

Synopsis
Skin ulceration is a major source of morbidity and is often difficult to manage. Ulcers due to an inflammatory etiology or microvascular occlusion are particularly challenging in terms of diagnosis and treatment. The management of such ulcers requires careful assessment of associated systemic conditions and a thorough analysis of the ulcer's clinical and histologic findings. In this report, we discuss several examples of inflammatory ulcers and the approach to their diagnosis and treatment.
Introduction
Skin ulceration represents a difficult clinical problem and a major source of morbidity for patients. The patient's quality of life is also significantly affected by pain, swelling, and management of wound drainage. Localized infection, wound colonization, and systemic symptoms add to the morbidity, and can lead to amputation and mortality. Wound care and therapy can be very challenging, and the use of wound dressings must rest on a rational basis and must not be too cumbersome or uncomfortable. Also, cutaneous ulcers take a considerable length of time to heal. The limitations to the patients' mobility, social interactions, and their ability to work result in feelings of helplessness and depression.1 Most cutaneous ulcers of the lower extremity are caused by venous insufficiency, arterial insufficiency, or neuropathy (especially of diabetic etiology), and in general such ulcers are not difficult to diagnose. However, ulcers associated with or due to systemic inflammatory conditions are often a major diagnostic and therapeutic challenge. We generally call these chronic ulcerations “inflammatory ulcers” (i.e., pyoderma gangrenosum, vasculitic ulcers, cryoglobulinemic ulcers, etc.) because a major and primary component of their pathophysiology indeed rests on inflammation and immunologic phenomena. However, this group of ulcers also includes conditions due to microcirculatory occlusion; a primary localized inflammatory component is less obvious in these conditions. Therefore, for the purpose of our discussion in this report, we will use a broad definition of “inflammatory ulcers”, which include these two aspects of chronic ulcers that are not due to classical vascular diseases or neuropathy. As with most complex conditions, inflammatory ulcers require a careful multidisciplinary consultation and treatment approach. The internist, dermatologist, surgeon, and rheumatologist are often called upon to contribute their expertise in order to establish the diagnosis and coordinate care.
Basic approach to patients with inflammatory ulcers
The diagnosis of inflammatory ulcers begins with a detailed history. Several important questions need to be to be asked (from the patient/records, or elicited by physical exam), and the following is a reasonable list. What was the primary lesion? How did the lesion progress? How fast was the progression to ulceration? Was the lesion painful? What management and treatment interventions took place and have they improved or worsened the condition? Has there been a similar problem in the past? Were any new medications started over the last couple of months? Was a surgical procedure performed in the last several months? Have there been any changes in the patient's general health? A thorough review of systems provides clues to diagnosis. A past medical history of connective tissue diseases, diabetes, heart disease, kidney disease, inflammatory bowel disease, hepatitis, hypertension, coagulopathies, prior pregnancy, and malignancies help support or suggest a particular etiology and diagnosis.
Physical examination needs to be comprehensive. Physical exam and attention to details must not be focused on the ulcer alone. Rather, careful observation of the surrounding skin and attention to other areas of the integument such as the oral mucosa and nails are essential. Cutaneous findings, such as livedo reticularis (a netlike violaceous discoloration surrounding a central paler area), palpable purpura, petechiae, nail splinter hemorrhages, and/or oral ulcers support an inflammatory cause of the ulceration. Lipodermatosclerosis, commonly presenting as redness, induration, and hyperpigmentation of the skin in the lower extremity, supports a diagnosis of venous insufficiency. Examination of the peripheral vascular system and testing for the presence of neuropathy are performed to exclude arterial insufficiency, venous disease, and/or neuropathy as causes of the ulceration. Examination of the ulcer involves recognizing features that are characteristic of certain types of ulcers and to identify problems that one can treat. For example, areas of necrosis and the presence of an eschar suggest a thrombotic disorder. Violaceous undermined borders are suggestive of pyoderma gangrenosum; A reddish yellow plaque surrounding the ulcer is characteristic of necrobiosis lipoidica diabeticorum (NLD), which is typically associated with diabetes. Ulcers are often complicated by swelling, infection, irritant contact dermatitis from the wound drainage or dressings, or an allergic contact dermatitis from topical medications and dressings. An ulcer with foul odor and purulent drainage is most likely secondarily infected or heavily colonized and will benefit from systemic/topical antibiotics or topical antiseptic treatment. Areas with necrosis and eschar formation in a patient with good pulses would benefit from surgical debridement, most importantly in diabetic patients. Leg swelling should be addressed with compression, when this will not compromise blood supply. Use of a scoring system in addition to ulcer size may be a good objective way to track improvement (Figure 1). As always, the wound bed must be optimized according to the established principles of wound bed preparation (WBP) to promote healing and to make available treatments more effective.2 Pain management is important to ensure compliance with treatment; referral to a pain management service may sometimes be necessary. Therefore, as just discussed, the workup and approach to inflammatory ulcers still requires careful attention to the principles of general wound care and management.
Figure 1. Wound Bed score (WBS) and its individual features.

The total WBS adds each individual score for each characteristic to give a total score.  Percent of eschar present(>25, 1–25%, none);
Percent of eschar present(>25, 1–25%, none);  Severity of peri-ulcer dermatitis (severe, moderate, none or mild);
Severity of peri-ulcer dermatitis (severe, moderate, none or mild);  Depth of the wound (severely depressed or raised compared to peri-wound skin);
Depth of the wound (severely depressed or raised compared to peri-wound skin);  Severity of callus/fibrosis (severe, moderate, none or minimal);
Severity of callus/fibrosis (severe, moderate, none or minimal);  Percent of pink granulation tissue present (<50%, 50–75%, >75%);
Percent of pink granulation tissue present (<50%, 50–75%, >75%);  Severity of edema (severe, moderate, none/mild);
Severity of edema (severe, moderate, none/mild);  Percent of healing edges (<25%, 25–75%, >75%);
Percent of healing edges (<25%, 25–75%, >75%);  Frequency of dressing changes (severe, moderate, none/mild)
Frequency of dressing changes (severe, moderate, none/mild)
Some of the main causes of inflammatory ulcers may be classified into the following primary or associated categories: immunologic conditions (i.e., collagen vascular diseases and vasculitis); thrombotic or hypercoagulable states (i.e., cholesterol embolization and the antiphospholipid syndrome); pyoderma gangrenosum and necrobiosis lipoidica (Figure 2A–I).
Figure 2. Examples of some inflammatory ulcers.

(A) Pyoderma gangrenosum in a patient with rheumatoid arthritis. (B) The more `granulomatous' appearance of Pyoderma gangrenosum. (C) Typical Pyoderma gangrenosum with purple edges. (D) Undermined ulcer of Pyoderma gangrenosum. (E) The undulating borders of a rheumatoid ulcer. (F) Eschar in an ulcer due to polyarteritis nodosa. (G) Same patient shown in (F), after successful use of immunosuppressants. (H) The `angular' ulcers seen in patients with collagen vascular diseases, often mimicking factitial ulcers. (I) Livedo reticularis and small ulcers in a patient with cholesterol embolization.
