

Summary
The protozoan parasite Cryptosporidium parvum is a major cause of gastrointestinal disease; no effective drug therapy exists to treat this infection. Curiously, C. parvum IMPDH (CpIMPDH) is most closely related to prokaryotic IMPDHs, suggesting that the parasite obtained its IMPDH gene via horizontal transfer. We previously identified inhibitors of CpIMPDH that do not inhibit human IMPDHs. Here we show that these compounds also inhibit IMPDHs from Helicobacter pylori, Borrelia burgdorferi, and Streptococcus pyogenes, but not from Escherichia coli. Residues Ala165 and Tyr358 comprise a structural motif that defines susceptible enzymes. Importantly, a second generation CpIMPDH inhibitor has bacteriocidal activity on H. pylori but not Escherichia coli. We propose that CpIMPDH-targeted inhibitors can be developed into a new class of antibiotics that will spare some commensal bacteria.
Introduction
Microbial infections are now the second leading cause of death worldwide. Many commonly used antibiotics have been rendered ineffective by the upsurge of drug resistance, and years of neglect have left a mere trickle of new antibiotics in the pipeline, creating an urgent need for novel scaffolds and targets (Fischbach and Walsh, 2009). The re-purposing of other drug development programs for antibiotic discovery is a promising strategy to address this problem (Miller, et al., 2009; Walsh and Fischbach, 2009). Inosine 5’-monophosphate dehydrogenase (IMPDH) presents an intriguing opportunity for such re-purposing. This enzyme catalyzes the pivotal step in guanine nucleotide biosynthesis, the conversion of IMP to XMP with the concomitant reduction of NAD+ in a reaction that involves a covalent intermediate E-XMP* (Figure 1a). The guanine nucleotide pool controls proliferation, so IMPDH inhibitors such as mycophenolic acid, merimepodib, mizoribine, tiazofurin and ribavirin are used in immunosuppressive, cancer and antiviral therapy. As yet IMPDH inhibitors have not been exploited in antimicrobial applications, in part because no bacterial-selective IMPDH inhibitors have been identified (Hedstrom, 2009).
Figure 1. The IMPDH reaction.

a. Chemical mechanism: a conserved Cys attacks C2 of IMP and hydride is transferred to NAD+ producing the covalent intermediate E-XMP*. E-XMP* is hydrolyzed with a conserved Arg residue acting as a general base to produce XMP. b. The hydride transfer reaction proceeds in an open enzyme conformation. After NADH departs, a mobile flap folds into the NAD site, carrying the catalytic Arg into the active site. Inhibitors compete with the flap, so the equilibrium between open and closed states is a determinant of inhibitor affinity. c. Phylogenetic tree of IMPDHs. Constructed as described in (Min, et al., 2008).
The development of IMPDH-based therapy is further complicated by the conformational gymnastics of the catalytic cycle (Hedstrom, 2009). IMPDHs have two conformational states, an open conformation that allows NAD to bind, and a closed conformation where a mobile flap binds in the NAD site (Figure 1b; (Hedstrom, 2009)). The closed conformation is required for the hydrolysis of the covalent intermediate E-XMP*. Thus the flap competes with inhibitors that bind in the NAD site, and this competition is an important determinant of inhibitor potency (Hedstrom, 2009). For example, Tritrichomonas foetus IMPDH is 400-fold more resistant to mycophenolic acid than human IMPDH2; ~10-fold of this difference is due to changes in the structure of the inhibitor binding site while ~40-fold can be attributed to a preference for the closed conformation (Digits and Hedstrom, 2000). Therefore, for TfIMPDH, the conformational equilibrium is a more important determinant of inhibitor susceptibility than the residues that contact the inhibitor. This example illustrates the difficulty in predicting inhibitor action from IMPDH sequence.
The protozoan parasite Cryptosporidium parvum is a major cause of diarrhea and malnutrition and a potential bioterrorism agent (Fayer, 2004). The parasite has a streamlined purine salvage pathway that depends on IMPDH for the production of guanine nucleotides (Abrahamsen, et al., 2004; Striepen, et al., 2004). Surprisingly, CpIMPDH is closely related to prokaryotic IMPDHs (e.g., Helicobacter pylori IMPDH, Figure 1c), suggesting that its IMPDH gene was obtained by horizontal transfer (Striepen, et al., 2004; Striepen, et al., 2002)). Prokaryotic and eukaryotic IMPDHs differ in both structural features and kinetic properties (Zhang, et al., 1999), which suggests that selective inhibition should be possible. We have recently identified eight selective inhibitors of CpIMPDH in a high throughput screen targeting the NAD site (compounds A–H, Table 1) (Kirubakaran, et al.; MacPherson, et al.; Maurya, et al., 2009; Umejiego, et al., 2008). Several of these inhibitors display antiparasitic activity in a tissue culture model of C. parvum infection (Umejiego, et al., 2008). A program of medicinal chemistry optimization has yielded inhibitors with nanomolar affinity in several structurally distinct frameworks (Kirubakaran, et al.; MacPherson, et al.; Maurya, et al., 2009).
