

Abstract
Wnt-signalling plays an important role in Wilms tumorigenesis. Upon activation, intracellular signal transduction results in stabilization, accumulation and nuclear translocation of β-catenin. Nuclear β-Catenin then acts in conjunction with members of the TCF/Lef family to cause transcriptional upregulation of specific proliferation-associated target genes such as c-myc or cyclin D1. Constitutive activation of β-Catenin through mutations in CTNNB1 has been found in about 15% of Wilms tumors. Nuclear β-catenin protein has been detected by immunohistochemistry in an even higher proportion of Wilms tumors suggesting alternative genetic pathways leading to β-Catenin activation.
Nephroblastomas induced in rats by either N-ethylnitrosourea or methyl(methoxymethyl)nitrosamine are histologically similar to Wilms tumors and provide a valuable rodent model. To study the involvement of the wnt-signalling pathway in rat nephroblastomas we examined 25 chemically induced rat nephroblastomas for nuclear accumulation of β-Catenin protein and for mutations in Ctnnb1. 16 of 25 tumors showed nuclear accumulation of immunoreactive β-catenin protein although no mutation was found in any of the tumors analyzed. These findings support the idea that active wnt-signalling contributes to tumorigenesis in carcinogen-induced nephroblastomas.
Introduction
Wilms tumor is the most common pediatric kidney cancer and among the most common childhood malignancies in Europe and North America [1;2]. One of 10000 children is affected. Most Wilms tumors occur sporadically and are unilateral, but there are also a few cases (1%) with a family history that are often bilateral and manifest earlier [3–5]. Chemotherapy, surgery and radiation in advanced stages of disease are the current therapeutic strategies and result in overall cure of about 85% of patients. However, acute toxicity, such as growth inhibition of haematopoietic, intestinal and germ cells, as well as unwanted long-term side effects, like secondary tumors, scoliosis and others, still warrant the development of novel improved therapeutical approaches.
Wilms tumors are thought to arise from metanephric blastemal cells that normally differentiate into the epithelial components of the mature nephron. Wilms tumors recapitulate metanephrogenesis and have a histological architecture showing varying proportions of epithelial, mesenchymal and blastemal components. Some Wilms tumors show heterologous cartilaginous or rhabdomyoblastoid differentiation. Only little is known about the genetic basis of Wilms tumor development. Loss of heterozygosity studies implicated several tumor suppressor genes, which have been mapped to chromosomes 11p13, 11p15, 16q and 1p [6–9].
One of these, the Wilms tumor suppressor gene WT1, has been molecularly characterized. WT1 has been mapped to chromosome 11p13 [10;11]. It encodes a zinc-finger protein capable of binding DNA as a transcription factor [12]. WT1 regulates the expression of several important target genes like E-cadherin, insulin-like-growth-factor-2 and insulin-like-growth-factor-1 receptor. Up to 25% of human nephroblastomas may have WT1 inactivating mutations [13–15].
In about 15% of Wilms tumors, β-catenin mutations have been found and a linkage between β-catenin mutations and WT1 mutations has been observed [16;17]. β-catenin is the main effector molecule of the wnt-signalling pathway and, in the absence of wnt ligands, is being phosphorylated by glycogen synthase kinase 3β, APC and axin which leads to β-catenin’s rapid ubiquitin-mediated degradation [18–20]. Activation of the wnt-signalling pathway is initiated by binding of wnt proteins to their receptors on the cell surface, which trigger the intracellular inhibition of β-catenin phosphorylation. This in turn leads to accumulation and nuclear translocation of β-Catenin protein where it interacts with members of the Lef-1/TCF family. As heterodimeric transcription factors they cause transcriptional upregulation of specific target genes, e.g. cyclin D1 or c-myc [21;22].
Besides Wilms tumors, activating mutations of β-catenin have been reported in several human childhood malignancies like hepato-, medullo- and pancreatoblastoma [23]. Also other human tumors such as colon, brain, prostate and skin cancer are containing such mutations [24–27]. Since then, the wnt-signalling pathway has become the focus of novel molecular therapeutic targets. Drugs like non-steroidal anti-inflammatory drugs (NSAIDs), including selective cyclooxygenase (COX)-2 inhibitors, recombinant biomolecules and virus-mediated tumor cell killing represent promising new strategies [28–34]. Some of these therapies may eventually turn out to be helpful in the treatment of Wilms tumors. Translational research in Wilms tumors is, however, hampered by the fact that the incidence of Wilms tumors is relatively low. Therefore, it is difficult and very time-consuming to test the efficacy of novel therapies in Wilms tumor patients. Preclinical animal or in vitro models could in principal substitute for larger clinical studies but they are lacking. To date there are no transgenic mouse models and no established Wilms tumor cell lines available for preclinical research.
The rat offers unique opportunities in the study of nephroblastoma. Rat strains such as Noble [35] and Sprague-Dawley [36], are predisposed to spontaneous nephroblastoma development and can be induced with direct alkylating agents, e.g., N-ethylnitrosourea (ENU) to form nephroblastomas at high incidence with transplacental exposures [37;38]. These neoplasms show a triphasic histotype typical of Wilms tumors with favorable histology. Other carcinogens, e.g., the nitrosamines, induce primarily stromal tumors, that is, renal mesenchymal tumors [37], which resemble congenital mesoblastic nephroma. These variations allow for unparalled opportunities for comparative studies into mechanisms of renal tumorigenesis. The aim of our study was therefore to characterize the only available rodent model, chemically-induced rat nephroblastomas, for alterations affecting the wnt-signalling pathway.