Specific Inflammatory Conditions
Pyoderma Gangrenosum
Pyoderma Gangrenosum (PG) was first described in patients with rapidly progressive, painful ulcers with violaceous undermined borders.3 It is a disorder characterized by pathergy and the development of a larger ulcer upon debridement or with trauma.4 When already present, it can also develop in other, distant, seemingly unrelated areas of the skin; hence autologous grafting is a relative contraindication in these patients. Several clinical variants of PG have been recognized. The classic variant starts as a pustule that develops into a rapidly enlarging ulcer with undermined borders, surrounding erythema, and a purulent base. Among other systemic conditions, PG is associated with rheumatoid arthritis, inflammatory bowel disease, Bechet's, a monoclonal gammopathy (typically IgA), myeloproliferative disease; many cases are idiopathic.5 The pustular variant of PG manifests as painful pustules with an inflamed border and may not necessarily progress to frank ulceration. Very importantly, the skin manifestations of PG often parallel the severity of the underlying systemic disease, and often resolve with control of their acute flare.6 The bullous variant of PG is characterized by painful bullae, which progress to superficial erosions and are associated with myeloproliferative disorders.7 The vegetative variant is a superficial painless ulcer with undermined edges. It slowly enlarges and may be exophytic with no clear pattern of specific associated disease.8 Parastomal PG presents as a non healing ulcer next to a colostomy site. It should be considered in all patients with inflammatory bowel disease and a peristomal ulceration; the ulcer is typically quite refractory to local wound care approaches.9–10 Extra-cutaneous involvement of PG has been reported as cavitary lung lesions, pulmonary infiltrates, episcleritis, development of a psoas muscle abscess, and splenic abscesses.11–14
The onset of the systemic condition may precede, follow, or occur simultaneously with PG; they also may run an independent course. Asymmetric seronegative monoarticular inflammation involving large joints is also associated with PG. A monoclonal IgA gammopathy can be present in the setting of PG and is usually benign, although the development of multiple myeloma has been reported.5 Systemic malignancy has been estimated to occur in 7% of PG patients, with leukemia being the most common neoplasm.7 Certain drugs have also been implicated in the development of PG (GM- CSF, interferons, and antipsychotic drugs).15–18 The pathogenesis of PG is poorly understood. However, it is postulated to be the result of neutrophil dysfunction, which may either consist of a defect in chemotaxis or be due to neutrophil hyper- reactivity. Also, clonality of T- cells has been detected in patients with PG in the absence of myeloproliferative disease, possibly indicating an excessive response to antigenic stimuli.19–20
Vascular and other types of ulcers may be misdiagnosed as PG. This can be a serious clinical problem because treatment of PG with immunosuppressants may ultimately worsen ulcers caused or complicated by infection or malignancy. For example, ulcers caused by atypical mycobacteria and deep fungal infections may be misdiagnosed as PG. Initial (and temporary) improvements in an ulcer upon immunosuppressive treatment do not prove that it represents PG, and should be analyzed with caution. Similarly, it is important to reevaluate the diagnosis of PG when the ulcers are recalcitrant to immunosuppressive therapy. Other inflammatory ulcers improve with immunosuppression, such as those associated with leukocytoclastic vasculitis, Wegener's granulomatosis, cutaneous polyarteritis nodosa, or the antiphospholipid antibody syndrome.4
PG is a diagnosis of exclusion, a fact that is not generally appreciated by many clinicians. Critical diagnostic procedures, including biopsy, and other laboratory work up are used to exclude other causes of the ulceration such as infections, malignancies, connective tissue diseases, or vaso-occlusive conditions. A skin biopsy is important to rule out other diagnoses more than to confirm a diagnosis of PG; the histology is never truly diagnostic and still requires exclusion of an infectious etiology. An adequate biopsy would be an excisional biopsy down to subcutaneous tissue, obtaining enough tissue for histology and culture for bacteria, fungi, and mycobacteria. Laboratory tests should include an analysis of and detection of abnormalities in CBC, ESR, c- and p-ANCA, basic metabolic panel, serum protein electrophoresis, coagulation panel, antiphospholipid antibody, cryoglobulins, cryofibrinogen, chest x-ray, colonoscopy, as well as venous and arterial studies.4, 21 A patient with inflammatory bowel disease may have minimal or no symptoms, but still have abnormalities upon colonoscopy.
Biopsy findings in PG are variable, depending on the area biopsied and stage of the ulcer. A biopsy taken from the erythematous zone peripheral to the ulcer might show thrombosis and a lymphocytic infiltrate which, when present around blood vessels, might qualify as a lymphocytic vasculitis; in general, the term lymphocytic vasculitis is somewhat controversial. The necrotic undermined border and the ulcer be would typically show a dense mixed infiltrate and abscess formation. Concomitant leukocytoclastic vasculitis (with destruction of dermal blood vessel walls) is not uncommon but may be secondary to severe inflammation of the skin or even infection.22
No prospective randomized trials have been performed to determine the optimal treatment of PG, and only a few studies with case numbers of more than 15 patients have been published. High oral doses of systemic corticosteroids (>60 mg/day) and cyclosporine are the best documented treatments and are often used as first line therapy. Both medications are useful for immediate control of the disease but are not suitable for long term use because of their side effects. It has been reported that methylprednisolone, 0.5 to 1.0 mg/kg/day, or cyclosporine at 5 mg/kg/day alone or in combination can be of benefit within 24 hours.23–27 Methylprednisolone might be preferable to prednisone, especially since the latter needs to be converted to the first in the liver for optimal effects. Patients with PG have to be on oral corticosteroids for at least several weeks, as short tapering courses of corticosteroids (over a week, for example, are ineffective.24 Pulse therapy with intravenous (I.V.) corticosteroids can lead to rapid improvement (Figure 3A–D). Therapy with I.V. methylprednisolone (1g/day) for 3–5 days is the typical regimen. We prefer a duration of 5 days. In our opinion, methylprednisolone I.V. pulse therapy is most safely performed with the patient on telemetry, for unpredictable electrolyte imbalances may lead to fatal arrhythmias and even sudden death. Concomitant use of a diuretic is a relative contraindication to the use of pulse methylprednisolone therapy.25, 28–29
Figure 3. Patient with pyoderma gangrenosum treated with pulse steroids overlapped with mycophenolate mofetil as corticosteroid sparing agent.

(A) The appearance of a PG ulcer prior to treatment. (B) Dramatic impovement after second day of pulse methylprednisolone. (C) Six days after pulse steroids the ulcers decreased markedly in size. (D) Healed ulcer 3 weeks after treatment.
Complications of therapy for PG need to be carefully considered. Patients on long term systemic corticosteroids are at a risk of developing adrenal suppression, osteoporosis, peptic ulcers, steroid psychosis, among other complications; avascular necrosis of the femoral head can occur very rapidly and at the onset of therapy. Nephrotoxicity, worsening of hypertension, congestive heart failure, increased risk of malignancy are associated with cyclosporine. We prefer not to use cyclosporine in patients with hypertension or evidence of coronary artery disease.
Tumor necrosis factor- α antibodies or inhibitors have been used for treatment of PG. The most studied anti-TNF-α drug for PG is infliximab. Infliximab is a humanized monoclonal antibody against TNF-α that prevents the activation of pro-inflammatory cytokines resulting from the recruitment of inflammatory cells to tissue sites.30,31 Generally, infliximab is administered with an induction dosing regimen infused as 5 mg/kg at weeks 0, 2, and 6. The optimal dosing of infliximab for PG is not known. There is controversy about whether infliximab is effective in patients with PG but without underlying inflammatory bowel disease (IBD).32–36 Immune-related side effects to infliximab, such as fever, chills, pruritus, urticaria, low or high blood pressure, and dyspnea are most common reported. Infliximab therapy is associated with an increased risk of infections, including tuberculosis (TB). Reactivation of latent TB and hepatitis has been reported. It is not known whether infliximab increases the risk of solid tumors and malignancies; similar biologic drugs do increase the risk of lymphoma. Infliximab treatment may not be suitable for patients with congestive heart failure. Rare cases of neurologic side effects, such as optic neuritis, demyelinating disorders, and multiple sclerosis have been reported with infliximab. Furthermore, a lupus-like drug reaction has been associated with this biologic agent. Development of antibodies to infliximab increases the frequency of infusion reactions and shortens the duration of remission.31
Etanercept is a dimeric soluble form of the TNF-α receptor that prevents ligand binding to TNF receptors on the cell, thus rendering TNFα inactive. In several publications, etanercept has been reported to be beneficial in the treatment of PG, at dosages of 25 to 50 mg weekly.37–40 An advantage of using etanercept (over infliximab, for example) is that it can be self-administered subcutaneously by the patient. Side effects are of etenercept are similar to those observed with infliximab. However, etanercept has been linked to the development of lymphomas in children treated with this drug before the age of 18.
Other therapies for PG, such as mycophenolate mofetil, dapsone, thalidomide,and I.V. immunoglobulins have been used alone or in combination with systemic corticosteroids.41–45 For patients with limited and superficial PG, topical or intralesional corticosteroids and topical tacrolimus have been reported to be effective.46–47 Goals of local wound care are relief of pain, prevention of colonization/infection, and to support rapid reepithelialization. Bioengineered skin has been successfully used to stimulate reepithelialization and reduce pain, while circumventing the problem of pathergy associated with the use of autologous grafts.48
Vasculitis
Inflammatory processes involving blood vessels can lead to skin ulceration. Vasculitis results from inflammation of the blood vessel wall leading to hemorrhage, ischemia, and infarction. This sequence of events may be associated with connective tissue diseases (collagen vascular diseases), malignancy, or a drug reaction. Systemic symptoms of fevers, malaise, and joint pains necessitate further workup for a connective tissue disease.49–50 Differently sized blood vessels may be affected. Vasculitis of the small dermal vessels manifests as palpable purpura, vesicobullous lesions, and superficial ulcers with regular borders. Vasculitis of the muscular arteries presents as painful red nodules, punched-out irregularly shaped ulcers, or gangrene51 (Figure 4). Often, patients with vasculitis will have systemic symptoms. The lower extremities are most commonly affected; involvement of the upper extremity, trunk and head is an indication of more severe systemic disease.52
Figure 4. Diagrammatic representation of the difference in small versus medium-sized (deeper) vessel vasculitis and the impact on skin findings.

A superficial vasculitis (left side of diagram) leads to a wedge-shaped area of necrosis and, thus, a well defined and regular skin purpura or necrosis. Conversely, occlusion of a deep vessel (right side of diagram) leaves open the chance for anastomosing vessels to alter the effect at the skin.
