa T. J Org Chem. 1979;44:3734. [Google Scholar]
- 15.(a) Xifra R, Ribas X, Llobet A, Poater A, Duran M, Sola M, Stack TDP, Benet-Buchholz J, Donnadieu B, Mahia J, Parella T. Chem Eur J. 2005;11:5146. doi: 10.1002/chem.200500088. [DOI] [PubMed] [Google Scholar]; (b) Ribas X, Jackson DA, Donnadieu B, Mahía J, Parella T, Xifra R, Hedman B, Hodgson KO, Llobet A, Stack TDP. Angew Chem Intl Ed. 2002;41:2991. doi: 10.1002/1521-3773(20020816)41:16<2991::AID-ANIE2991>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 16.(a) Huffman LM, Stahl SS. J Am Chem Soc. 2008;130:9196. doi: 10.1021/ja802123p. [DOI] [PubMed] [Google Scholar]; (b) King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. J Am Chem Soc. 2010;132:12068. doi: 10.1021/ja1045378. [DOI] [PubMed] [Google Scholar]
- 17.A referee asked if [CuNAr2]n could be directly observed in this system. The 19F NMR spectrum of the reaction of a p-fluoro analog of complex 1 with CuI in DMF-d7 at −50 °C contained a new signal, but the identity of the new species could not be determined unambigously.
- 18.This complex likely contains DMSO solvent as a ligand. Reactions in tBuS(O)Me occur in lower yield than those in DMSO, suggesting that coordination of DMSO controls the reactivity of the unsaturated copper amide.
- 19.Tetrameric CuNR2 complexes in which R = alkyl groups are known (see ref 9), and we found that they do not react with iodoarenes. The CuNPh2 species generated in situ would have weaker bridging ligands and would be generated initially as a lower-coordinate species.
- 20.Annunziata A, Galli C, Marinelli M, Pau T. Eur J Org Chem. 2001;2001:1323. [Google Scholar]
- 21.(a) Melzer M, Mossin S, Dai X, Bartell A, Kapoor P, Meyer K, Warren T. Angew Chem Intl Ed. 2010;49:904. doi: 10.1002/anie.200905171. [DOI] [PubMed] [Google Scholar]; (b) Mankad NP, Antholine WE, Szilagyi RK, Peters JC. J Am Chem Soc. 2009;131:3878. doi: 10.1021/ja809834k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones GO, Liu P, Houk KN, Buchwald SL. J Am Chem Soc. 2010;132:6205. doi: 10.1021/ja100739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newcomb M, Le Tadic MH, Putt DA, Hollenberg PF. J Am Chem Soc. 1995;117:3312. [Google Scholar]
- 24.Previous DFT calculations have shown that the barrier for oxidative addition of ArI to anionic copper [Cu(N(phth)2]− and [Cu(NHAc)2]− is higher than that for oxidative addition of ArI to three-coordinate L2Cu(Nphth) and L2Cu(NHAc). See ref 10a and 25.
- 25.Zhang SL, Liu L, Fu Y, Guo QX. Organometallics. 2007;26:4546. [Google Scholar]
- 26.DFT calculations suggested that both Cu(I) and Cu(III) species, [Cu(I)(NPh2)] and [Cu(III)(NPh2)(Ph)(I)], bind a single DMSO though oxygen.
- 27.(a) Aalten HL, Vankoten G, Grove DM, Kuilman T, Piekstra OG, Hulshof LA, Sheldon RA. Tetrahedron. 1989;45:5565. [Google Scholar]; (b) Couture C, Paine AJ. Can J Chem. 1985;63:111. [Google Scholar]; (c) Arai S, Hida M, Yamagishi T. Bull Chem Soc Jpn. 1978;51:277. [Google Scholar]
- 28.Weingarten H. J Org Chem. 1964;29:3624. [Google Scholar]; Lindley J. Tetrahedron. 1984;40:1433. [Google Scholar]
- 29.Bethell D, Jenkins IL, Quan PM. J Chem Soc Perkin Trans 2. 1985:1789. [Google Scholar]; Cohen T, Cristea I. J Am Chem Soc. 1976;98:748. [Google Scholar]
- 30.Because LCuNHMe is more electron-rich than LCuNPh2, LCuNHMe could be more prone to engage in single electron transfer processes.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.