

Abstract
Chronic, plaque-associated inflammation of the gingiva and the periodontium are among the most common oral diseases. Periodontitis (PD) is characterized by the inflammatory destruction of the periodontal attachment and alveolar bone, and its clinical appearance can be influenced by congenital as well as acquired factors. The existence of a rheumatic or other inflammatory systemic disease may promote PD in both its emergence and progress. However, there is evidence that PD maintains systemic diseases. Nevertheless, many mechanisms in the pathogenesis have not yet been examined sufficiently, so that a final explanatory model is still under discussion, and we hereby present arguments in favor of this. In this review, we also discuss in detail the fact that oral bacterial infections and inflammation seem to be linked directly to the etiopathogenesis of rheumatoid arthritis (RA). There are findings that support the hypothesis that oral infections play a role in RA pathogenesis. Of special importance are the impact of periodontal pathogens, such as Porphyromonas gingivalis on citrullination, and the association of PD in RA patients with seropositivity toward rheumatoid factor and the anti-cyclic citrullinated peptide antibody.
Introduction
Periodontitis (PD), the most common oral disease, is a destructive inflammatory disease of the supporting tissues of the teeth and is caused by specific microorganisms [1]. As a rule, PD develops through gingivitis, an inflammation of the marginal periodontium. However, not every gingivitis develops further into PD. Both the amount and virulence of the microorganisms and the resistance factors of the host (risk factors and immune status) are crucial for the progression of the periodontal destruction (Figure 1). PD has been proposed as having an etiologic or modulating role in cardiovascular and cerebrovascular disease, diabetes, and respiratory disease and adverse pregnancy outcome, and several mechanisms have been proposed to explain or support such theories. Moreover, oral lesions are indicators of disease progression, and the oral cavity can be a window to overall health and body systems. In recent years, remarkable epidemiological and pathological relationships between periodontal diseases and rheumatic diseases, especially rheumatoid arthritis (RA), have been presented.
Figure 1.
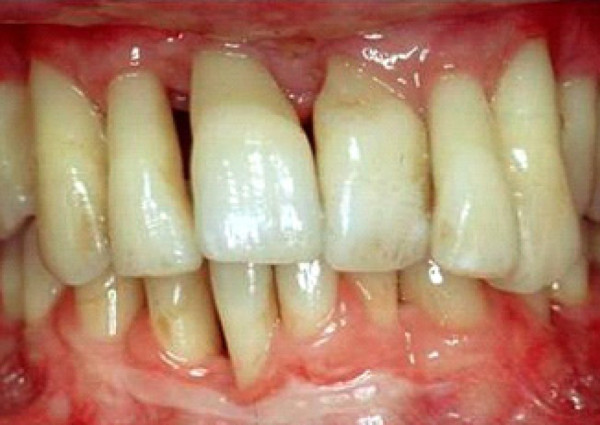
Severe periodontitis with loss of periodontal attachment and alveolar bone.
Pathogenesis of periodontal diseases
Chronic, plaque-associated inflammation of the periodontium is among the most common oral diseases and has a prevalence of 80% to 90% [1], resulting in soft and hard periodontal tissue destruction and ultimately in tooth loss [2]. Both the amount and virulence of the microorganisms and the resistance factors of the host (risk factors and immune status) are crucial for the initiation and progression of the periodontal destruction [3]. Besides detailed concepts about microbiological, molecular, and cellular mechanisms, which determine the strength and balance of the cellular and humoral host response in tissues, it became apparent that a complex and primarily endogenous periodontal microflora is responsible for disease initiation and progression.
Bacterial oral infection
PD is to be understood as an opportunistic infection [4]. It results directly in tissue injury or provokes excessive, autodestructive inflammatory responses, depending on the pathogenicity of the agents or the performance of the immune defense. In particular, Gram-negative anaerobic bacteria, which form a bacterial plaque biofilm on the tooth surface, initiate this tissue-destroying process [5]. Among a complex and still largely unknown microflora, about 20 bacteria species, which live in the subgingival environment, have been identified as periodontal pathogens and are linked to several forms of PD. The best analyzed of these bacteria are Porphyromonas gingivalis, Prevotella intermedia, Tannerella forsythia, and Aggregatibacter actinomycetem-comitans [6].
Biofilm infection
In the periodontal pocket of periodontal disease, there exists a condition in which periodontopathic bacteria form a film-like colony by adherence and aggregation [7]. After a few hours, resident microorganisms - most of which are mostly Gram-positive - stick to the membranaceous layer of the smooth surface of the teeth (pellicle layer), which settles within minutes up to a few hours after thorough mechanical tooth cleaning. By means of fimbriae, pili, and so-called glycocalyx, Gram-negative bacteria can attach to those microorganisms [8]. A complex and extremely resistant biofilm with a decisive evolutionary advantage for the bacteria develops. They can cooperate metabolically [8], and because of the complexity and subgingival location, the bacteria are protected from immunologic defense mechanisms of the host as well as from antibiotic agents. The enhancement of the bacterial pathogenicity is a result thereof [8]. Continuous stimulation by bacteria inflicts injury within the gingiva, destroys the local immune system, and activates osteoclasts in the tissue, so that the PD can progress [9].