The key steps in the diagnosis of vasculitis are to determine blood vessel damage and the vessel size involved. The histologic pattern, supported by immunofluorescence findings, ANCA status, and workup for systemic disease, are the essential elements for diagnosis and for appropriate treatment. Skin biopsies for routine studies as well as immunofluorescence should be performed on the most recent lesions, preferably those present for <48 hours. Since many ulcers due to vasculitis involve the medium sized vessels, a skin biopsy extending to the subcutaneous fat is necessary. In skin lesions presenting with a livedoid pattern, the skin biopsy must be taken from the paler center. Non-ulcerated areas are preferred for biopsy. However, when biopsies are taken from the ulcer bed, it is important to include the ulcer's edge49, 53–54 (Figure 5). In addition to the biopsy, a complete blood count (CBC) with differential, renal and liver function tests, urinalysis, fecal occult blood testing, hepatitis B and C serologies, antiphospholipid antibody testing, immunoglobulin levels, complement levels, ANA, RF and p- and c-ANCA testing are recommended. Blood cultures and echocardiography may be considered in patients with a history of intravenous drug use or a high fever and a heart murmur.51 Vasculitis may present as a single episode 7–10 days after exposure to a systemic drug or from infection; the vasculitis may resolve in 1–3 months, but an intermittent and relapsing course with ulceration is seen in connective tissue disease (CTD) associated vasculitis, cryoglobulinemia, and malignancy.49
Figure 5. Example of a biopsy for an inflammatory ulcer.
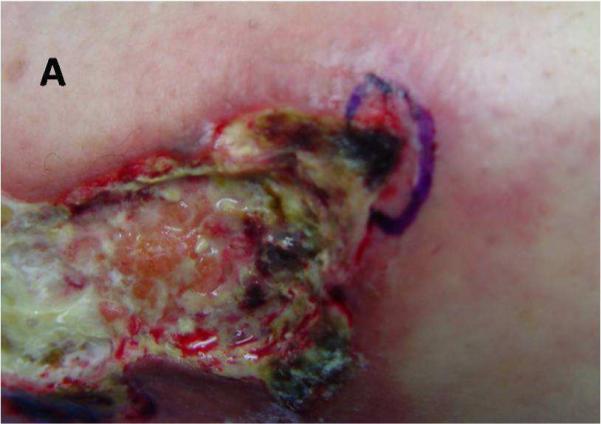
Excisional biopsy marked on edge of ulcer including surrounding erythema.
Microvascular occlusive processes due to cryofibrinogenemia, the antiphospholipid antibody syndrome, hypercoagulable states from protein C and S deficiency, or caused by medication (such as warfarin) are important differential diagnoses to consider when livedoid skin lesions are observed. In many cases, the red or hyperpigmented cutaneous streaks are subtle (microlivedo), and certainly don't resemble the full-blown net-like hyperpigmentation one sees in livedo reticularis.
Leukocytoclastic vasculitis may present as superficial ulcerations and with hemorrhagic bullae on the lower portion of the leg and often without extra cutaneous involvement. However, constitutional symptoms may be present. A minority of patients may have renal or gastrointestinal involvement. Skin biopsy will show a small vessel vasculitis, while immunofluorescence may reveal complement and immunoglobulin deposition on vessel walls. Trigger factors for vasculitis are drugs, infection, and, rarely, an underlying connective tissue disease. Septic vasculitis is generally a small vessel vasculitis, as in leukocytoclastic vasculitis, that is immune complex negative and is often caused by endocarditis and septicemia from a variety of microorganisms. Typical skin findings are purpura, pustules, hemorrhagic bullae and ulcers. Blood cultures and echocardiography are essential for diagnosis.55 Cutaneous small vessel vasculitis occurs frequently in SLE, RA, and Sjogren's. In general, there is multi-organ involvement. The vasculitis may also affect larger blood vessels, as shown by the histology. Physical findings vary from palpable purpura, to ulcers, to digital gangrene indicating significant arterial involvement.51
However, certain conditions affect larger blood vessels more commonly. Wegener's granulomatosis is a disease that presents with lung, kidney and skin involvement. It is associated with a destructive upper respiratory tract disease, saddle-nose deformities, erosive sinusitis, and subglottic stenosis. Patients will often have multiple lung nodules, infiltrates or alveolar hemorrhage, and segmental necrotizing glomerulonephritis. Skin findings may be palpable purpura, subcutaneous nodules, ulcers and digital infarcts, vesicobullous lesions, and gingival hyperplasia. Vasculitis in this systemic condition affects small to medium sized vessels. Biopsies from the upper respiratory tract may be diagnostic. A pattern of positive c-ANCA and PR-3- ANCA is strongly associated with WG. When untreated, the condition may approache a mortality of 80%.51 Cutaneous polyarteritis nodosa (PAN) manifests as painful nodules progressing to ulceration with neuropathy. The clinical manifestations of PAN represent a spectrum, varying from nodules, livedo reticularis, and polyneuropathy, to severe disease with cutaneous ulcers, pain, sensory neuropathy, fever, malaise and arthralgia and progressive systemic disease; acral gangrene, foot drop, mononeuropathy multiplex, and musculoskeletal involvement can occur.56 When the medium-sized blood vessel involvement of PAN is strongly suspected, multiple deep punch or incisional biopsies down to the fascia may be required to diagnose this disorder. Moreover, serial sections through the specimen are frequently necessary, because the vasculitis is focal and segmental.57 (Figure 6A)
Figure 6. Histologic findings in different inflammatory ulcers.
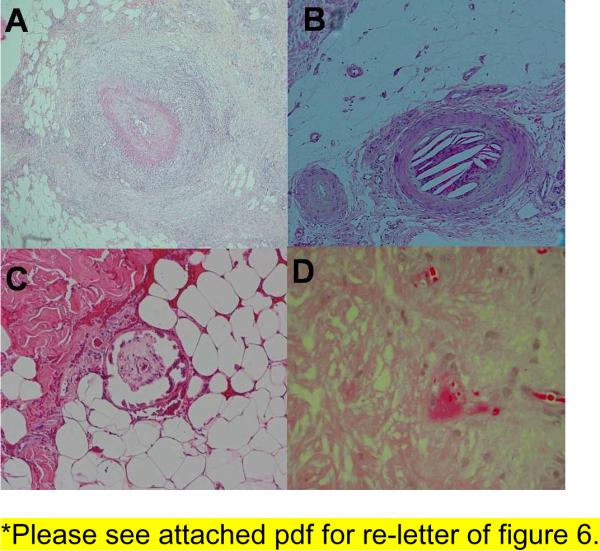
(A) Dense inflammation around a medium sized artery seen in polyarteritis nodosa. (B) Cleft like spaces inside a vessel seen in cholesterol emboli. (C) Fibrin thrombi inside a vessel seen in cryofibrinogenemia. (D) Calcification of medial layer of a blood vessel seen in calciphylaxis.