Materials and methods
Tumor samples
All tumors were histologically classified after standard hematoxylin and eosin (HE) staining of 2 μm thick sections.
Tumors induced with N-ethylnitrosourea
Male and female Noble rats were obtained from Charles River (Kingston, NY) at 6–8 wk of age. Dr. G. Muschik (Chemical Synthesis and Analysis Laboratory, Sciences Applications International Corporation, Frederick Cancer Research and Development Center, Frederick, MD) synthesized the N-ethylnitrosourea (ENU), which was stored at −20°C and was dissolved and diluted in saline just before use. Female and male Noble rats were paired overnight, and the females were examined the next morning (O-dpc) for vaginal plugs. Pregnancy was confirmed by palpation 10 d later. At 17 d post coitum, the pregnant rats received an intraperitoneal injection of 0.5 mmol/kg body weight ENU. They were allowed to deliver normally and to nurse their offspring, which were weaned at 4 wk of age. Rats were euthanized when tumors became palpable or the animals became moribund. Kidneys were either snap-frozen in liquid nitrogen or fixed in 10% buffered formalin and embedded in paraffin.
Tumors induced with Methyl(methoxymethyl)nitrosamine
Methyl(methoxymethyl)nitrosamine (DMN-OMe) was synthesized according to the method of Eiter et al. [39]. Neonatal F344/NCr rats received a single intraperitoneal injection of DMN-OMe (4.0 mmol/kg of body weight) in phosphate-buffered saline (pH 7.0) within 48 h after birth. Large palpable renal masses were excised from euthanized animals and fixed for histology or frozen at −70 C for DNA extraction.
Immunohistochemistry
Immunostaining with antibodies against β-catenin was performed on paraffin embedded tissue (2 μm thick sections). Antigen retrieval was achieved by microwave twice in 10mM citrate buffer (pH 6) for 5 min. Endogenous peroxidase was quenched with methanol and H2O2. Primary β-catenin antibody (C19220, Transduction Laboratories, Lexington, KY, USA) was diluted 1:200 and incubated over night (4°C). A standard peroxidase-antiperoxidase protocol was used for visualization, with diaminobenzidine as chromogen.
Cell culture
Cells used in the experiments were CC531, ENU-T1, HCT116 and 293T cells. HCT116 and 293T cells were kindly provided by Dr. S. Dihlmann, University of Heidelberg, Germany. The ENU-T1 cells were provided by Y. Nagashima, Department of Pathology, Yokohama City University, Yokohama, Japan. All cells were cultured under sterile condition in RPMI 1640 medium supplied with 10 % (v/v) fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5 % CO2. All cell culture reagents by Gibco BRL, Karlsruhe, Germany.
TOPflash/FOPflash transfection assays
For the TOPflash/FOPflash-reporter assays (Upstate Biotechnology, Lake Placid, NY, USA), 5 × 105 cells were seeded per well the day before transfection. Transfection was performed with QIAGEN Effectene Transfection Kit. Cells were transfected with 1 μg vector DNA (pTOPflash or pFOPflash) and 0,2 μg pRSV-lacZ as normalization vector [40]. As control, HCT116 and 293T cell lines were used. After 24h incubation, cells were harvested and lysed for standard luciferase assay (Promega GmbH, Mannheim, Germany) measured on a Sirius Luminometer (V2.2). For normalization, a standard galactosidase assay (Promega GmbH, Mannheim, Germany) was used. All transient transfection experiments were done in triplicate and repeated twice. To normalize the data of the different cell lines, the relative light counts per second (RLU/s) measured in the luminometer were correlated with the absorption measured with the galactosidase assay at 405 nm.
To get the fold activation the arithmetic mean was calculated of all the transfection data and TOPflash mean was divided through FOPflash mean. Standard deviation (σ) was calculated from the mean ratio of all transfections.
DNA purification
Genomic DNA was extracted from snap-frozen tumor tissue according to Sukumar et al. [41] or from paraffin-embedded tumor tissues after microdissection to remove contaminating normal tissue. Tumor tissue from three to five 6 μm thick HE stained sections was then directly transferred into 180 μl of tissue lysis buffer from the QIAmp DNA Mini Kit (Qiagen, Hilden, Germany) and incubated at 57°C over night. The procedure of DNA purification was according to the standard protocol supplied with the kit. The integrity of the purified DNA was checked on a 0,8% agarose gel. DNA of cell lines was purified according to the protocol supplied with the QIAmp DNA Mini Kit (Qiagen, Hilden, Germany).
PCR amplification
PCR and DNA sequencing was performed as described in Koesters et al. [42]: after DNA purification exon 3 of rat Ctnnb1 (β-catenin) was amplified by PCR using following primers: RNbcat-fwd1 (5′-GCTGACCTGATGGAGTTGGA-3′) and RNbcat-rev1 (5′-GCTACTTGCTCTTGCGTGAA-3′). Amplification was performed for 35 cycles in a reaction containing 2mM MgCl2 and 1U Platinum-Taq Polymerase (Life Technologies, Rockville, MD, USA). Cycling conditions were: initial denaturation for 5 min at 94°C, 35 cycles of denaturation for 30 s at 94°, 30 s annealing at 55°C and 30 s elongation at 72°C, followed by a final step of elongation for 7 min at 72°C. PCR fragments were purified using High Pure PCR Purification Kit (Roche Diagnostics, Basel, Switzerland), essentially as recommended by the manufacturer.