Virulence factors
Besides the ability to form biofilms, the synthesis of toxic substances is among the most important characteristics of dental pathogenic bacteria. Enzymes have a direct tissue-destroying effect (neutral phosphatases and collagenases). Leucotoxins and immunoglobulin-splitting substances elude defense mechanisms [10]. Moreover, osteoclast-activating alkaline and acid phosphatases lead indirectly to the loss of the periodontal attachment apparatus. Lipopolysaccharides (LPSs) and proteoglycans, which are both part of the cell wall of Gram-negative bacteria, play a key role in activating the immune system with the release of diverse cytokines, subsequently causing alveolar bone resorption.
The two phases of the immune response - the critical pathway
The pathogenetic model of the critical pathway assumes the simple cause-and-effect principle and permits a differentiated view of the processes of the marginal development of PD. According to this model, there are two phases [11].
First phase: initial immune response
Through the junctional epithelium, which adheres to the tooth surface, a constant immunologic interaction between periodontal-pathogenic bacteria and the cells of the immune system takes place [10]. Bacteria release metabolites and toxins and thereby activate the immune system. LPS is one of the strongest activators of immunologic inflammatory processes [10,12]. Apart from the formation of integrin ICAM-1 (intercellular adhesion molecule-1), numerous proinflammatory and inflammatory cytokines such as interleukin-1-beta (IL-1β) as well as IL-6, IL-8, IL-1, and tumor necrosis factor-alpha (TNF-α) [13] are involved in this process. In this way, the path of the neutrophil granulocytes via leucodiapedesis through the endothelium to the inflammatory center is established [10]. There, pathogenic bacteria, which are opsonized by complement factors (C3b and C4b) and immunoglobulins (IgG1 and IgG2), are phagocytosed [14]. This early inflammatory response to the biofilm is dominated by polymorph nuclear leucocytes (PMNLs), which have a modulatory effect on T and B lymphocytes and which increasingly penetrate the tissue [15]. An increased secretion of gingival sulcus fluid leads to the washout of proinflammatory mediators and the recruitment of inflammatory cells from the tissue. Together with immunoglobulins, PMNLs form an effective defense bank against pathogenic microorganisms [11]. If the microbial attack is successfully and sufficiently embanked in this first phase, the inflammation is - according to the model of the critical pathway - restricted to the marginal gingiva (resulting in gingivitis) [11]. The second phase of the immune response is activated if the described defense mechanisms are insufficient.
Second phase of the immune reaction
Current data show that PD develops preferably in disposed persons with an abnormal inflammatory immune reaction to microbial plaque. Monocytes or macrophages or both become active as an unspecific immune response [16]. Histologically, the longer-lasting inflammation manifests itself in the conversion of the junctional epithelium into the so-called pocket epithelium, which is characterized by its increased proliferation rate in the basal area and an increased permeability for larger molecules. Because of the stimulation of the CD14 receptors by LPS complex formation, macrophages secrete the prostaglandin E2 (PgE2) and the proinflammatory cytokines IL-1β and TNF-α, which convey the periodontal bone absorption. Matrix metalloproteinases (MMPs) participate in the destruction of the extracellular matrix [12]. Furthermore, secondary inflammatory mediators (for example, the platelet-activated factor and biogenic amines [bradykinin and histamin] [16]) support the adhesion of neutrophil granulocytes to endothelic cells by endothelial expression of adhesion molecules [12]. IL-1β and PgE2 both have an inhibiting effect on the collagen synthesis [12]. PgE2 leads to vasodilation, bone atrophy, and edema formation [12]. B and T cells and plasma cells are to be found in the inflammatory infiltrate and gingival sulcus fluid, respectively. Most of the plasma cells form immunoglobulins of the type IgG (approximately 60% to 80%). Significantly fewer of those formed are IgAs (approximately 10% to 40%), and only a few are IgMs [17]. The local antibody synthesis produces antibodies that are diverse in quantity and specificity. These antibodies can be more frequently local than systemic [17].
Rheumatic diseases such as rheumatoid arthritis and periodontitis
Many recent studies have identified statistically significant associations between established PD and rheumatic diseases. In particular, RA as a chronic inflammatory joint disease shows numerous characteristics and pathogenetic processes that have similarities to PD. RA and its relationship to PD are the best-studied topics, and there are numerous publications in this regard. Therefore, this review focuses on this disease. Interesting and important results that describe the importance of microcirculation, osteoporosis, and other common risk factors in respect to the relationship between the rheumatic diseases such as RA and PD are expected to appear in the future.
Association studies in rheumatoid arthritis and periodontitis
It has been reported that patients with longstanding active RA have a significantly increased incidence of PD when compared with healthy subjects [18-20] and that patients with PD have a higher prevalence of RA than patients without PD [21]. de Pablo and colleagues [22], using data from the Third National Health and Nutrition Examination Survey (NHANES III), show a significant association between RA and PD in the US population. In patients with RA, a significant correlation in teeth loss and alveolar bone loss was found, and this may well represent various aspects of periodontal health [23].