In PAN, treatment of the underlying disease often leads to resolution of the vasculitis. Mild disease may be treated with dapsone or colchicine. More severe or unresponsive PAN is managed with prednisone and/or with a corticosteroid sparing agent. Systemic vasculitis with internal organ involvement may require the use of prednisone and cyclophosphamide, frequently in combination. Once remission has been achieved, maintenance with methotrexate or azathioprine alone is recommended.58–59
Cholesterol embolism (CE) (Figure 6B[E1])
Patients present with varied skin manifestations such as livedo reticularis, purpura, cyanosis, blue toes, digital gangrene, painful subcutaneous nodules, renal failure; interestingly, arterial hypertension is very uncommon. Patients may also have fever, weight loss, myalgia, and anemia. Large vessels may be the source of the embolus, from which fragments separate and occlude the microcirculation. Several organ systems may be affected, with azotemia, abdominal pain from mucosal ulceration, gastrointestinal involvement from bleeding, ischemia, infarction, and perforation. Myocardial infarction, stroke, and transient ischemic attacks are not uncommon.60–64 Cholesterol embolism (CE) occurs spontaneously or may be iatrogenic. Pathophysiologically, CE is the result of shearing forces causing atherosclerotic plaque rupture. Iatrogenic CE is caused by an endovascular surgical procedure or the initiation of anticoagulant therapy (warfarin, heparin). However, in some cases CE can develop weeks to months from the time of surgery/endovascular procedures or initiation of anticoagulation.61, 65
Patients at high risk for CE are those with severe atherosclerotic disease. The formation of a fibrofatty plaque within the vessel wall, composed of a necrotic core of cell debris, foam cells, lipid and cholesterol crystals and an overlying fibrous cap, is the central pathologic entity in atherosclerosis. When a plaque ruptures, a thrombus forms and breaks up, leading to <100 μ cholesterol fragments that embolize and occlude vessels in distant sites and affect different organ systems, including the skin. After the cholesterol crystals occlude the microcirculation, inflammation ensues, followed by a foreign body reaction with thrombus formation and eventual fibrosis. This sequence of events results in ischemia and infarction.66 The presence of skin findings, such as livedo reticularis, microlivido, or the blue toe syndrome, in addition to progressive renal failure, is highly suggestive of cholesterol crystal embolization.65 However, a skin biopsy showing characteristic biconvex needle like cleft in vessels on histology is detected in up to 92% of patients and is specific for CE61(Figure 6C). An optimal biopsy site is a tender nodule. However, the livido area also has a high yield. CE has a high mortality rate; in one review of 221 patients mortality was 81%.62
There is no established treatment for CE. When possible, anticoagulants should be withdrawn and further vascular procedures should be avoided after a diagnosis is established.67–68 Aggressive supportive treatment has been reported to improve one year survival rates.65 In that report, this approach included maintaining the patient's nutritional status, treating or preventing cardiac failure, and using hemodialysis. Systemic corticosteroids were started with evidence of recurrent CE or inflammation. However, the use of systemic corticosteroids is not consistently successful. Care must be exercised when patients are treated with corticosteroids, as fluid retention and alterations in electrolyte imbalance will further complicate the patient's cardiac status.69 Statins, which can stabilize plaques, have been shown to decrease the risk of progression to end stage renal disease and have improved the one year cumulative survival.70 Surgical intervention (i.e., removing the thrombus) in CE patients is associated with significant morbidity, and its use is reserved for cases where immediate survival is at stake.65
Calciphylaxis
Calciphylaxis has been classically described as the sudden development of tender, violaceous, and reticulate lesions that progress to necrotic ulcerations of the lower extremities in patients with renal failure on chronic hemodialysis. These ulcers heal poorly and are very painful. Areas commonly affected are those with thick adipose tissue, such as the abdomen, breasts, and buttocks. There is often an elevated parathyroid (PTH) level (secondary to renal failure), with abnormal calcium and phosphate metabolism.71 Some authors suggest that proximal ulcerations involving the abdomen and buttocks have a worse prognosis.72 Recently, this classical concept and clinical context of calciphylaxis have been changing, as shown by several published case reports indicating a similar clinical course in the absence of renal failure, no obvious dysregulation of calcium and phosphate metabolism, and with normal PTH levels. However, a consistent histologic finding is calcification of the vessel walls, eventually leading to thrombosis and infarction of the skin. Other risk factors associated with the development of calciphylaxis include the following: obesity, liver disease, systemic corticosteroid use, and an elevated calcium phosphorus product.73 In addition, contributing factors are diabetes mellitus, warfarin use, decreased functional protein C and S activity, vitamin D supplementation, and low albumin levels. Thus, in the proper clinical context from physical examination and histologic findings, calciphylaxis should be considered even in the absence of kidney disease.74 Peripheral pulses are preserved, since only the smaller vessels are affected by calciphylaxis. Histology of the cutaneous lesions shows calcium deposits within the blood vessel walls, mainly in the media and intima of small arterioles and venules, frequently with a concentric, circumferential, and ring like pattern (Figure 6D). These findings are seen in the dermis and the subcutaneous fat. Septal and lobular fat necrosis and epidermal ulceration with dermal necrosis are also identified.75
Treatment is of calcyphylaxis is mainly supportive. The goals of local wound care are to manage drainage with wound dressings, improve pain, treat infection, and stimulate healing (Figure 7A–D). If altered calcium metabolism and hyperparathyroidism are identified, treatment is directed to normalize these abnormalities. Several therapeutic measures have been tried, and with varied success. Simple approaches, such as establishing a diet with low phosphate intake and initiating oral phosphate binders, can be started. Bisphosphonates have been used because of their inhibitory effect on macrophages and pro-inflammatory cytokines to prevent further calcification. For example, pamidronate and etidronate have been used with some success.76–78 Cinacalcet is a new calcimimetic agent that works by increasing the sensitivity of the calcium sensing receptor on the parathyroid gland, thereby decreasing PTH levels and normalizing calcium levels.79 This drug has been useful in patients with secondary hyperparathyroidism.80–81 Sodium thiosulfate is thought to dissolve calcium deposits in tissues by forming soluble calcium thiosulfate complexes. It is given I.V. and has been reported to improve pain and wound healing.82–84 Parathyroidectomy in patients with calciphylaxis and elevated PTH benefits wound healing and improves short term mortality; the long term benefits of this approach are unknown.85–87
Figure 7. Patient with calciphylaxis without renal failure, normal parathyroid levels, normal calcium and phosphate product.

(A) Painful ulcer with background livedo reticularis. (B) Worsening ulcer not responsive to pulse steroids, azathioprine, pentoxifylline, antibiotics and debridement. Skin biopsy showed calciphylaxis. (C) Repeat debridement, IV antibiotics, bioengineered skin applied to ulcer with good response; Azathioprine discontinued after one month, compression therapy, pain management, antiseptic dressings were used.(D) Ulcer healed after 3 months.
The prognosis of calcyphylaxis is generally poor, and the condition is associated with a mortality as high as 60–80%.73 Sepsis occurs commonly, with the ulceration as the probable portal of entry for infection.
Cryofibrinogenemia and Cryoglobulinemia
Signs and symptoms of cryofibrinogenemia are due to cutaneous ischemia. Patients present with livedo reticularis or subtle areas of microlivedo, purpura, ecchymoses, and painful ulcerations that, strangely, are not commonly precipitated by cold ambient temperatures. Systemic symptoms of malaise and fever are common. Often, there is an underlying inflammatory disease such as malignancy, diabetes mellitus, collagen vascular disease, or an infection. Essential cryofibrinogenemia occurs without a secondary cause and should be considered in healthy patients presenting with unexplained areas of cutaneous ischemia.88 Rarely, other thrombotic events such as myocardial infarction, thrombophlebitis, and cerebral infarction are seen. Paradoxical spontaneous bleeding due to consumption of clotting factors may occur.88
One needs to contrast cryofibrinogenemic versus cryoglobulinemic ulcers. The diagnosis of cryofibrinogenemic ulcers is made in a clinical setting of sudden onset of skin changes and ulcerations in the presence of elevated plasma cryofibrinogen and without serum cryoglobulins and evidence of other inflammatory causes of micro-occlusive diseases; the relationship of onset of pain, discomfort, and ulceration to cold exposure is uncommon. In contrast, cryoglobulinemic ulcers are definitely accommpanied by purpura triggered by cold exposure or prolonged standing plus arthralgia and weakness; cryoglobulins are detected in serum. Cryoglobulins are associated with connective tissue diseases, hematologic malignancies, and hepatitis. Cryofibrinogenemia is commonly idiopathic, but can be associated with myeloproliferative disorders and connective tissue diseases, Treatment of the underlying disease causing these cryoproteinemias leads to improvement of symptoms and physical findings.
On skin biopsy of ulcers due to cryoproteinemias, one sees small and medium sized vessel vasculitis. In terms of distinction, cryofibrinogens are a cold, insoluble complex of fibrin, fibrinogen and fibrin split products with albumin, plasma proteins and immunoglobulins. Cryofibrinogen forms a clot with thrombin and reversibly precipitates in plasma at 4° celsius89, whereas cryoglobulins are cold precipitating immunoglobulins in serum and dissolve when warmed. A skin biopsy showing fibrin occluding the dermal blood vessels, angiogram with abrupt occlusion of small to medium sized arteries, and elevation of serum levels of α-1 antitrypsin and α-2macroglobulin support a diagnosis of cryofibrinogenemia. High levels of α-1 antitrypsin and α2- macroglobulin inhibit plasmin and fibrinolysis, which leads to the thrombotic occlusion of small to medium arteries by the cryofibrinogens.90–91 The detection of cryofibrinogens in plasma requires special handling and attention. Blood must be collected in tubes containing oxalate, citrate, or ethylenediaminetetraacetic acid as the anticoagulant. After collection, the blood must be quickly centrifuged at 37° C. Blood should also be collected in a plain glass tube, allowed to clot, and then processed in a similar fashion in order to detect the presence of cryoglobulins.
Treatment of cryofibrinogenemic ulcers includes avoidance of cold exposure and local wound care. The anabolic steroid stanozolol has an established fibrinolytic effect and has been used with considerable success in the past.92–93 However, this drug has now been withdrawn from the market and is no longer easily available as a reliable preparation. However, we have now used danazol with success (V. Falanga, unpublished). Systemic corticosteroids alone do not appear to be an effective therapy. Treatment of the underlying condition may lead to improvement. Plasmapheresis has been effective in a few reports.93–97 We have had success with the use of combined pentoxifylline and colchicine in one patient with cryofibrinogenemic ulcers.98 (Figure 8A,B)
Figure 8. Patient with cryofibrinogenemia.

(A) Typical presentation of livedo reticularis. (B) Ulcers healed with compression pentoxifylline and colchicine.