Table 1.
Inhibition of IMPDHs by compounds A–H. These compounds (100 µM) do not inhibit EcIMPDH, TfIMPDH and LdIMPDH. The errors on the IC50 values were less than 10% unless otherwise noted. "Intrinsic" values (adjusted for the fraction of enzyme in the open conformation) are shown in parentheses.
| Compound | IC50(µM) | ||||
|---|---|---|---|---|---|
| CpIMPDHa | HpIMPDH | BbIMPDH | SpIMPDH |
EcIMPDH- S250A/L444Y |
|
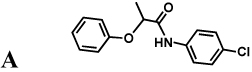 |
3.3 0.66 |
2.2 1.1 |
1.4 0.84 |
96 12 |
1.8 0.54 |
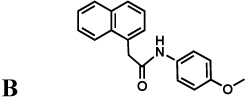 |
1.6 0.32 |
1.3 0.65 |
1.8 1.1 |
85 11 |
0.70 0.21 |
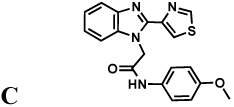 |
1.2 0.24 |
0.60 0.30 |
0.60 0.36 |
70 9.1 |
0.60 0.18 |
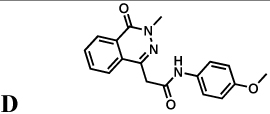 |
5.4 1.1 |
3.0 1.5 |
1.7 1.0 |
49 6.4 |
2.3 0.69 |
 |
1.6 0.32 |
1.5 0.75 |
0.90 0.54 |
15 2.0 |
1.8 0.54 |
 |
1.4 0.28 |
1.1 0.55 |
0.80 0.48 |
13 1.7 |
1.1 0.33 |
 |
0.1 0.02 |
0.30 0.15 |
0.22 0.13 |
0.55 0.07 |
0.40 0.12 |
 |
0.9 0.18 |
1.2 0.6 |
0.90 0.54 |
0.86 0.11 |
1.0 0.3 |
 |
0.008b, c 0.0016 |
0.0011b, e 0.0005 |
n.d. | 0.0054b, c 0.0007 |
0.0019b, f 0.0006 |
data from (Umejiego, et al., 2008);
determined with a tight-binding equation;
error ≤ 37%.
error = 73%; error = 52%. Supported by Figure S1.
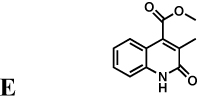
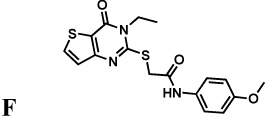
Mycophenolic acid and merimepodib bind in the nicotinamide subsite of the NAD site of human IMPDH, stacking against the purine ring of E-XMP* and within a single subunit of the tetramer (Sintchak, et al., 1996; Sintchak and Nimmesgern, 2000). In contrast, the x-ray crystal structure of inhibitor C64 revealed that this inhibitor interacts with the purine ring of IMP and then crosses the dimer interface to interact with Tyr358 in the adjacent subunit (Figure 2a,b; CpIMPDH numbering; (MacPherson, et al.)). At present, no structures are available of the other inhibitor complexes. However, with the exception of compound E, all of the inhibitors contain two aromatic groups linked by spacers of similar length (Table 1), and therefore may be able to recapitulate this binding mode.
Figure 2. Structures of IMPDH inhibitor binding sites.