DNA sequencing
Sequencing reactions were set up using 30 ng of purified PCR product and 10 pmol of sequencing primer in a total reaction volume of 10 μl following a dye terminator protocol (Big Dye, Applied Biosystems, Darmstadt, Germany). The sequencing reactions were run on an ABI Prism 310 DNA Sequencer (Applied Biosystems). Sequencing primers were Rnbcat-fwd1 and Rnbcat-rev2 (5′-CTTGCTCCCACTCATAAAGG-3′). Cycling conditions were according to the dye terminator protocol with an annealing temperature of 52°C.
Results
In this study, we analysed a total number of 25 chemically-induced rat nephroblastomas. 18 of the analysed tumors were primary nephroblastomas (15 induced with ENU and 3 induced with DMN-OMe), whereas 7 of the analysed tumors were syngeneic transplants of ENU-induced primary nephroblastomas. All tumors were composed of blastemal, mesenchymal, and epithelial components as is typically seen in triphasic Wilms tumor. The different histologic sub-compartments were, however, unevenly distributed. Most of the tumors presented with a mainly undifferentiated blastemal component. Some of the tumors showed rhabdomyoblastoid differentiation, which is also commonly seen in Wilms tumors. One nephroblastoma showed diffuse anaplasia (table I).
Table I.
Immunohistology and mutational analysis of carcinogen induced rat nephroblastomas and mesenchymal tumors
| Sample no. | Carcinogen | Histology | Immuno-reactivity Score | DNA Sequencing Ctnnb1 exon 3 | |
|---|---|---|---|---|---|
| ca | /na | ||||
| normal rat kidney | |||||
| NK | none | normal rat kidney | + | − | wtd |
| primary nephroblastomas | |||||
| N1 | ENUb | Triphasic | ++ | + | wt |
| N2 | ENU | Triphasic, predominant blastemal | ++ | + | wt |
| N3 | ENU | Triphasic, predominant blastemal | + | + | wt |
| N4 | ENU | Triphasic, predominant blastemal | + | − | wt |
| N5 | ENU | Triphasic, predominant blastemal/stromal | ++ | − | wt |
| N6 | ENU | Triphasic, weak differentiated | + | + | wt |
| N7 | ENU | Triphasic, predominant blastemal | + | + | wt |
| N8e | ENU | Triphasic | ++ | − | wt |
| N9e | ENU | Triphasic | ++ | − | wt |
| N10e | ENU | Triphasic | + | − | wt |
| N11e | ENU | Triphasic, mainly blastemal | ++ | + | wt |
| N12e | ENU | Triphasic | + | + | wt |
| N13e | ENU | Triphasic | + | + | wt |
| N14e | ENU | Triphasic | + | − | wt |
| N15e | ENU | Triphasic | − | − | wt |
| N16 | DMN-OMec | Triphasic, predominant blastemal | + | + | wt |
| N17 | DMN-OMe | Triphasic, predominant stromal | ++ | + | wt |
| N18 | DMN-OMe | Triphasic, predominant stomal, anaplastic | ++ | + | wt |
| transplanted nephroblastomas | |||||
| HT1e | ENU | Triphasic, predominant blastemal with focal epithelium | ++ | + | wt |
| HT2e | ENU | Triphasic, predominant bastemal | ++ | + | wt |
| HT3e | ENU | Triphasic, predominant blastemal | + | + | wt |
| HT4e | ENU | Triphasic, predominant blastemal | + | − | wt |
| HT5e | ENU | Triphasic, predominant mesenchymal | − | + | wt |
| HT6e | ENU | Triphasic, predominant blastemal/stromal | − | + | wt |
| HT7e | ENU | Triphasic | + | − | wt |
| cell lines | |||||
| ENU-T1 | ENU | adherent cells | wt | ||
| CC531 | adherent cells | Codon 41, ACC→ATC Thr→Ile |
|||
c/n, cytoplasmatic/nuclear staining
ENU, transplacental ENU treated animals
DMN, neonatal DMN-Ome treated animals
wt, wild type
DNA extracted by microdissection
The β-catenin immunophenotype of all tumors and of normal rat kidney was assessed by immunohistochemistry.
β-catenin protein in normal rat kidney was mainly localized to tubular epithelial membranes. The strongest membranous β-catenin staining was found laterally and basally. Additionally, tubular epithelial cells showed also weak cytosolic β-catenin staining. Distal tubules always showed stronger cytosolic and membranous immunoreactivity than proximal tubules. Nuclear staining was never observed in normal rat kidney. All non-epithelial components of the kidney, like the capillary loops of the glomeruli or the stroma or interstitial cells never showed β-catenin protein expression (Fig 1 a).
Figure 1. Immunohistological expression of β-catenin in carcinogen induced rat nephroblastomas.