Necrobiosis Lipoidica Diabeticorum
Necrobiosis lipoidica diabeticorum (NLD) is characterized by single or multiple asymptomatic red to yellow shiny plaques that gradually enlarge and within which, due to atrophy, dermal blood vessels are readily apparent. Ulceration is a complication of NLD, and commonly develops after trauma to the lesions. Almost all patients with NLD have diabetes mellitus, or will develop it, or have a strong family history of it. However, less than 1% of diabetic patients have NLD.99–100 The pathogenic steps leading to NLD are unknown. Skin biopsy of the lesions shows an atrophic epidermis and dermal areas of necrobiotic collagen surrounded by palisading histiocytes and extending to the subcutaneous fat. Spontaneous resolution of NLD has been reported to occur in about 20% of cases. A variety of treatments have been used for treatment of NLD, including topical and intralesional corticosteroids, topical tacrolimus, psoralens plus UVA (PUVA), and antimalarials. In patients with severe ulcerations within NLD, infliximab, etanercept, pentoxifylline, cyclosporine and thalidomide have been used. Interestingly, tight control of the patient's diabetes does not improve NL.101–108
Warfarin Induced Skin Necrosis
Sudden development of paresthesia, edema, petechiae, and ecchymoses progressing to full-thickness necrosis and deep ulcers within 3–10 days after the initiation of warfarin therapy should alert the clinician to the possibility of warfarin- induced skin necrosis (WISN).109–111 WISN has been described in patients on anticoagulation, typically several days after therapy is stopped or on re-treatment.111–113 WISN is thought to be the result of a paradoxical hypercoagulable state, possibly due to inhibition of vitamin K dependent coagulation factors relative to other factors, thus producing a transient imbalance that predisposes to clotting. Protein C deficiency, protein S deficiency, and anti-thrombin III deficiency are considered a risk factor in the development of WISN. However, not all patients with protein C, S and anti-thrombin III deficiencies will develop WISN.111, 114–116
The diagnosis of WISN is made by history and/or with a skin biopsy showing histologic findings of microthrombi and thrombi within the blood vessels of the dermis and subcutaneous tissue, including capillaries and venules; typically, there is no vasculitis. WISN needs to be differentiated from heparin-induced necrosis associated with heparin-induced thrombocytopenia and thrombosis (HITT), since both warfarin and heparin are frequently used concurrently. Both conditions present with skin necrosis. HITT is associated with a 50% reduction in platelet levels from baseline or a platelet count less than 150. However, platelet counts are frequently normal in HITT with skin ulceration. Symptoms begin at 4–12 days after starting heparin. The heparin PF-4 antibody is frequently present.109, 117 In contrast to WISN, the thrombus in HITT is mostly composed of platelets; true arterial thrombosis is common.118
Early diagnosis and immediate withdrawal of warfarin improves the prognosis of WISN. Replacement of clotting factors with fresh frozen plasma and the use of an alternate anticoagulant such as heparin may be required. Thus, it is important to distinguish between WISN and HITT, because the mainstay of treatment of HITT is discontinuation of heparin and switching to a different anticoagulant such as argatroban; warfarin use alone is contraindicated in HITT.119 Aggressive wound care and surgical intervention to remove devitalized tissue are needed in many patients. Antibiotics are also frequently used but given with caution, since antibiotics may alter or decrease the vitamin K-producing flora in the gut. Treatment with systemic corticosteroids or vasodilators has not improved WISN.109, 115 Useful measures to prevent warfarin necrosis are to avoid initial loading with warfarin and starting anticoagulation first with unfractionated heparin and overlapping with warfarin for about five days.114–115
Conclusion
The sudden development of painful ulcerations with systemic symptoms in the context of an underlying disease, change in medications, and/or recent surgical intervention points to an inflammatory cause of the ulceration. A broad differential diagnosis exists for these ulcers. History, physical examination, and laboratory workup should be done, with careful consideration of an autoimmune, infectious, and or occlusive process as the cause of ulceration. Providing wound care with the goals of optimizing wound healing by decreasing swelling, controlling the bacterial burden, and avoiding an irritant or allergic contact dermatitis are equally important. Treatment of the underlying illness will often improve the ulceration as well. Since these diseases are not so common, no specific guidelines have been developed. Treatment should be carefully chosen, with emphasis on minimizing possible complications with its use. A multi-disciplinary approach is necessary to manage these patients, and consultations with different specialties should be the rule. Re-evaluation of the diagnosis and management are advised if the ulcers fail to respond to treatment.
Acknowledgments
This work was supported by the NIH Center of Biomedical Research Excellence (P20RR018757) at Roger Williams Medical Center (PI: V. Falanga)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure and Products Page The authors have no conflict of interest to declare.
References
- 1.Herber OR, Schnepp W, Rieger MA. A systematic review on the impact of leg ulceration on patients' quality of life. Health Qual Life Outcomes. 2007;5:44. doi: 10.1186/1477-7525-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panuncialman J, Falanga V. The science of wound bed preparation. Clin Plast Surg. 2007 Oct;34(4):621–632. doi: 10.1016/j.cps.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Brunsting LA, Goeckerman WH, O'Leary PA. Pyoderma (echthyma) gangrenosum: clinical and experimental observations in 5 cases occuring in adults. Arch Dermatol Syphilol. 1930;22:655–680. [Google Scholar]
- 4.Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002 Oct 31;347(18):1412–1418. doi: 10.1056/NEJMoa013383. [DOI] [PubMed] [Google Scholar]
- 5.Powell FC, Schroeter AL, Su WP, Perry HO. Pyoderma gangrenosum: a review of 86 patients. Q J Med. 1985 May;55(217):173–186. [PubMed] [Google Scholar]
- 6.Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996 Mar;34(3):395–409. doi: 10.1016/s0190-9622(96)90428-4. quiz 410–392. [DOI] [PubMed] [Google Scholar]
- 7.Duguid CM, O'Loughlin S, Otridge B, Powell FC. Paraneoplastic pyoderma gangrenosum. Australas J Dermatol. 1993;34(1):17–22. doi: 10.1111/j.1440-0960.1993.tb00841.x. [DOI] [PubMed] [Google Scholar]
- 8.Langan SM, Powell FC. Vegetative pyoderma gangrenosum: a report of two new cases and a review of the literature. Int J Dermatol. 2005 Aug;44(8):623–629. doi: 10.1111/j.1365-4632.2005.02591.x. [DOI] [PubMed] [Google Scholar]
- 9.Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000 Sep 27;284(12):1546–1548. doi: 10.1001/jama.284.12.1546. [DOI] [PubMed] [Google Scholar]
- 10.Sheldon DG, Sawchuk LL, Kozarek RA, Thirlby RC. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000 May;135(5):564–568. doi: 10.1001/archsurg.135.5.564. discussion 568–569. [DOI] [PubMed] [Google Scholar]
- 11.Hubbard VG, Friedmann AC, Goldsmith P. Systemic pyoderma gangrenosum responding to infliximab and adalimumab. Br J Dermatol. 2005 May;152(5):1059–1061. doi: 10.1111/j.1365-2133.2005.06467.x. [DOI] [PubMed] [Google Scholar]
- 12.Field S, Powell FC, Young V, Barnes L. Pyoderma gangrenosum manifesting as a cavitating lung lesion. Clin Exp Dermatol. 2008 Jul;33(4):418–421. doi: 10.1111/j.1365-2230.2008.02756.x. [DOI] [PubMed] [Google Scholar]