Role of oral infections and immune response
Successful treatment of RA with antibiotics against bacterial anaerobic infections points to the involvement of bacteria in the etiopathogenesis of RA [24]. The hypothesis that oral infections play a role in RA pathogenesis may be supported by the detection of bacterial DNA of anaerobes and high antibody titers against these bacteria in both the serum and the synovial fluid of RA patients in the early and later stages of the disease [25]. The highly pathogenic bacteria of the oral flora can maintain a chronic bacteremia that may damage distant organs (joints and endocardium) [26]. Periodontal pathogens, as P. gingivalis, have the ability to impair epithelium integrity, invade human endothelium cells, and influence both transcription and protein synthesis [27]. By these means, periodontal pathogenes have a direct systemic access to the blood circulation [28]. Examinations of patients with RA show an increased number of specific antibodies and the DNA of these bacteria in the blood and synovial liquid [29]. Recently, it was shown that P. gingivalis is able to invade primary human chondrocytes that were isolated from knee joints and to induce cellular effects [30]. As a consequence of this invasion, P. gingivalis delayed cell cycle progression and increased cell apoptosis in these chondroncytes [30].
P. gingivalis and gingipains
P. gingivalis produces arginine-specific (gingipain R) and lysine-specific (gingipain K) cystein endopeptidase [24], which play a role in bacterial housekeeping and infection, including amino acid uptake from host proteins and fimbriae maturation [31,32]. Gingipains are proteolytic enzymes [32] responsible for the expression of the virulence. Proteinases such as MMP-1, MMP-3, and MMP-9 are activated and extracellular matrix host proteins such as laminin, fibronectin, and collagen are degraded by P. gingivalis gingipains [33]. Furthermore, gingipains are responsible for an increased vascular permeability and for the degradation of complement factors [33].
P. gingivalis and the enzyme peptidylarginine deiminase
P. gingivalis is currently the only known bacterium with expression of peptidyl arginine deiminase (PAD), which represents an important pathogenic factor of RA [20,34]. The enzymatic deimination of arginine residuals to citrulline through the enzyme PAD is a form of post-translational protein modification [35]. The consequence is a modification of the structure of the protein, by which its biochemical and antigen characteristics are changed. However, the PAD expressed by P. gingivalis is not entirely homologue to human PAD but leads to an irreversible, post-translational conversion of arginine to citrulline [20,35]. So far, citrullination has been found particularly in proteins of the cytoskeleton (for example, cytokeratin, vimentin, and filaggrin) in the course of the apoptosis. Diseases like RA result in the (patho)physiological citrullination of structure proteins and in an increased accumulation of citrullinated proteins (for example, mutated citrullinated vimentin) [26]. The reduced immunotolerance of these patients to citrullinated proteins seems to be a key problem, so that - especially for RAs of high severity - an increased formation of autoantibodies develops [26]. For P. gingivalis, the by-product ammoniac serves as a neutralizer of the acid milieu and thereby ensures the growth of the bacterium [34]. Interestingly, patients with PD show a high concentration of ammonia in the sulcus fluid [34]. Possibly, periodontal infections with pathogens such as P. gingivalis in association with a genetic predisposition support inflammatory diseases like RA or have an immunoregulating effect on the course of the disease or both [10,20]. In this context, the P. gingivalis titer in patients with RA correlated significantly with the concentration of anti-citrullinated protein/peptide antibody (ACPA) [10,20]. It is thus hypothesized that in a genetically susceptible (shared epitope-positive) individual, such citrullinated peptides may interrupt tolerance to endogenous citrullinated antigens, resulting in the generation of an immune response to citrullinated self-antigens. Hitchon and colleagues [36] reported an association between immune responses to the oral pathogen P. gingivalis and the presence of ACPA in a population with a high background prevalence of RA-predisposing HLA-DRB1 alleles. This gene-environment interaction may result in breaking self-tolerance to citrullinated antigens or amplification of these autoimmune responses or both and may ultimately lead to the development of RA [36].
P. gingivalis and the rheumatoid factor
The rheumatoid factor (RF) has been found in RA and in other chronic inflammation diseases, including PD [20]. The RF could be verified in the gingiva, in the subgingival plaque, and in the serum of patients with PD [34]. Seropositive patients with PD showed increased titers of IgG and IgM antibodies against oral microorganisms when compared with seronegative patients with PD [34]. The RF of seropositive patients shows a cross-reaction with oral bacterial epitopes [37]. The P. gingivalis proteinase is responsible for the epitope development in the RF-Fc region. The proteinases are regarded as important virulence factors since they make the growth of P. gingivalis possible and lead to the degradation of the host tissue [38]. Bonagura and colleagues [39] identified the lysine and arginine amino acid sequences for these Fc regions of the IgG molecule. Since P. gingivalis decomposes lysine and arginine in particular and the IgG3 CH2 and CH3 domains are processed by the P. gingivalis proteinase, they take over a key function in the RF production of rheumatoid cells [40]. A current study (n = 69) of patients with RA evaluated the prevalence and severity of PD and their relationship to RA disease activity and severity [41]. Patients with osteoarthritis (OA) served as controls (n = 35) [41]. PD was more common and severe in patients with RA when compared with patients with OA [41]. Though apparently unrelated to disease activity, the presence itself of these auto-antibodies does seem to be highly relevant in association to disease pathogenesis in RA and the occurrence of poor outcomes.