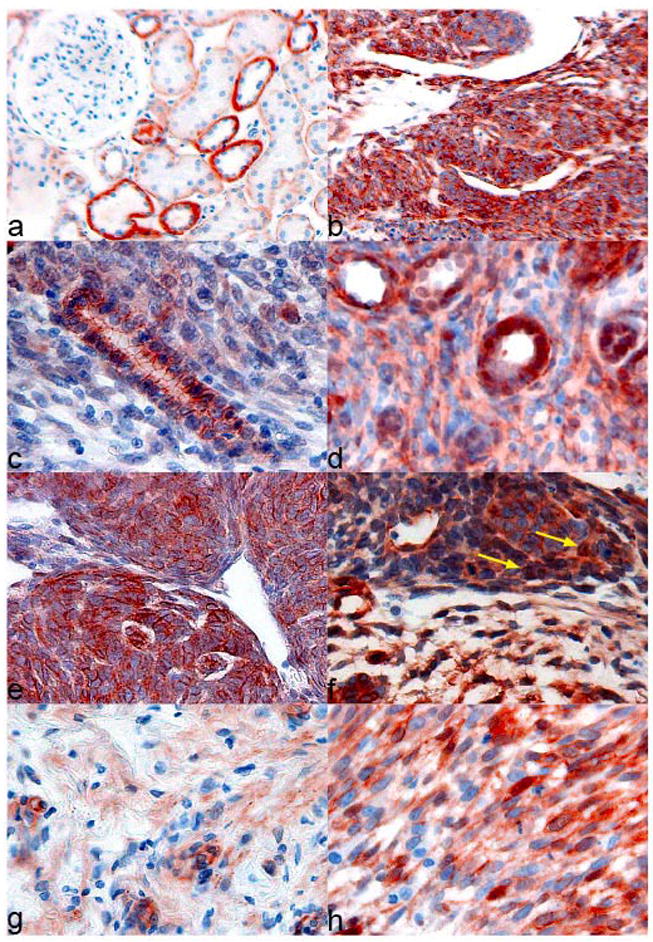
Normal rat kidney shows focal membranous expression on the lateral site of the tubular epithelial cells, distal tubules show stronger expression than proximal tubules, no nuclear expression can be found (a), tumor tissue (b) presented on the same slide often shows much more cytosolic β-Catenin expression; epithelial tumor cells without (c) and with nuclear expression of β-Catenin (d); blastemal component of a rat nephroblastoma with strong membranous and cytosolic expression (e), some blastemal components also reveal nuclear β-Catenin expression (f); mesenchymal differentiated cells with (h) and without (g) nuclear β-Catenin expression.
Since the intensity of the cytosolic β-catenin staining in normal rat kidney tissue always correlated well with the intensity of the membranous staining, both were combined in a cytosolic score of (+), rating both the staining intensity and the relative frequency (about 25 %) of the highly expressing distal tubular epithelial cells in normal rat kidney. Tumor tissue was scored as showing less (−), the same (+), or more (++) cytosolic β-catenin immunoreactivity than normal kidney, which in case of most primary tumors (n=15) was present within the same section. Normal kidney tissue from an untreated animal stained within the same series was used as reference for the remaining primary tumors and for the syngeneic transplants. Nuclear β-catenin staining was found at different frequencies and scored positve (+), when found in at least three cells per section. Normal kidney never showed any nuclear staining.
In the present study, 10 of 25 (40 %) rat nephroblastomas showed a much stronger cytoplasmatic β-catenin immunoreactivity than normal kidney and this was mostly due to the very strong staining of the blastematous sub-compartment in these tumors (Fig 1 b). 7 of these tumors also showed nuclear positvity. 12 of 25 (48%) tumors analyzed showed the same cytosolic β-catenin immunoreactivity as normal kidney and again 7 of these presented with additional nuclear positivity. Only three tumors showed less overall β-Catenin immunoreactivity than normal kidney, but two of these tumors presented with nuclear positive cells even though.
Together, nuclear staining of β-catenin was observed in addition to membranous and cytosolic localization in 16 of 25 (64,0 %) tumors analyzed, in 11 of 18 (61,1%) primary and in 5 of 7 (71,4%) transplanted tumors. The immunohistochemical results are summarized in table I.
As has been found in Wilms tumors [43], nuclear β-Catenin staining was not observed in every histological sub-compartment of a tumor when it showed nuclear accumulation in one compartment. Epithelial compartments that mimic normal tubular architecture showed a prominent membranous staining (Fig 1c). Nuclear β-catenin expression, however, was observed in epithelial tumor compartments in two cases (Fig 1 d). Cells of blastematous appearance showed mostly a membranous and cytoplasmatic staining (Fig 1 e), but some tumors displayed nuclear positivity also there (Fig 1 f). The most frequently found nuclear immunoreactivity was, however, in cells with mesenchymal differentiation, such as spindle or rhabdomyoblastoid cells. Although a few mesenchymal tumor sub-compartments were without any nuclear β-catenin immunoreactivity (Fig 1 g), the percentage of nuclear positive cells in mesenchymal/stromal areas (Fig 1 h) sometimes ranged up to 20% of the tumor cells (not shown). Similarly, a strong cytosolic staining was observed in mesenchymal sub-compartments (not shown).
The ENU-T1 cell line was derived from an ENU induced rat nephroblastoma by Nagashima et al [44]. In order to assay wnt-signalling activity in these cells, the TOP-/FOPflash reporter assay was used. The TOPflash plasmid contains a luciferase reporter gene under the control of a minimum promoter containing multiple TCF binding sites which become activated in response to Wnt/β-catenin pathway stimulation [45]. As a negative control pFOPflash was used which contains mutant instead of normal TCF-binding sites. pRSVlacZ was used for normalization [40]. As shown in figure 2, ENU-T1 cells showed lower activation of wnt signalling (TOPflash/FOPflash ratio 1,38 (σ: 0,47)) than 293T cells [46] which are known to have a low intrinsic wnt-activity (TOPflash/FOPflash ratio 1,79 (σ: 0,25)) whereas HCT116 cells, a human colorectal cancer cell line harbouring an oncogenic mutation of β-catenin [47] showed a nearly nine fold higher expression of TOPflash than of FOPflash (TOPflash/FOPflash ratio 8,86 (σ: 2,46)). These findings do not support the idea that wnt-signalling is significantly activated in ENU-T1 cells.