- 13.Kanoh S, Kobayashi H, Sato K, Motoyoshi K, Aida S. Tracheobronchial pulmonary disease associated with pyoderma gangrenosum. Mayo Clin Proc. 2009 Jun;84(6):555–557. doi: 10.4065/84.6.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kruger S, Piroth W, Amo Takyi B, Breuer C, Schwarz ER. Multiple aseptic pulmonary nodules with central necrosis in association with pyoderma gangrenosum. Chest. 2001 Mar;119(3):977–978. doi: 10.1378/chest.119.3.977. [DOI] [PubMed] [Google Scholar]
- 15.Ross HJ, Moy LA, Kaplan R, Figlin RA. Bullous pyoderma gangrenosum after granulocyte colony-stimulating factor treatment. Cancer. 1991 Jul 15;68(2):441–443. doi: 10.1002/1097-0142(19910715)68:2<441::aid-cncr2820680239>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Montoto S, Bosch F, Estrach T, Blade J, Nomdedeu B, Nontserrat E. Pyoderma gangrenosum triggered by alpha2b-interferon in a patient with chronic granulocytic leukemia. Leuk Lymphoma. 1998 Jun;30(1–2):199–202. doi: 10.3109/10428199809050944. [DOI] [PubMed] [Google Scholar]
- 17.Sanders S, Busam K, Tahan SR, Johnson RA, Sachs D. Granulomatous and suppurative dermatitis at interferon alfa injection sites: report of 2 cases. J Am Acad Dermatol. 2002 Apr;46(4):611–616. doi: 10.1067/mjd.2002.119087. [DOI] [PubMed] [Google Scholar]
- 18.Srebrnik A, Shachar E, Brenner S. Suspected induction of a pyoderma gangrenosum-like eruption due to sulpiride treatment. Cutis. 2001 Mar;67(3):253–256. [PubMed] [Google Scholar]
- 19.Brooklyn TN, Williams AM, Dunnill MG, Probert CS. T-cell receptor repertoire in pyoderma gangrenosum: evidence for clonal expansions and trafficking. Br J Dermatol. 2007 Nov;157(5):960–966. doi: 10.1111/j.1365-2133.2007.08211.x. [DOI] [PubMed] [Google Scholar]
- 20.Magro CM, Kiani B, Li J, Crowson AN. Clonality in the setting of Sweet's syndrome and pyoderma gangrenosum is not limited to underlying myeloproliferative disease. J Cutan Pathol. 2007 Jul;34(7):526–534. doi: 10.1111/j.1600-0560.2006.00654.x. [DOI] [PubMed] [Google Scholar]
- 21.Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004 Nov;43(11):790–800. doi: 10.1111/j.1365-4632.2004.02128.x. [DOI] [PubMed] [Google Scholar]
- 22.Su WP, Schroeter AL, Perry HO, Powell FC. Histopathologic and immunopathologic study of pyoderma gangrenosum. J Cutan Pathol. 1986 Oct;13(5):323–330. doi: 10.1111/j.1600-0560.1986.tb00466.x. [DOI] [PubMed] [Google Scholar]
- 23.Matis WL, Ellis CN, Griffiths CE, Lazarus GS. Treatment of pyoderma gangrenosum with cyclosporine. Arch Dermatol. 1992 Aug;128(8):1060–1064. [PubMed] [Google Scholar]
- 24.Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB, Jr., White WL, Callen JP. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore) 2000 Jan;79(1):37–46. doi: 10.1097/00005792-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Chow RK, Ho VC. Treatment of pyoderma gangrenosum. J Am Acad Dermatol. 1996 Jun;34(6):1047–1060. doi: 10.1016/s0190-9622(96)90285-6. [DOI] [PubMed] [Google Scholar]
- 26.Vidal D, Puig L, Gilaberte M, Alomar A. Review of 26 cases of classical pyoderma gangrenosum: clinical and therapeutic features. J Dermatolog Treat. 2004 Jun;15(3):146–152. doi: 10.1080/09546630410031909. [DOI] [PubMed] [Google Scholar]
- 27.Elgart G, Stover P, Larson K, et al. Treatment of pyoderma gangrenosum with cyclosporine: results in seven patients. J Am Acad Dermatol. 1991 Jan;24(1):83–86. doi: 10.1016/0190-9622(91)70016-u. [DOI] [PubMed] [Google Scholar]
- 28.Prystowsky JH, Kahn SN, Lazarus GS. Present status of pyoderma gangrenosum. Review of 21 cases. Arch Dermatol. 1989 Jan;125(1):57–64. [PubMed] [Google Scholar]
- 29.Johnson RB, Lazarus GS. Pulse therapy. Therapeutic efficacy in the treatment of pyoderma gangrenosum. Arch Dermatol. 1982 Feb;118(2):76–84. doi: 10.1001/archderm.118.2.76. [DOI] [PubMed] [Google Scholar]
- 30.Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005 Aug;53(2):273–283. doi: 10.1016/j.jaad.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Wolverton S. Comprehensive Dermatologic Drug Therapy. second ed. Elsevier; Philadelphia: 2007. [Google Scholar]
- 32.Adisen E, Oztas M, Gurer MA. Treatment of idiopathic pyoderma gangrenosum with infliximab: induction dosing regimen or on-demand therapy? Dermatology. 2008;216(2):163–165. doi: 10.1159/000111515. [DOI] [PubMed] [Google Scholar]
- 33.Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006 Apr;55(4):505–509. doi: 10.1136/gut.2005.074815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regueiro M, Valentine J, Plevy S, Fleisher MR, Lichtenstein GR. Infliximab for treatment of pyoderma gangrenosum associated with inflammatory bowel disease. Am J Gastroenterol. 2003 Aug;98(8):1821–1826. doi: 10.1111/j.1572-0241.2003.07581.x. [DOI] [PubMed] [Google Scholar]
- 35.Romero-Gomez M, Sanchez-Munoz D. Infliximab induces remission of pyoderma gangrenosum. Eur J Gastroenterol Hepatol. 2002 Aug;14(8):907. doi: 10.1097/00042737-200208000-00021. [DOI] [PubMed] [Google Scholar]
- 36.Mimouni D, Anhalt GJ, Kouba DJ, Nousari HC. Infliximab for peristomal pyoderma gangrenosum. Br J Dermatol. 2003 Apr;148(4):813–816. doi: 10.1046/j.1365-2133.2003.05294.x. [DOI] [PubMed] [Google Scholar]
- 37.Roy DB, Conte ET, Cohen DJ. The treatment of pyoderma gangrenosum using etanercept. J Am Acad Dermatol. 2006 Mar;54(3 Suppl 2):S128–134. doi: 10.1016/j.jaad.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 38.Goldenberg G, Jorizzo JL. Use of etanercept in treatment of pyoderma gangrenosum in a patient with autoimmune hepatitis. J Dermatolog Treat. 2005;16(5–6):347–349. doi: 10.1080/09546630500424722. [DOI] [PubMed] [Google Scholar]
- 39.Pastor N, Betlloch I, Pascual JC, Blanes M, Banuls J, Silvestre JF. Pyoderma gangrenosum treated with anti-TNF alpha therapy (etanercept) Clin Exp Dermatol. 2006 Jan;31(1):152–153. doi: 10.1111/j.1365-2230.2005.01972.x. [DOI] [PubMed] [Google Scholar]
- 40.Charles CA, Leon A, Banta MR, Kirsner RS. Etanercept for the treatment of refractory pyoderma gangrenosum: a brief series. Int J Dermatol. 2007 Oct;46(10):1095–1099. doi: 10.1111/j.1365-4632.2007.03286.x. [DOI] [PubMed] [Google Scholar]
- 41.Nousari HC, Lynch W, Anhalt GJ, Petri M. The effectiveness of mycophenolate mofetil in refractory pyoderma gangrenosum. Arch Dermatol. 1998 Dec;134(12):1509–1511. doi: 10.1001/archderm.134.12.1509. [DOI] [PubMed] [Google Scholar]
- 42.Daniels NH, Callen JP. Mycophenolate mofetil is an effective treatment for peristomal pyoderma gangrenosum. Arch Dermatol. 2004 Dec;140(12):1427–1429. doi: 10.1001/archderm.140.12.1427. [DOI] [PubMed] [Google Scholar]
- 43.Hecker MS, Lebwohl MG. Recalcitrant pyoderma gangrenosum: treatment with thalidomide. J Am Acad Dermatol. 1998 Mar;38(3):490–491. doi: 10.1016/s0190-9622(98)70513-4. [DOI] [PubMed] [Google Scholar]
- 44.Hagman JH, Carrozzo AM, Campione E, Romanelli P, Chimenti S. The use of high-dose immunoglobulin in the treatment of pyoderma gangrenosum. J Dermatolog Treat. 2001 Mar;12(1):19–22. doi: 10.1080/095466301750163527. [DOI] [PubMed] [Google Scholar]
- 45.Altman J, Mopper C. Pyoderma gangrenosum treated with sulfone drugs. Minnesota medicine. 1966;49(1):22–26. [PubMed] [Google Scholar]
- 46.Lally A, Hollowood K, Bunker CB, Turner R. Penile pyoderma gangrenosum treated with topical tacrolimus. Arch Dermatol. 2005 Sep;141(9):1175–1176. doi: 10.1001/archderm.141.9.1175. [DOI] [PubMed] [Google Scholar]
- 47.Kontos AP, Kerr HA, Fivenson DP, Remishofsky C, Jacobsen G. An open-label study of topical tacrolimus ointment 0.1% under occlusion for the treatment of pyoderma gangrenosum. Int J Dermatol. 2006 Nov;45(11):1383–1385. doi: 10.1111/j.1365-4632.2006.03133.x. [DOI] [PubMed] [Google Scholar]