Periodontitis, rheumatoid arthritis, and genetic factors
In 1987, a successful demonstration of the connection between HLA-DR4 and rapidly progressive periodontitis (RPP) by specific typing of the HLA gene loci HLA-A, HLA-B, HLA-C, and HLA-D was achieved. In that study, a DR4 frequency of 80% in patients with RPP as opposed to 38% in the control group was observed [42]. Another study [43] confirmed these results by an examination of the DRB1*04 alleles that code HLA-DR4. In patients with RPP, a significantly higher frequency (42%) of one of the DRB1 subtypes *0401, *0404, *0405, or *0408 could be detected, whereas the control group showed a frequency of these subtypes to be only 7%. These DRB1 subtypes are part of the so-called shared epitope genotypes, which also play a role in other inflammatory diseases like RA [26].
Superantigens and heat shock proteins
Heat-resistant, hydrophilic molecules with a molecular weight of 24 to 30 kDa are referred to as superantigens. They are able to virtually glue together T-cell receptors (TCRs) and major histocompatibility complex II molecules [44], which trigger a permanent signal in T cells. The region V beta (VβV) has been identified as the binding position for superantigens and is located in the variable part of the beta chain of the TCR. TCRs of the Vβ gene (Vβ 6, 8, 14, and 17) are more frequent in patients with RA than in the control groups [45]. These superantigens of RA can be influenced by oral bacteria, although the P. intermedia stimulates the expression of Vβ 8 and Vβ 17 genes in CD4 (+) T cells, and both bacteria P. gingivalis and P. intermedia can also increase the expression of Vβ 6 and Vβ 8 [46].
Heat shock proteins (HSPs) protect the cell from stress by reversibly interacting with abnormal proteins and peptides and by participating in their backfolding and decomposition. Furthermore, HSPs fulfill a function in the hereditary and acquired immunity and are associated with the pathogenesis of RA [47].
In the serum of patients with RA, high levels of oral bacterial HSP were found. Seventy-kilodalton Prevotella melaninogenica HSP and P. intermedia HSP have also been identified in periodontal disease [24]. However, HSP 70 antibodies are also found in the synovium of patients with RA and occur in the synovialis if the HSP 70 expression is triggered by specific stress factors (for example, heat, trauma, endotoxins, and anti-inflammatory drugs) and by proinflammatory cytokines (TNF-α, IL-1, and IL-6) [24]. Therefore, superantigens and HSP in patients with RA are not specific to oral bacteria [24].
Autoantigens
The citrullinated form of the α-enolase is an autoantigen that plays a role in the glycolysis. α-Enolase operates as a receptor and activator of plasminogen, as an HSP, and as a Myc-binding protein [26]. The citrullinated α-enolase has been detected together with other citrullinated antigens in the synovial tissue of patients with early RA [26]. However, the finding of a specificity of 97.1% in this cohort is remarkable. Lundberg and colleagues [27] identified an immunodominant epitope of the citrullinated α-enolase. The data on the sequence similarity and cross-reactivity let us assume that this immunodominant epitope of the citrullinated α-enolase plays a role in the primary autoimmunity of a subgroup of RA patients with bacterial infections, especially P. gingivalis [20,27].
It is well known that immunoglobulins of the class IgG act as antigens. Interestingly, IgG is glycolized differently in patients with RA. In 60% of the patients but not in healthy control groups matched by age, the terminal galactose is missing in the carbohydrate groups of the Fc part. Anti-agalactosyl IgG antibodies (CARF) showed a slightly higher sensitivity of 73.9% but a specificity as low as that for RF. This lack of terminal galactose is associated with a poor prognosis in the course of the disease [48]. Under these conditions, the saccharolytic bacterium P. melaninogenica is able to bind at the Fc region of the IgG molecule and to metabolize galactose with its enzyme [49]. Variations in the composition of the sugar moiety can influence antibody activity in autoimmune disorders. Furthermore, there are bacteria (Escherichia coli) that are inhibited by galactose (Gal) or N-acetyl-galactosamines (GalNAc) and other carbohydrates [50].
The pathogenic periodontal bacteria produce enzymes (collagenases, hyaluronidases, neuraminidases, and others) that degenerate the intercellular matrix and the collagenous skeleton, thereby facilitating the infiltration of further microorganisms into the tissue [38]. In patients with RA, the presence of autoantibodies against collagen II (CII), a main component of the hyaline cartilage, has been verified [51]. P. gingivalis expresses a lysine-specific proteinase, shows collagenase activity, and reduces all collagen molecules except from those for CII [52]. Lysine in position 270 of CII 263 to 370 can be hydroxylized and further on glycolized to monosaccharides or disaccharides (for example, with a beta-D-galactopyranosyl or with an alpha-glycopyranosyl-(1,2)-beta-galactopyranosyl residue).
T helper 17 cells and interleukin-17
The role of the T helper 17 (Th17) cells in the host defense is not completely known. It was able to be shown that IL-17 stimulates the generation and mobilization of neutrophils and plays an important role in the defense of extracellular bacteria [53]. Th17 cells and IL-17 play an important role in the pathogenesis of RA. On the other hand, Th17 cells are also present in chronic periodontal disease [54]. IL-17 can be found in periodontal lesions and potentially plays a role in the etiopathogenesis of periodontal disease. The P. gingivalis antigen stimulates the T cells to express IL-17 [54].