Figure 2. TOPflash/FOPflash ratio.
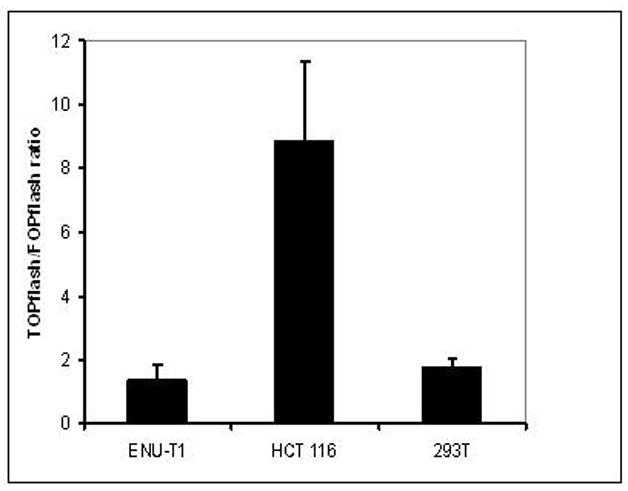
Expression ratio of the N-ethylnitrosourea generated ENU-T1 cell line after transfection with reporter plasmids pTOPflash and pFOPflash to examine the activation grade of wnt-signalling pathway in these cells. HCT116 cell line with activated wnt-signalling shows a nearly nine fold higher TOPflash tah FOPflash activity (TOP/FOP ratio 8,86; σ = 2,46). 293 T cells are presenting themselves with low activated wnt signalling pathway (TOP/FOP ratio 1,79; σ = 0,25). ENU-T1 cells are showing low, but not significant activation (TOP/FOP ratio 1,38; σ = 0,47).
Finally, genomic DNA from all tumors and from ENU-T1 cells was analyzed for mutations in exon 3 of the β-catenin gene Ctnnb1 by sequencing analysis. In 15 cases (see also table I) where there was no frozen tumor material available genomic DNA was extracted from paraffin-embedded, haematoxylin/eosin stained tissue after microdissection to remove contaminating normal kidney tissue.
For sequencing analysis of the region encoding the regulatory phosphorylation sites within the amino-terminus of β-catenin protein exon 3 of Ctnnb1 was amplified by PCR and subjected to direct sequencing. However, we did not find a single mutation in 25 of 25 samples analyzed. As a positive control, CC531 cells were analysed which harbors a known mutations in codon 41 (ACC→ATC) of Ctnnb1 [48]. Using direct sequencing analysis of PCR products this mutation was clearly identified in our study (not shown). The results of the sequencing analysis are included in table I.
Discussion
Aberrant activation of the wnt-signalling pathway results in stabilization and nuclear accumulation of β-catenin protein. This phenomenon has been recognized as an important oncogenic step in various human tumor entities. Because of its widespread involvement in human cancer the wnt-signalling pathway has become a major focus of novel targeted therapeutical strategies. Since a significant proportion of Wilms tumors bear mutations of CTNNB1 [16;17], or show an otherwise activated wnt-signalling pathway [23;43]. Any such novel targeted therapy may turn out to be of important future impact in Wilms Tumor patients. To test the efficacy of any novel therapy, however, appropriate preclinical model systems are required. In the present study, we have analyzed the only currently available animal model of Wilms tumors, chemically induced rat nephroblastomas, for alterations in the wnt-signalling pathway similar to those which have been found in human Wilms tumors. A total number of 25 rat nephroblastomas induced by either ENU or DMN-OMe were studied. All tumors showed a conventional triphasic architecture consistent with Wilms tumor irrespective of if they were primary tumors or syngeneic transplants. Furthermore, the rat nephroblastoma cell line ENU-T1 was assayed for β-Catenin mediated transcription using Topflash/Fopflash reporter gene assays.
In normal rat kidney we found β-catenin protein mainly localized to the tubular epithelial membranes, starting from the epithelium of the Bowman’s capsule down to the collecting duct. The analyzed sections never showed β-catenin staining of non-epithelial components like capillary loops or stroma. Furthermore, no nuclear β-catenin protein was ever observed in normal rat kidney. These findings probably reflect the important role of β-catenin in cell-cell adhesion and are well in accordance with what is known about β-Catenin expression in normal human kidney. The stronger staining in the distal tubules might probably reflect the fact that there are more junctions in the distal tubules.
Using immunohistochemical analysis of β-Catenin expression in chemically induced rat nephroblastomas we found 16 of 25 (64%) tumors with nuclear accumulation of β-Catenin protein. Interestingly, also 24 of 36 (66%) human Wilms tumors previously showed the same phenomenon [43]. In any case, mainly blastemal or mesenchymal rat nephroblastoma cells, but always only a subset of these, showed nuclear accumulation of β-catenin protein. Only rarely, also epithelial subcompartments showed nuclear localization of β-catenin which is very similar to the human situation: In Wilms tumors nuclear positivity in epithelial-like tumor cells has been reported in only a single case [43].