- 48.de Imus G, Golomb C, Wilkel C, Tsoukas M, Nowak M, Falanga V. Accelerated healing of pyoderma gangrenosum treated with bioengineered skin and concomitant immunosuppression. J Am Acad Dermatol. 2001 Jan;44(1):61–66. doi: 10.1067/mjd.2001.107962. [DOI] [PubMed] [Google Scholar]
- 49.Carlson JA, Cavaliere LF, Grant-Kels JM. Cutaneous vasculitis: diagnosis and management. Clin Dermatol. 2006 Sep–Oct;24(5):414–429. doi: 10.1016/j.clindermatol.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 50.Carlson JA, Ng BT, Chen KR. Cutaneous vasculitis update: diagnostic criteria, classification, epidemiology, etiology, pathogenesis, evaluation and prognosis. Am J Dermatopathol. 2005 Dec;27(6):504–528. doi: 10.1097/01.dad.0000181109.54532.c5. [DOI] [PubMed] [Google Scholar]
- 51.Chen KR, Carlson JA. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol. 2008;9(2):71–92. doi: 10.2165/00128071-200809020-00001. [DOI] [PubMed] [Google Scholar]
- 52.Ioannidou DJ, Krasagakis K, Daphnis EK, Perakis KE, Sotsiou F, Tosca AD. Cutaneous small vessel vasculitis: an entity with frequent renal involvement. Arch Dermatol. 2002 Mar;138(3):412–414. doi: 10.1001/archderm.138.3.412. [DOI] [PubMed] [Google Scholar]
- 53.Grzeszkiewicz TM, Fiorentino DF. Update on cutaneous vasculitis. Semin Cutan Med Surg. 2006 Dec;25(4):221–225. doi: 10.1016/j.sder.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Sais G, Vidaller A, Jucgla A, Servitje O, Condom E, Peyri J. Prognostic factors in leukocytoclastic vasculitis: a clinicopathologic study of 160 patients. Arch Dermatol. 1998 Mar;134(3):309–315. doi: 10.1001/archderm.134.3.309. [DOI] [PubMed] [Google Scholar]
- 55.Carlson JA, Chen KR. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopathol. 2006 Dec;28(6):486–506. doi: 10.1097/01.dad.0000246646.45651.a2. [DOI] [PubMed] [Google Scholar]
- 56.Carlson JA, Chen KR. Cutaneous vasculitis update: neutrophilic muscular vessel and eosinophilic, granulomatous, and lymphocytic vasculitis syndromes. Am J Dermatopathol. 2007 Feb;29(1):32–43. doi: 10.1097/01.dad.0000245198.80847.ff. [DOI] [PubMed] [Google Scholar]
- 57.Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989 Dec;16(6):429–442. doi: 10.1111/j.1346-8138.1989.tb01582.x. [DOI] [PubMed] [Google Scholar]
- 58.Lapraik C, Watts R, Scott DG. Modern management of primary systemic vasculitis. Clin Med. 2007 Jan–Feb;7(1):43–47. doi: 10.7861/clinmedicine.7-1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bosch X, Guilabert A, Espinosa G, Mirapeix E. Treatment of antineutrophil cytoplasmic antibody associated vasculitis: a systematic review. JAMA. 2007 Aug 8;298(6):655–669. doi: 10.1001/jama.298.6.655. [DOI] [PubMed] [Google Scholar]
- 60.Keon WJ, Heggtveit HA, Leduc J. Perioperative myocardial infarction caused by atheroembolism. J Thorac Cardiovasc Surg. 1982 Dec;84(6):849–855. [PubMed] [Google Scholar]
- 61.Falanga V, Fine MJ, Kapoor WN. The cutaneous manifestations of cholesterol crystal embolization. Arch Dermatol. 1986 Oct;122(10):1194–1198. [PubMed] [Google Scholar]
- 62.Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987 Oct;38(10):769–784. doi: 10.1177/000331978703801007. [DOI] [PubMed] [Google Scholar]
- 63.Moolenaar W, Lamers CB. Cholesterol crystal embolisation to the alimentary tract. Gut. 1996 Feb;38(2):196–200. doi: 10.1136/gut.38.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rushovich AM. Perforation of the jejunum: a complication of atheromatous embolization. Am J Gastroenterol. 1983 Feb;78(2):77–82. [PubMed] [Google Scholar]
- 65.Belenfant X, Meyrier A, Jacquot C. Supportive treatment improves survival in multivisceral cholesterol crystal embolism. Am J Kidney Dis. 1999 May;33(5):840–850. doi: 10.1016/s0272-6386(99)70415-4. [DOI] [PubMed] [Google Scholar]
- 66.Gore I, McCombs HL, Lindquist RL. OBSERVATIONS ON THE FATE OF CHOLESTEROL EMBOLI. J Atheroscler Res. 1964 Nov–Dec;4:527–535. doi: 10.1016/s0368-1319(64)80055-7. [DOI] [PubMed] [Google Scholar]
- 67.Bruns FJ, Segel DP, Adler S. Control of cholesterol embolization by discontinuation of anticoagulant therapy. Am J Med Sci. 1978 Jan–Feb;275(1):105–108. doi: 10.1097/00000441-197801000-00013. [DOI] [PubMed] [Google Scholar]
- 68.Kawakami Y, Hirose K, Watanabe Y, et al. Management of multiple cholesterol embolization syndrome--a case report. Angiology. 1990 Mar;41(3):248–252. doi: 10.1177/000331979004100311. [DOI] [PubMed] [Google Scholar]
- 69.Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol. 2003 Sep;17(5):504–511. doi: 10.1046/j.1468-3083.2003.00710.x. [DOI] [PubMed] [Google Scholar]
- 70.Scolari F, Ravani P, Pola A, et al. Predictors of renal and patient outcomes in atheroembolic renal disease: a prospective study. J Am Soc Nephrol. 2003 Jun;14(6):1584–1590. doi: 10.1097/01.asn.0000069220.60954.f1. [DOI] [PubMed] [Google Scholar]
- 71.Dauden E, Onate MJ. Calciphylaxis. Dermatol Clin. 2008 Oct;26(4):557–568. ix. doi: 10.1016/j.det.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 72.Oh DH, Eulau D, Tokugawa DA, McGuire JS, Kohler S. Five cases of calciphylaxis and a review of the literature. J Am Acad Dermatol. 1999 Jun;40(6 Pt 1):979–987. doi: 10.1016/s0190-9622(99)70087-3. [DOI] [PubMed] [Google Scholar]
- 73.Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007 Apr;56(4):569–579. doi: 10.1016/j.jaad.2006.08.065. [DOI] [PubMed] [Google Scholar]
- 74.Kalajian AH, Malhotra PS, Callen JP, Parker LP. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009 Apr;145(4):451–458. doi: 10.1001/archdermatol.2008.602. [DOI] [PubMed] [Google Scholar]
- 75.Essary LR, Wick MR. Cutaneous calciphylaxis. An underrecognized clinicopathologic entity. Am J Clin Pathol. 2000 Feb;113(2):280–287. doi: 10.1309/AGLF-X21H-Y37W-50KL. [DOI] [PubMed] [Google Scholar]
- 76.Monney P, Nguyen QV, Perroud H, Descombes E. Rapid improvement of calciphylaxis after intravenous pamidronate therapy in a patient with chronic renal failure. Nephrol Dial Transplant. 2004 Aug;19(8):2130–2132. doi: 10.1093/ndt/gfh305. [DOI] [PubMed] [Google Scholar]
- 77.Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis. 2006 Jul;48(1):151–154. doi: 10.1053/j.ajkd.2006.04.062. [DOI] [PubMed] [Google Scholar]
- 78.Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol. 2007 Dec;57(6):1021–1025. doi: 10.1016/j.jaad.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 79.Byrnes CA, Shepler BM. Cinacalcet: a new treatment for secondary hyperparathyroidism in patients receiving hemodialysis. Pharmacotherapy. 2005 May;25(5):709–716. doi: 10.1592/phco.25.5.709.63595. [DOI] [PubMed] [Google Scholar]
- 80.Sharma A, Burkitt-Wright E, Rustom R. Cinacalcet as an adjunct in the successful treatment of calciphylaxis. Br J Dermatol. 2006 Dec;155(6):1295–1297. doi: 10.1111/j.1365-2133.2006.07540.x. [DOI] [PubMed] [Google Scholar]
- 81.Velasco N, MacGregor MS, Innes A, MacKay IG. Successful treatment of calciphylaxis with cinacalcet-an alternative to parathyroidectomy? Nephrol Dial Transplant. 2006 Jul;21(7):1999–2004. doi: 10.1093/ndt/gfl114. [DOI] [PubMed] [Google Scholar]
- 82.Brucculeri M, Cheigh J, Bauer G, Serur D. Long-term intravenous sodium thiosulfate in the treatment of a patient with calciphylaxis. Semin Dial. 2005 Sep–Oct;18(5):431–434. doi: 10.1111/j.1525-139X.2005.00082.x. [DOI] [PubMed] [Google Scholar]
- 83.Hayden MR, Tyagi SC, Kolb L, Sowers JR, Khanna R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005;4(1):4. doi: 10.1186/1475-2840-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guerra G, Shah RC, Ross EA. Rapid resolution of calciphylaxis with intravenous sodium thiosulfate and continuous venovenous haemofiltration using low calcium replacement fluid: case report. Nephrol Dial Transplant. 2005 Jun;20(6):1260–1262. doi: 10.1093/ndt/gfh825. [DOI] [PubMed] [Google Scholar]