Metallomatrix proteinases
Under a clinically healthy gingival situation, the continuous cellular composition and decomposition processes in the periodontium are balanced, so that collagen decomposing MMP and tissue inhibitors of MMP (TIMPs), for example, are always to be found. In PD, TIMPs are overbalanced in favor of the MMPs, which consequently have an increased active concentration. A key enzyme for the tissue destruction in the context of PD, MMP-8 in its active form decomposes fibrillar collagen structures and also is associated with alveolar bone destruction [12,55]. Consequently, the detection of mediators such as MMP-8 in the gingival sulcus fluid may be a method to monitor inflammatory activities, and this adds to classical periodontal diagnostics (probing depths, clinical attachment level, and bleeding on probing). A recent study showed lower MMP-8 levels in the healthy control group than in the RA group with gingivitis, in the RA group with PD, or in the systemically healthy PD group (P < 0.05) [55]. In contrast, MMP-13 levels were similar in all groups (P > 0.05). RA patients with gingivitis or PD had similar MMP-8, MMP-13, and TIMP-1 levels as did the systemic healthy control group (P > 0.05). This study indicates that the simultaneous appearance of RA and PD has no influence on the investigated parameters. Increased MMP-8 levels in the gingival sulcus fluid can be found in periodontal inflammation. The long-term application of glucocorti-coids and nonsteroidal anti-inflammatory drugs causes similar high MMP-8 and MMP-13 levels in RA patients and systemic healthy probands and possibly results in an overproduction of those enzymes.
Risk factors in periodontitis or rheumatoid arthritis or both
The clinical appearance of PD (Figure 1) and RA can be influenced by congenital as well as acquired factors, which can increase the probability of the development and progression of the disease. From an etiologic and pathogenetic point of view, risk factors other than pathogenic bacteria and oral hygiene play a crucial role. These risk factors can influence every sub-step of the pathogenesis, and this explains the individually different manifestations of the disease [3]. The individual risk factors such as age, gender, body mass, and genetic factors (IL-1β polymorphism and HLA gene associations) are the focus of attention. Other exogenic risk factors such as nutritive factors, socioeconomic status, psychological factors (for example, stress), and lifestyle (cigarette smoking and alcohol consumption) as well as systemic diseases may influence the pathogenesis [56,57]. However, it has also been known for some years now that patients with PD not only suffer from local loss of the connective tissue and the hard tissue but also have an increased risk of developing systemic diseases [58]. This interrelation is referred to as 'periodontal medicine'.
Smoking
The best documented environmental factor that contributes to RA susceptibility is smoking. Importantly, smoking appears to contribute to disease susceptibility only in individuals who develop autoantibody-positive RA characterized by the presence of ACPAs. There is a clustering of RA risk associated with smoking, presence of shared epitope alleles and the presence of ACPA [58]. Hitchon and colleagues [36] tried to determine whether oral hygiene and smoking are associated with RA, ACPA, and immune responses to P. gingivalis. It is known that smoking and poor oral health habits both increase the risk of PD [36]. Smoking is a risk factor for PD possibly through the effects of nicotine on inflammatory cytokine profiles [59] and MMP-3 activity [60] or potentially even through direct effects on P. gingivalis gene expression. Hitchon and colleagues [36] did find a high prevalence of both smoking and poor oral health habits in the study population on the basis of self-report questionnaire data; however, they could not define a clear association between these factors and the presence of either RA or ACPA.
Conclusions
We conclude that there is some evidence for the relationship between the presence of PD and the development of RA. The existence of an inflammatory systemic disease may promote PD in both its emergence and progress. Periodontal pathogens have direct systemic access to the blood circulation. Therefore, treatment with antibiotics in patients with RA can be effective. The enzyme PAD represents an important pathogenetic factor for RA. P. gingivalis is currently the only known bacterium with expression of PAD and plays a role in the humoral immune response and in the presence of APCA in patients with RA. Oral hygiene and smoking represent environmental factors influencing the risk for the development of RA. Other risk factors (for example, nutritive factors, stress, and lifestyle factors) should also be examined in the future with regard to their relationship to PD and RA. Further basic and clinical studies are needed. In particular, longitudinal studies are needed in order to clarify the temporal relationship between PD and RA. These studies will need to include high numbers of patients but a low number of confounding factors in order to derive firm conclusions. For clinical studies, it is necessary to create a network of cooperation with rheumatologists and dentists.
Abbreviations
ACPA: anti-citrullinated protein/peptide antibody; CII: collagen II; HSP: heat shock protein; Ig: immunoglobulin; IL: interleukin; LPS: lipopolysaccharide; MMP: matrix metalloproteinase; OA: osteoarthritis; PAD: peptidyl arginine deiminase; PD: periodontitis; PgE2: prostaglandin E2; PMNL: polymorph nuclear leucocyte; RA: rheumatoid arthritis; RF: rheumatoid factor; RPP: rapidly progressive periodontitis; TCR: T-cell receptor; Th17: T helper 17; TIMP: tissue inhibitor of metalloproteinase; TNF-α: tumor necrosis factor-alpha.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Jacqueline Detert, Email: jacqueline.detert@charite.de.