The intratumoral heterogeneity in nuclear β-catenin immunoreactivity is intriguing. It is, however, unlikely that this was due to technical reasons. Normal rat kidney tissue never showed nuclear localization and was often present on the same section as the tumor which showed nuclear s-catenin. Intratumoral heterogeneity in immunostaining has been also described in a variety of human malignancies [43;49–51]. In Wilms tumors, genetic heterogeneity has been previously ruled out as an alternative explanation for the observed heterogeneity in nuclear immunoreactivity by showing that regions lacking nuclear β-catenin accumulation had the same mutational state as regions with nuclear β-catenin positivity [43]. From these studies it is apparent that mutational activation of the wnt-signalling pathway is required but not sufficient on its own for nuclear accumulation of β-Catenin protein and that other, epigenetic factors are likely to contribute to this phenomenon.
Although the immunohistochemical findings on β-Catenin protein expression and intracellular distribution in rat nephroblastomas are well in accordance with previous studies in Wilms tumors it was surprising not to find any mutation in Ctnnb1 whereas 12.5% of Wilms tumors harbour such lesions. Since we used a rat colon cancer cell line, CC531, as a control we can exclude technical difficulties as being responsible for the failure of detecting Ctnnb1 mutations. CC531 cells harbour a known mutation in codon 41 of Ctnnb1 (ACC→ATC) [48] and this mutation was readily confirmed in the present study. Tumor DNA, however, could have been contaminated by DNA from normal adjacent tissue present in the samples used here. To exclude this, in 15 cases we used DNA extracted from tumor tissue obtained after microdissection but we found no mutation in these cases either. We conclude that activation of the wnt signalling pathway in rat nephroblastomas probably occurs through mutations of other members of the wnt-signalling pathway, which may include AXIN-2, GSK-3s, TCF-4, APC [52], or the recently discovered WTX gene [53;54]. As an important member of the β-catenin-destruction-complex it supports ubiquitinylation and degradation of β-catenin protein and therefore prevents β-catenin localization in the nucleus. So WTX functions as a negative regulator of wnt-signaling and, if mutated, might therefore lead to nuclear accumulation of β-catenin. Mutations of WTX have been found in about one third of human Wilms tumors and could explain a high proportion of tumors showing nuclear β-Catenin protein in the absence of CTNNB1 mutations. Similarly, rat WTX might be involved in the pathogenesis of rat nephroblastoma although no studies have yet been carried out to resolve this issue.
We also assayed a cell line derived from ENU-treated rats, ENU-T1, by the TOPflash/FOPflash reporter gene assay. This assay is well suited to detect any activation of the wnt-signalling pathway irrespectively of what exact member of the wnt signaling cascade is activated, however, it can do so only in cultured cells. Unfortunately, ENU-T1 represents the only one established cell line derived from a chemically-induced rat nephroblastoma and these cells showed weak but not significant activation of wnt-signalling pathway activity in our assays. Together, our findings of nuclear β-Catenin protein found in the majority of tumors analyzed indicate that activation of the wnt-signalling pathway may play a significant role in the development of chemically-induced rat nephroblastomas. Hence, chemically-induced rat nephroblastomas may provide a valuable platform for future preclinical studies targeting the wnt-signalling pathway. However, unlike in Wilms tumors [16], β-catenin exon 3 mutations do not appear to play a significant role. CMT and nuclear β-Catenin were used as substitute markers of an active wnt-signalling pathway.
Acknowledgments
We would like to thank Jeanette Charon Alvarez for excellent technical assistance and Susanne Dihlmann, University of Heidelberg, Germany for providing 293T and HCT116 cells and helpfull hints with the transfection. We would also like to thank Yoji Nagashima, Department of Pathology, Yokohama City University, Japan, for providing ENU-T1 cells. We greatly appreciate the work of Prof. Jakob Briner, Aarau, Switzerland for his expert advice on tumor histology. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Reference List
- 1.Miller RW, Young JL, Jr, Novakovic B. Childhood cancer. Cancer. 1995;75:395–405. doi: 10.1002/1097-0142(19950101)75:1+<395::aid-cncr2820751321>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 2.Pastore G, Znaor A, Spreafico F, Graf N, Pritchard-Jones K, Steliarova-Foucher E. Malignant renal tumours incidence and survival in European children (1978–1997): report from the Automated Childhood Cancer Information System project. Eur J Cancer. 2006;42:2103–2114. doi: 10.1016/j.ejca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Breslow N, Beckwith JB, Ciol M, Sharples K. Age distribution of Wilms’ tumor: report from the National Wilms’ Tumor Study. Cancer Res. 1988;48:1653–1657. [PubMed] [Google Scholar]