- 85.Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995 Dec;33(6):954–962. doi: 10.1016/0190-9622(95)90286-4. [DOI] [PubMed] [Google Scholar]
- 86.Arch-Ferrer JE, Beenken SW, Rue LW, Bland KI, Diethelm AG. Therapy for calciphylaxis: an outcome analysis. Surgery. 2003 Dec;134(6):941–944. doi: 10.1016/j.surg.2003.07.001. discussion 944–945. [DOI] [PubMed] [Google Scholar]
- 87.Dereure O, Leray H, Barneon G, Canaud B, Mion C, Guilhou JJ. Extensive necrotizing livedo reticularis in a patient with chronic renal failure, hyperparathyroidism and coagulation disorder: regression after subtotal parathyroidectomy. Dermatology. 1996;192(2):167–170. doi: 10.1159/000246350. [DOI] [PubMed] [Google Scholar]
- 88.Amdo TD, Welker JA. An approach to the diagnosis and treatment of cryofibrinogenemia. Am J Med. 2004 Mar 1;116(5):332–337. doi: 10.1016/j.amjmed.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 89.Nash JW, Ross P, Jr., Neil Crowson A, et al. The histopathologic spectrum of cryofibrinogenemia in four anatomic sites. Skin, lung, muscle, and kidney. Am J Clin Pathol. 2003 Jan;119(1):114–122. doi: 10.1309/KB5A-RGWV-L1R2-BPBN. [DOI] [PubMed] [Google Scholar]
- 90.Smith SB, Arkin C. Cryofibrinogenemia: incidence, clinical correlations, and a review of the literature. Am J Clin Pathol. 1972 Nov;58(5):524–530. doi: 10.1093/ajcp/58.5.524. [DOI] [PubMed] [Google Scholar]
- 91.Blain H, Cacoub P, Musset L, et al. Cryofibrinogenaemia: a study of 49 patients. Clin Exp Immunol. 2000 May;120(2):253–260. doi: 10.1046/j.1365-2249.2000.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Falanga V, Kirsner RS, Eaglstein WH, Katz MH, Kerdel FA. Stanozolol in treatment of leg ulcers due to cryofibrinogenaemia. Lancet. 1991 Aug 10;338(8763):347–348. doi: 10.1016/0140-6736(91)90483-6. [DOI] [PubMed] [Google Scholar]
- 93.Revenga F, Aguilar C, Gonzalez R, Paricio JF, Sanz P, Santos I. Cryofibrinogenaemia with a good response to stanozolol. Clin Exp Dermatol. 2000 Nov;25(8):621–623. doi: 10.1046/j.1365-2230.2000.00722.x. [DOI] [PubMed] [Google Scholar]
- 94.Copeman PW. Cryofibrinogenaemia and skin ulcers: treatment with plasmapheresis. Br J Dermatol. 1979 Jul;101(Suppl 17):57–58. [PubMed] [Google Scholar]
- 95.Kirsner RS, Eaglstein WH, Katz MH, Kerdel FA, Falanga V. Stanozolol causes rapid pain relief and healing of cutaneous ulcers caused by cryofibrinogenemia. J Am Acad Dermatol. 1993 Jan;28(1):71–74. doi: 10.1016/0190-9622(93)70012-i. [DOI] [PubMed] [Google Scholar]
- 96.Rubegni P, Flori ML, Fimiani M, Andreassi L. A case of cryofibrinogenaemia responsive to stanozolol. Br J Haematol. 1996 Apr;93(1):217–219. doi: 10.1046/j.1365-2141.1996.4791016.x. [DOI] [PubMed] [Google Scholar]
- 97.Williamson AE, Cone LA, Huard GS., 2nd Spontaneous necrosis of the skin associated with cryofibrinogenemia, cryoglobulinemia, and homocystinuria. Ann Vasc Surg. 1996 Jul;10(4):365–369. doi: 10.1007/BF02286781. [DOI] [PubMed] [Google Scholar]
- 98.Chartier M, Falanga V. Healing of ulcers due to cryofibrinogenemia with colchicine and high-dose pentoxifylline. Am J Clin Dermatol. 2009;10(1):39–42. doi: 10.2165/0128071-200910010-00007. [DOI] [PubMed] [Google Scholar]
- 99.Braverman IM. Skin Signs of Systemic Disease. 3rd ed. W.B. Saunders; Philadelphia: 1997. [Google Scholar]
- 100.Peyri J, Moreno A, Marcoval J. Necrobiosis lipoidica. Semin Cutan Med Surg. 2007 Jun;26(2):87–89. doi: 10.1016/j.sder.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 101.Zeichner JA, Stern DW, Lebwohl M. Treatment of necrobiosis lipoidica with the tumor necrosis factor antagonist etanercept. J Am Acad Dermatol. 2006 Mar;54(3 Suppl 2):S120–121. doi: 10.1016/j.jaad.2005.11.1042. [DOI] [PubMed] [Google Scholar]
- 102.Noz KC, Korstanje MJ, Vermeer BJ. Ulcerating necrobiosis lipoidica effectively treated with pentoxifylline. Clin Exp Dermatol. 1993 Jan;18(1):78–79. doi: 10.1111/j.1365-2230.1993.tb00977.x. [DOI] [PubMed] [Google Scholar]
- 103.Harth W, Linse R. Topical tacrolimus in granuloma annulare and necrobiosis lipoidica. Br J Dermatol. 2004 Apr;150(4):792–794. doi: 10.1111/j.0007-0963.2004.05899.x. [DOI] [PubMed] [Google Scholar]
- 104.Reinhard G, Lohmann F, Uerlich M, Bauer R, Bieber T. Successful treatment of ulcerated necrobiosis lipoidica with mycophenolate mofetil. Acta Derm Venereol. 2000 Jul-Aug;80(4):312–313. doi: 10.1080/000155500750012315. [DOI] [PubMed] [Google Scholar]
- 105.Stinco G, Parlangeli ME, De Francesco V, Frattasio A, Germino M, Patrone P. Ulcerated necrobiosis lipoidica treated with cyclosporin A. Acta Derm Venereol. 2003;83(2):151–153. doi: 10.1080/00015550310007616. [DOI] [PubMed] [Google Scholar]
- 106.Stanway A, Rademaker M, Newman P. Healing of severe ulcerative necrobiosis lipoidica with cyclosporin. Australas J Dermatol. 2004 May;45(2):119–122. doi: 10.1111/j.1440-0960.2004.00059.x. [DOI] [PubMed] [Google Scholar]
- 107.De Rie MA, Sommer A, Hoekzema R, Neumann HA. Treatment of necrobiosis lipoidica with topical psoralen plus ultraviolet A. Br J Dermatol. 2002 Oct;147(4):743–747. doi: 10.1046/j.1365-2133.2002.04856.x. [DOI] [PubMed] [Google Scholar]
- 108.Durupt F, Dalle S, Debarbieux S, Balme B, Ronger S, Thomas L. Successful treatment of necrobiosis lipoidica with antimalarial agents. Arch Dermatol. 2008 Jan;144(1):118–119. doi: 10.1001/archderm.144.1.118. [DOI] [PubMed] [Google Scholar]
- 109.Miura Y, Ardenghy M, Ramasastry S, Kovach R, Hochberg J. Coumadin necrosis of the skin: report of four patients. Ann Plast Surg. 1996 Sep;37(3):332–337. doi: 10.1097/00000637-199609000-00017. [DOI] [PubMed] [Google Scholar]
- 110.Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg. 2000 Mar;87(3):266–272. doi: 10.1046/j.1365-2168.2000.01352.x. [DOI] [PubMed] [Google Scholar]
- 111.Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy. 2006 Aug;26(8):1175–1179. doi: 10.1592/phco.26.8.1175. [DOI] [PubMed] [Google Scholar]
- 112.Wynn SS, Jin DK, Essex DW. Warfarin-induced skin necrosis occurring four days after discontinuation of warfarin. Haemostasis. 1997 Sep–Oct;27(5):246–250. doi: 10.1159/000217463. [DOI] [PubMed] [Google Scholar]
- 113.Franson TR, Rose HD, Spivey MR, Maroney J, Libnoch JA. Late-onset, warfarin-caused necrosis occurring in a patient with infectious mononucleosis. Arch Dermatol. 1984 Jul;120(7):927–931. [PubMed] [Google Scholar]
- 114.DeFranzo AJ, Marasco P, Argenta LC. Warfarin-induced necrosis of the skin. Ann Plast Surg. 1995 Feb;34(2):203–208. doi: 10.1097/00000637-199502000-00017. [DOI] [PubMed] [Google Scholar]
- 115.Tai CY, Ierardi R, Alexander JB. A case of warfarin skin necrosis despite enoxaparin anticoagulation in a patient with protein S deficiency. Ann Vasc Surg. 2004 Mar;18(2):237–242. doi: 10.1007/s10016-003-0080-4. [DOI] [PubMed] [Google Scholar]
- 116.Rose VL, Kwaan HC, Williamson K, Hoppensteadt D, Walenga J, Fareed J. Protein C antigen deficiency and warfarin necrosis. Am J Clin Pathol. 1986 Nov;86(5):653–655. doi: 10.1093/ajcp/86.5.653. [DOI] [PubMed] [Google Scholar]
- 117.Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol. 2009 Aug;61(2):325–332. doi: 10.1016/j.jaad.2008.12.039. [DOI] [PubMed] [Google Scholar]
- 118.Hunter JB, Lonsdale RJ, Wenham PW, Frostick SP. Heparin induced thrombosis: an important complication of heparin prophylaxis for thromboembolic disease in surgery. BMJ. 1993 Jul 3;307(6895):53–55. doi: 10.1136/bmj.307.6895.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Warkentin TE, Elavathil LJ, Hayward CP, Johnston MA, Russett JI, Kelton JG. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Intern Med. 1997 Nov 1;127(9):804–812. doi: 10.7326/0003-4819-127-9-199711010-00005. [DOI] [PubMed] [Google Scholar]