Nicole Pischon, Email: nicole.pischon@charite.de.
Gerd R Burmester, Email: gerd.burmester@charite.de.
Frank Buttgereit, Email: frank.buttgereit@charite.de.
References
- Saini R. Periodontitis a true infection. J Glob Infect Dis. 2009;1:149–151. doi: 10.4103/0974-777X.56251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten MA. Bacteria and periodontitis. J Can Dent Assoc. 1996;62:12–13. [PubMed] [Google Scholar]
- Detert J, Pischon N, Burmester GR, Buttgereit F. Pathogenesis of periodontitis in rheumatic diseases. Z Rheumatol. 2010;69:109–112. doi: 10.1007/s00393-009-0560-1. [DOI] [PubMed] [Google Scholar]
- Lovegrove JM. Dental plaque revisited: bacteria associated with periodontal disease. J N Z Soc Periodontol. 2004. pp. 7–21. [PubMed]
- Socransky SS, Haffajee AD. Dental biofilms: difficult therapeutic targets. Periodontol 2000. 2002;28:12–55. doi: 10.1034/j.1600-0757.2002.280102.x. Review. [DOI] [PubMed] [Google Scholar]
- Moore WE, Moore LV. The bacteria of periodontal diseases. Periodontol 2000. 1994;5:66–77. doi: 10.1111/j.1600-0757.1994.tb00019.x. [DOI] [PubMed] [Google Scholar]
- Kolenbrander PE, London J. Adhere today, here tomorrow: oral bacterial adherence. J Bacteriol. 1993;175:3247–3252. doi: 10.1128/jb.175.11.3247-3252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker CJ, Klier CM, Kolenbrander PE. Mechanisms of adhesion by oral bacteria. Annu Rev Microbiol. 1996;50:513–552. doi: 10.1146/annurev.micro.50.1.513. [DOI] [PubMed] [Google Scholar]
- Wang PL, Ohura K. Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors. Crit Rev Oral Biol Med. 2002;13:132–142. doi: 10.1177/154411130201300204. [DOI] [PubMed] [Google Scholar]
- Haffajee AD, Socransky SS. Microbial etiological agents of destructive periodontal diseases. Periodontol 2000. 1994;5:78–111. doi: 10.1111/j.1600-0757.1994.tb00020.x. [DOI] [PubMed] [Google Scholar]
- Kornman KS, Page RC, Tonetti S. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14:33–53. doi: 10.1111/j.1600-0757.1997.tb00191.x. [DOI] [PubMed] [Google Scholar]
- Taba M Jr, Kinney J, Kim AS, Giannobile WV. Diagnostic biomarkers for oral and periodontal diseases. Dent Clin North Am. 2005;49:551–571. doi: 10.1016/j.cden.2005.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moughal NA, Adonogianaki E, Thornhill MH, Kinane DF. Endothelial cell leukocyte adhesion molecule-1 (ELAM-1) and intercellular adhesion molecule-1 (ICAM-1) expression in gingival tissue during health and experimentally induced gingivitis. J Periodont Res. 1992;27:623–630. doi: 10.1111/j.1600-0765.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- Sanders LAM, Feldman RG, Voorhorts-Ogink MM, De Haas M, Rijkers GT, Capel PJ, Zegers BJ, Van de Winkel JG. Human immunglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2 - mediated bacterial phagocytosis by neutrophils. Infect Immun. 1995;63:73–81. doi: 10.1128/iai.63.1.73-81.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodontol Res. 1991;26(3 Pt 2):230–242. doi: 10.1111/j.1600-0765.1991.tb01649.x. [DOI] [PubMed] [Google Scholar]
- Offenbacher S. Periodontal disease: pathogenesis. Ann Periodontol. 1996;1:821–878. doi: 10.1902/annals.1996.1.1.821. [DOI] [PubMed] [Google Scholar]
- Ebersole JL, Taubman MA. The protective nature of host responses in periodontal diseases. Periodontol 2000. 1994;5:112–141. doi: 10.1111/j.1600-0757.1994.tb00021.x. [DOI] [PubMed] [Google Scholar]
- Pischon N, Pischon T, Kröer J, Gülmez E, Kleber BM, Bernimoulin JP, Landau H, Brinkmann PG, Schlattmann P, Zernicke J, Buttgereit F, Detert J. Association among rheumatoid arthritis, oral hygiene, and periodontitis. J Periodontol. 2008;79:979–986. doi: 10.1902/jop.2008.070501. [DOI] [PubMed] [Google Scholar]
- Mercado FB, Marshall RI, Klestov AC, Bartold PM. Relationship between rheumatoid arthritis and periodontitis. J Periodontol. 2001;72:779–787. doi: 10.1902/jop.2001.72.6.779. [DOI] [PubMed] [Google Scholar]
- Rosenstein ED, Greenwald RA, Kushner LJ, Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. 2004;28:311–318. doi: 10.1007/s10753-004-6641-z. [DOI] [PubMed] [Google Scholar]
- Georgiou TO, Marshall RI, Bartold PM. Prevalence of systemic diseases in Brisbane general and periodontal practice patients. Aust Dent J. 2004;49:177–184. doi: 10.1111/j.1834-7819.2004.tb00070.x. [DOI] [PubMed] [Google Scholar]
- de Pablo P, Dietrich T, McAlindon TE. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J Rheumatol. 2008;35:70–76. [PubMed] [Google Scholar]