- 4.Breslow NE, Langholz B. Childhood cancer incidence: geographical and temporal variations. Int J Cancer. 1983;32:703–716. doi: 10.1002/ijc.2910320609. [DOI] [PubMed] [Google Scholar]
- 5.Matsunaga E. Genetics of Wilms’ tumor. Hum Genet. 1981;57:231–246. doi: 10.1007/BF00278936. [DOI] [PubMed] [Google Scholar]
- 6.Grundy PE, Telzerow PE, Breslow N, Moksness J, Huff V, Paterson MC. Loss of heterozygosity for chromosomes 16q and 1p in Wilms’ tumors predicts an adverse outcome. Cancer Res. 1994;54:2331–2333. [PubMed] [Google Scholar]
- 7.Mannens M, Slater RM, Heyting C, Bliek J, de KJ, Coad N, de Pagter-Holthuizen P, Pearson PL. Molecular nature of genetic changes resulting in loss of heterozygosity of chromosome 11 in Wilms’ tumours. Hum Genet. 1988;81:41–48. doi: 10.1007/BF00283727. [DOI] [PubMed] [Google Scholar]
- 8.Maw MA, Grundy PE, Millow LJ, Eccles MR, Dunn RS, Smith PJ, Feinberg AP, Law DJ, Paterson MC, Telzerow PE. A third Wilms’ tumor locus on chromosome 16q. Cancer Res. 1992;52:3094–3098. [PubMed] [Google Scholar]
- 9.Steenman M, Redeker B, de MM, Wiesmeijer K, Voute PA, Westerveld A, Slater R, Mannens M. Comparative genomic hybridization analysis of Wilms tumors. Cytogenet Cell Genet. 1997;77:296–303. doi: 10.1159/000134602. [DOI] [PubMed] [Google Scholar]
- 10.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343:774–778. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 11.Rose EA, Glaser T, Jones C, Smith CL, Lewis WH, Call KM, Minden M, Champagne E, Bonetta L, Yeger H. Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms’ tumor gene. Cell. 1990;60:495–508. doi: 10.1016/0092-8674(90)90600-j. [DOI] [PubMed] [Google Scholar]
- 12.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 13.Gessler M, Konig A, Arden K, Grundy P, Orkin S, Sallan S, Peters C, Ruyle S, Mandell J, Li F. Infrequent mutation of the WT1 gene in 77 Wilms’ Tumors. Hum Mutat. 1994;3:212–222. doi: 10.1002/humu.1380030307. [DOI] [PubMed] [Google Scholar]
- 14.Huff V. Wilms tumor genetics. Am J Med Genet. 1998;79:260–267. doi: 10.1002/(sici)1096-8628(19981002)79:4<260::aid-ajmg6>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 15.Varanasi R, Bardeesy N, Ghahremani M, Petruzzi MJ, Nowak N, Adam MA, Grundy P, Shows TB, Pelletier J. Fine structure analysis of the WT1 gene in sporadic Wilms tumors. Proc Natl Acad Sci USA. 1994;91:3554–3558. doi: 10.1073/pnas.91.9.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koesters R, Ridder R, Kopp-Schneider A, Betts D, Adams V, Niggli F, Briner J, von Knebel DM. Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res. 1999;59:3880–3882. [PubMed] [Google Scholar]
- 17.Maiti S, Alam R, Amos CI, Huff V. Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res. 2000;60:6288–6292. [PubMed] [Google Scholar]
- 18.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Noort M, Meeldijk J, van der ZR, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- 20.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 21.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 22.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 23.Koesters R, von Knebel DM. The Wnt signaling pathway in solid childhood tumors. Cancer Lett. 2003;198:123–138. doi: 10.1016/s0304-3835(03)00367-7. [DOI] [PubMed] [Google Scholar]
- 24.Larue L, Delmas V. The WNT/Beta-catenin pathway in melanoma. Front Biosci. 2006;11:733–742. doi: 10.2741/1831. [DOI] [PubMed] [Google Scholar]
- 25.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 26.Koch A, Waha A, Tonn JC, Sorensen N, Berthold F, Wolter M, Reifenberger J, Hartmann W, Friedl W, Reifenberger G, Wiestler OD, Pietsch T. Somatic mutations of WNT/wingless signaling pathway components in primitive neuroectodermal tumors. Int J Cancer. 2001;93:445–449. doi: 10.1002/ijc.1342. [DOI] [PubMed] [Google Scholar]
- 27.Verras M, Sun Z. Roles and regulation of Wnt signaling and beta-catenin in prostate cancer. Cancer Lett. 2006;237:22–32. doi: 10.1016/j.canlet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Dihlmann S, Siermann A, von Knebel DM. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene. 2001;20:645–653. doi: 10.1038/sj.onc.1204123. [DOI] [PubMed] [Google Scholar]
- 29.Ring CJ. Cytolytic viruses as potential anti-cancer agents. J Gen Virol. 2002;83:491–502. doi: 10.1099/0022-1317-83-3-491. [DOI] [PubMed] [Google Scholar]
- 30.Roh H, Green DW, Boswell CB, Pippin JA, Drebin JA. Suppression of beta-catenin inhibits the neoplastic growth of APC-mutant colon cancer cells. Cancer Res. 2001;61:6563–6568. [PubMed] [Google Scholar]
- 31.Smith ML, Hawcroft G, Hull MA. The effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: evidence of different mechanisms of action. Eur J Cancer. 2000;36:664–674. doi: 10.1016/s0959-8049(99)00333-0. [DOI] [PubMed] [Google Scholar]