a. Structure of C64. b. Structure of the CpIMPDH•IMP•C64 complex (salmon; PDB accession 3KHJ (MacPherson, et al.)) and resistant IMPDHs from T. foetus (green; 1LRT (Gan, et al., 2002)) and Chinese hamster (blue; 1JR1, nearly identical to human IMPDH2 (Sintchak, et al., 1996)). Residues within 5 Å of C64 are displayed. C64 is shown in gray with a transparent surface; CpIMPDH residues are labeled; residue from the adjacent monomer are denoted with a '. Residues Thr221 and Glu329 are hidden under C64. c. The surface of the NAD binding site rendered by conservation of residues in prokaryotic IMPDHs from T. foetus, C. parvum, H. pylori, B. burgodorferi, S. pyogenes and EcIMPDH. Dark magenta, 100% conserved; tan, 63%; dark cyan, 25%. The NAD analog tiazofurin adenine dinucleotide (TAD) is light gray. The tiazofurin binding site is circled in blue and the ADP binding site is circled in red. d. The surface of the C64 binding site rendered by conservation of residues in sensitive IMPDHs from C. parvum, H. pylori, and B. burgdorferi, as well as the S250A/L444Y variant of EcIMPDH. Ser164, Met326 and Ser354 are within 5 Å of C64; Val143 is 8 Å away. Dark magenta, 100% conserved; tan, 60%; dark cyan, 20%. C64 depicted in ball-and-stick in light gray and IMP is shown in stick in dark gray. e. The surface of the C64 binding site rendered by conservation of residues in CpIMPDH and IMPDHs from 19 pathogenic bacteria: Acinetobacter baumannii (wound infection), Bacillus anthracis (anthrax), Bacteroides fragilis (peritoneal infections), Brucella abortus/melitensis/suis (brucellosis), B. burgdorferi (Lyme disease), Burkholderia cenocepacia (infection in cystic fibrosis), Bu. mallei (glanders), Bu. pseudomallei (melioidosis), Campylobacter jejuni (food poisoning), C. lari (food poisoning), Coxiella burnetii (Q fever), Francisella tularensis (tularemia), H. pylori (gastric ulcer/stomach cancer), Listeria monocytogenes (listeriosis), Staphylococcus aureus (major cause of nosocomial infection), S. pyogenes (major cause of nosocomial infections) and S. pneumoniae (pneumonia). Dark magenta, 100% conserved; tan, 63%; dark cyan, 25%. Alignments were constructed with CLUSTALW2 and molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) (Pettersen, et al., 2004). Supported by Figure S3.
Here we demonstrate that Tyr358 together with Ala165 comprise a structural motif that defines susceptibility to all eight CpIMPDH inhibitors. This motif is found in a wide variety of pathogenic bacteria. Importantly, the CpIMPDH inhibitor C91 blocks H. pylori growth, but not the growth of Escherichia coli. These observations suggest that CpIMPDH-targeted inhibitors can be developed into a new class of broader spectrum antibiotics that will spare commensal bacteria.
Results and Discussion
Expression, purification and characterization of recombinant IMPDHs
We expressed and purified prokaryotic IMPDHs from representative organisms: H. pylori (Gram negative ε proteobacteria), E. coli (Gram-negative γ proteobacteria), B. burgdorferi (spirochete), S. pyogenes (Gram-positive) and the protozoan parasite T. foetus, which also appears to have obtained its IMPDH gene from a prokaryote (Bapteste and Philippe, 2002). We also expressed an additional eukaryotic IMPDH from the protozoan parasite Leishmania donovani. EcIMPDH, BbIMPDH, TfIMPDH, SpIMPDH and LdIMPDH have been characterized previously (Digits and Hedstrom, 1999; Dobie, et al., 2007; Kerr and Hedstrom, 1997; Zhang, et al., 1999; Zhou, et al., 1997). The kinetic parameters of HpIMPDH are very similar to those of CpIMPDH, and are generally characteristic of bacterial IMPDHs (Table S2; (Hedstrom, 2009; Umejiego, et al., 2004; Zhang, et al., 1999)). Importantly, structures are available for TfIMPDH, SpIMPDH and BbIMPDH as well as for CpIMPDH and the human enzymes (Colby, et al., 1999; Gan, et al., 2002; Gan, et al., 2003; MacPherson, et al.; McMillan, et al., 2000; Prosise and Luecke, 2003; Prosise, et al., 2002; Sintchak, et al., 1996; Whitby, et al., 1997; Zhang, et al., 1999). CpIMPDH is most closely related to HpIMPDH with 60% sequence identity (Figure 1c; Table S1). The sequence of CpIMPDH is ~50% identical to EcIMPDH, BbIMPDH and SpIMPDH and ~30% identical to TfIMPDH and the eukaryotic enzymes (Table S1)(Min, et al., 2008; Striepen, et al., 2002). Importantly, HpIMPDH, BbIMPDH and SpIMPDH contain Tyr358 and are therefore expected to be susceptible to the C series inhibitors and perhaps also to the other CpIMPDH inhibitors.