- Lagervall M, Jansson L, Bergstrom J. Systemic disorders in patients with periodontal disease. J Clin Periodontol. 2003;30:293–299. doi: 10.1034/j.1600-051X.2003.00325.x. [DOI] [PubMed] [Google Scholar]
- Ogrendik M. Rheumatoid arthritis is linked to oral bacteria: etiological association. Mod Rheumatol. 2009;19:453–456. doi: 10.1007/s10165-009-0194-9. Review. [DOI] [PubMed] [Google Scholar]
- Moen K, Brun JG, Valen M, Skartveit L, Eribe EK, Olsen I, Jonsson R. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin Exp Rheumatol. 2006;24:656–663. [PubMed] [Google Scholar]
- Wegner N, Lundberg K, Kinloch A, Fisher B, Malmstrom V, Feldmann M, Venables PJ. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. 2010;233:34–54. doi: 10.1111/j.0105-2896.2009.00850.x. [DOI] [PubMed] [Google Scholar]
- Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, Mikuls TR, Venables PJ. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008;58:3009–3019. doi: 10.1002/art.23936. [DOI] [PubMed] [Google Scholar]
- Lu J, Zhang W, Hao Y, Zhu Y. Defect of cell wall construction may shield oral bacteria's survival in bloodstream and cause infective endocarditis. Med Hypotheses. 2009;73:1055–1057. doi: 10.1016/j.mehy.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Mikuls TR, Payne JB, Reinhardt RA, Thiele GM, Maziarz E, Cannella AC, Holers VM, Kuhn KA, O'Dell JR. Antibody responses to Porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int Immunopharmacol. 2009;9:38–42. doi: 10.1016/j.intimp.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischon N, Roehner E, Hocke A, N'guessan P, Mueller HC, Matziolis G, Kanitz V, Purucker P, Kleber BM, Bernimoulin JP, Burmester GR, Buttgereit F, Detert J. Effects of Porphyromonas gingivalis on cell cycle progression and apoptosis of primary human chondrocytes. Ann Rheum Dis. 2008;68:1902–1907. doi: 10.1136/ard.2008.102392. [DOI] [PubMed] [Google Scholar]
- Imamura T. The role of gingipains in the pathogenesis of periodontal disease. J Periodontol. 2003;74:111–118. doi: 10.1902/jop.2003.74.1.111. [DOI] [PubMed] [Google Scholar]
- Stathopoulou PG, Galicia JC, Benakanakere MR, Garcia CA, Potempa J, Kinane DF. Porphyromonas gingivalis induce apoptosis in human gingival epithelial cells through a gingipain-dependent mechanism. BMC Microbiol. 2009;9:107. doi: 10.1186/1471-2180-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jie Bao G, Kari K, Tervahartiala T, Sorsa T, Meurman JH. Proteolytic activities of oral bacteria on ProMMP-9 and the effect of synthetic proteinase inhibitors. Open Dent J. 2008;2:96–102. doi: 10.2174/1874210600802010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw WT, Potempa J, Farley D, Travis J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect Immun. 1999;67:3248–3256. doi: 10.1128/iai.67.7.3248-3256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Hitchon CA, Chandad F, Ferucci ED, Willemze A, Ioan-Facsinay A, van der Woude D, Markland J, Robinson D, Elias B, Newkirk M, Toes RM, Huizinga TW, El-Gabalawy HS. Antibodies to Porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J Rheumatol. 2010;37:1105–1112. doi: 10.3899/jrheum.091323. [DOI] [PubMed] [Google Scholar]
- Thé J, Ebersole JL. Rheumatoid factor from periodontitis patients crossreacts with epitopes on oral bacteria. Oral Dis. 1996;2:253–262. doi: 10.1111/j.1601-0825.1996.tb00235.x. [DOI] [PubMed] [Google Scholar]
- Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000. 2000;24:153–192. doi: 10.1034/j.1600-0757.2000.2240108.x. [DOI] [PubMed] [Google Scholar]
- Bonagura VR, Artandi SE, Davidson A, Randen I, Agostino N, Thompson K, Natvig JB, Morrison SL. Mapping studies reveal unique epitopes on IgG recognized by rheumatoid arthritis-derived monoclonal rheumatoid factors. J Immunol. 1993;151:3840–3852. [PubMed] [Google Scholar]
- Martin T, Crouzier R, Weber JC, Kipps TJ, Pasquali JL. Structure-function studies on a polyreactive (natural) autoantibody. Polyreactivity is dependent on somatically generated sequences in the third complementaritydetermining region of the antibody heavy chain. J Immunol. 1994;152:5988–5996. [PubMed] [Google Scholar]
- Dissick A, Redman RS, Jones M, Rangan BV, Reimold A, Griffiths GR, Mikuls TR, Amdur RL, Richards JS, Kerr GS. Association of periodontitis with rheumatoid arthritis: a pilot study. J Periodontol. 2010;81:223–230. doi: 10.1902/jop.2009.090309. [DOI] [PubMed] [Google Scholar]