- 32.Zhou L, An N, Haydon RC, Zhou Q, Cheng H, Peng Y, Jiang W, Luu HH, Vanichakarn P, Szatkowski JP, Park JY, Breyer B, He TC. Tyrosine kinase inhibitor STI-571/Gleevec down-regulates the beta-catenin signaling activity. Cancer Lett. 2003;193:161–170. doi: 10.1016/s0304-3835(03)00013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dihlmann S, von Knebel DM. Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int J Cancer. 2005;113:515–524. doi: 10.1002/ijc.20609. [DOI] [PubMed] [Google Scholar]
- 34.Francis SO, Mahlberg MJ, Johnson KR, Ming ME, Dellavalle RP. Melanoma chemoprevention. J Am Acad Dermatol. 2006;55:849–861. doi: 10.1016/j.jaad.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 35.Hard GC, Noble RL. Spontaneous rat nephroblastoma: ultrastructure of a transplant line. Arch Pathol Lab Med. 1982;106:418–422. [PubMed] [Google Scholar]
- 36.Chandra M, Carlton WW. Incidence, histopathologic and electron microscopic features of spontaneous nephroblastomas in rats. Toxicol Lett. 1992;62:179–190. doi: 10.1016/0378-4274(92)90020-k. [DOI] [PubMed] [Google Scholar]
- 37.Hard GC. Differential renal tumor response to N-ethylnitrosourea and dimethylnitrosamine in the Nb rat: basis for a new rodent model of nephroblastoma. Carcinogenesis. 1985;6:1551–1558. doi: 10.1093/carcin/6.11.1551. [DOI] [PubMed] [Google Scholar]
- 38.Diwan BA, Rice JM. Effect of stage of development on frequency and pathogenesis of kidney tumors induced in Noble (Nb) rats exposed prenatally or neonatally to N-nitrosoethylurea. Carcinogenesis. 1995;16:2023–2028. doi: 10.1093/carcin/16.9.2023. [DOI] [PubMed] [Google Scholar]
- 39.Eiter K, Hebenbrock HF, Kabbe HJ. Neue offenkettige and cyclische alpha-nitrosaminalkylather. Liebigs Ann Chem. 1972;765:55–77. [Google Scholar]
- 40.Gorman C, Padmanabhan R, Howard BH. High efficiency DNA-mediated transformation of primate cells. Science. 1983;221:551–553. doi: 10.1126/science.6306768. [DOI] [PubMed] [Google Scholar]
- 41.Sukumar S, Notario V, Martin-Zanca D, Barbacid M. Induction of mammary carcinomas in rats by nitroso-methylurea involves malignant activation of H-ras-1 locus by single point mutations. Nature. 1983;306:658–661. doi: 10.1038/306658a0. [DOI] [PubMed] [Google Scholar]
- 42.Koesters R, Hans MA, Benner A, Prosst R, Boehm J, Gahlen J, Doeberitz MK. Predominant mutation of codon 41 of the beta-catenin proto-oncogene in rat colon tumors induced by 1,2-dimethylhydrazine using a complete carcinogenic protocol. Carcinogenesis. 2001;22:1885–1890. doi: 10.1093/carcin/22.11.1885. [DOI] [PubMed] [Google Scholar]
- 43.Koesters R, Niggli F, von Knebel DM, Stallmach T. Nuclear accumulation of beta-catenin protein in Wilms’ tumours. J Pathol. 2003;199:68–76. doi: 10.1002/path.1248. [DOI] [PubMed] [Google Scholar]
- 44.Nagashima Y, Ohaki Y, Umeda M, Oshimura M, Misugi K. Establishment and characterization of an immature epithelial cell line (ENU-T-1) derived from a rat nephroblastoma. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989;57:383–392. doi: 10.1007/BF02899105. [DOI] [PubMed] [Google Scholar]
- 45.Korinek V, Barker N, Morin PJ, van WD, de WR, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 46.Numa F, Hirabayashi K, Tsunaga N, Kato H, O’Rourke K, Shao H, Stechmann-Lebakken C, Varani J, Rapraeger A, Dixit VM. Elevated levels of syndecan-1 expression confer potent serum-dependent growth in human 293T cells. Cancer Res. 1995;55:4676–4680. [PubMed] [Google Scholar]
- 47.Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopoulou A, Bicknell D, Bodmer WF, Tomlinson IP. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci USA. 2000;97:3352–3357. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Germann A, Dihlmann S, Hergenhahn M, Doeberitz MK, Koesters R. Expression profiling of CC531 colon carcinoma cells reveals similar regulation of beta-catenin target genes by both butyrate and aspirin. Int J Cancer. 2003;106:187–197. doi: 10.1002/ijc.11215. [DOI] [PubMed] [Google Scholar]
- 49.Wei Y, Fabre M, Branchereau S, Gauthier F, Perilongo G, Buendia MA. Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene. 2000;19:498–504. doi: 10.1038/sj.onc.1203356. [DOI] [PubMed] [Google Scholar]
- 50.Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol. 1999;155:703–710. doi: 10.1016/s0002-9440(10)65168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eberhart CG, Tihan T, Burger PC. Nuclear localization and mutation of beta-catenin in medulloblastomas. J Neuropathol Exp Neurol. 2000;59:333–337. doi: 10.1093/jnen/59.4.333. [DOI] [PubMed] [Google Scholar]
- 52.Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 53.Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, Kim JC, Feinberg AP, Gerald WL, Vargas SO, Chin L, Iafrate AJ, Bell DW, Haber DA. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315:642–645. doi: 10.1126/science.1137509. [DOI] [PubMed] [Google Scholar]
- 54.Major MB, Camp ND, Berndt JD, Yi X, Goldenberg SJ, Hubbert C, Biechele TL, Gingras AC, Zheng N, Maccoss MJ, Angers S, Moon RT. Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science. 2007;316:1043–1046. doi: 10.1126/science/1141515. [DOI] [PubMed] [Google Scholar]