- Katz J, Goultschin J, Benoliel R, Brautbar C. Human leukocyte antigen (HLA) DR4. Positive association with rapidly progressing periodontitis. J Periodontol. 1987;58:607–610. doi: 10.1902/jop.1987.58.9.607. [DOI] [PubMed] [Google Scholar]
- Bonfil JJ, Dillier FL, Mercier P, Reviron D, Foti B, Sambuc R, Brodeur JM, Sedarat C. A ''case control'' study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis. Identification of types and subtypes using molecular biology (PCR.SSO) J Clin Periodontol. 1999;26:77–84. doi: 10.1034/j.1600-051X.1999.260203.x. [DOI] [PubMed] [Google Scholar]
- Gascoigne NR, Ames KT. Direct binding of secreted T-cell receptor beta chain to superantigen associated with class II major histocompatibility complex protein. Proc Natl Acad Sci USA. 1991;88:613–616. doi: 10.1073/pnas.88.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta IA, Sud S, Song Z, Affholter JA, Karvonen RL, Fernández-Madrid F, Wooley PH. T-cell receptor (Vbeta) bias in the response of rheumatoid arthritis synovial fluid T cells to connective tissue antigens. Scand J Rheumatol. 1997;26:166–173. doi: 10.3109/03009749709065676. [DOI] [PubMed] [Google Scholar]
- Leung KP, Torres BA. Prevotella intermedia stimulates expansion of Vbetaspecific CD4(+) T cells. Infect Immun. 2000;68:5420–5424. doi: 10.1128/IAI.68.9.5420-5424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragno S, Colston MJ, Lowrie DB, Winrow VR, Blake DR, Tascon R. Protection of rats from adjuvant arthritis by immunization with naked DNA encoding for mycobacterial heat shock protein 65. Arthritis Rheum. 1997;40:277–283. doi: 10.1002/art.1780400212. [DOI] [PubMed] [Google Scholar]
- Lacki JK, Porawska W, Mackiewicz U, Mackiewicz S, Muller W. Changes in agalactosyl IgG levels correlate with radiological progression in early rheumatoid arthritis. Ann Med. 1996;28:265–269. doi: 10.3109/07853899609033129. [DOI] [PubMed] [Google Scholar]
- Haraldsson G, Meurman JH, Kononen E, Holbrook WP. Properties of hemagglutination by Prevotella melaninogenica. Anaerobe. 2005;11:285–289. doi: 10.1016/j.anaerobe.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Eshdat Y, Sharon N. Recognitory bacterial surface lectins which mediate its mannose-specific adherence to eukaryotic cells. Biol Cell. 1984;51:259–266. doi: 10.1111/j.1768-322x.1984.tb00307.x. [DOI] [PubMed] [Google Scholar]
- Ronnelid J, Lysholm J, Engstrom-Laurent A, Klareskog L, Heyman B. Local antitype II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA-DR4-restricted IgG response. Arthritis Rheum. 1994;37:1023–1029. doi: 10.1002/art.1780370707. [DOI] [PubMed] [Google Scholar]
- Bedi GS, Williams T. Purification and characterization of a collagendegrading protease from Porphyromonas gingivalis. J Biol Chem. 1994;269:599–606. [PubMed] [Google Scholar]
- Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso CR, Garlet GP, Crippa GE, Rosa AL, Junior WM, Rossi MA, Silva JS. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. 2009;24:1–6. doi: 10.1111/j.1399-302X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- Biyikoğlu B, Buduneli N, Kardeşler L, Aksu K, Pitkala M, Sorsa T. Gingival crevicular fluid MMP-8 and -13 and TIMP-1 levels in patients with rheumatoid arthritis and inflammatory periodontal disease. J Periodontol. 2009;80:1307–1314. doi: 10.1902/jop.2009.090130. [DOI] [PubMed] [Google Scholar]
- Heasman LSF, Preshaw PM, McCracken GI, Hepburn S, Heasman PA. The effect of smoking on periodontal treatment response: a review of clinical evidence. J Clin Periodontol. 2006;33:241–253. doi: 10.1111/j.1600-051X.2006.00902.x. Review. [DOI] [PubMed] [Google Scholar]
- Tonetti MS. Periodontitis and risk for atherosclerosis: an update on intervention trials. J Clin Periodontol. 2009;36(Suppl 10):15–19. doi: 10.1111/j.1600-051X.2009.01417.x. [DOI] [PubMed] [Google Scholar]
- Lundstrom E, Kallberg H, Alfredsson L, Klareskog L, Padyukov L. Geneenvironment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 2009;60:1597–1603. doi: 10.1002/art.24572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Heens GL, Kikkert R, Aarden LA, van der Velden U, Loos BG. Effects of smoking on the ex vivo cytokine production in periodontitis. J Periodontal Res. 2009;44:28–34. doi: 10.1111/j.1600-0765.2007.01047.x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Song F, Windsor LJ. Cigarette smoke condensate affects the collagen-degrading ability of human gingival fibroblasts. J Periodontal Res. 2009;44:704–713. doi: 10.1111/j.1600-0765.2008.01179.x. [DOI] [PubMed] [Google Scholar]