Spectrum of inhibition of C series inhibitors
Compound C inhibits HpIMPDH and BbIMPDH with similar values of IC50 to CpIMPDH (Table 1). C is a noncompetitive (mixed) inhibitor of HpIMPDH with respect to NAD+ (data not shown), as observed with CpIMPDH (Umejiego, et al., 2008). Importantly, the values of Ki and IC50 are similar, as expected for noncompetitive inhibition. C also inhibits SpIMPDH, although in this case the value of IC50 is increased by a factor of ~60 relative to CpIMPDH. No inhibition is observed at 100 µM C for TfIMPDH, EcIMPDH and LdIMPDH, as expected since these enzymes do not contain Tyr358.
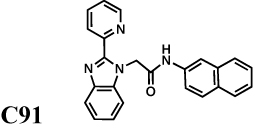
We also evaluated the spectrum of a second generation inhibitor, C91, arising from our medicinal chemistry optimization program (Kirubakaran, et al.); this compound is a potent inhibitor of CpIMPDH with IC50 = 8 nM. C91 is also a potent inhibitor of both HpIMPDH and SpIMPDH, demonstrating that potent broader spectrum inhibitors can be obtained (Table 1). C91 fails to inhibit EcIMPDH, TfIMPDH, LdIMPDH or human IMPDHs (IC50 > 10 µM).
The conformational contribution to inhibitor selectivity
As noted in the introduction, IMPDH undergoes a conformational change in the middle of its catalytic cycle that brings a mobile flap into the NAD site (Figure 1b). The competition of the flap for this site can be an important determinant of inhibitor susceptibility, and might explain the low susceptibility of SpIMPDH to C despite the presence of Tyr358 (Table 2; (Guillén Schlippe and Hedstrom, 2005; Guillén Schlippe and Hedstrom, 2005; Guillén Schlippe, et al., 2004; Hedstrom, 2009; Kohler, et al., 2005; Riera, et al., 2008; Umejiego, et al., 2004)). Therefore we determined the equilibrium between open and closed conformations (Kc) for the various IMPDHs (Table 2 and Figure S3; see Supplemental Material for description (Hedstrom, 2009)). The values of Kc vary considerably among the prokaryotic IMPDHs, from 0.7 for BbIMPDH to 140 for TfIMPDH.
Table 2. The conformational contribution to potency of IMPDH inhibitors.
As shown in Figure 1b, inhibitors compete with the mobile flap for the vacant NADH site. Therefore the observed affinity will be determined by the intrinsic affinity of the inhibitor for the NAD site and the affinity of the flap, i.e., Kint = Ki /(1 + Kc), where Kint is the intrinsic affinity, Ki is the measured inhibition constant and Kc is the conformational equilibrium determined as described in Materials and Methods.
| Enzyme |
Ki Tzf (mM) |
Ki ADP (mM) |
Kc |
Kint Tzf (mM) |
Kint ADP (mM) |
|---|---|---|---|---|---|
| CpIMPDH a | 1.5 ± 0.1 | 42 ± 6 | 4 | 0.3 | 8 |
| HpIMPDH | 1.3 ± 0.1 | 6.0 ± 0.5 | 1 | 0.7 | 3 |
| BbIMPDH | 1.1 ± 0.2 | 7.4 ± 0.6 | 0.7 | 0.7 | 4 |
| SpIMPDH | 8 ± 1 | 13 ± 2 | 7 | 0.7 | 2 |
| TfIMPDH a | 69 ± 9 | 31 ± 2 | 140 | 0.5 | 0.2 |
| EcIMPDH | 5.3 ± 0.6 | 10 ± 2 | 9 | 0.5 | 1 |
| EcIMPDH-S250A | 2.5 ± 0.3 | 0.10 ± 0.01 | 1.5 | 1 | 0.04 |
| EcIMPDH- S250A/L444Y | 1.1 ± 0.2 | 4.1 ± 0.7 | 2.3 | 0.3 | 1 |
| hIMPDH2 a | 1.3 ± 0.1 | 9 ± 2 | ≤ 0.2 | 1.3 | 9 |
| LdIMPDH | 8.3 ± 0.7 | 15 ± 2 | 2.3 | 2.5 | 5 |
data from (Riera, et al., 2008). Supported by Figure S2.
Tiazofurin inhibition illustrates the validity of this approach and the magnitude of the conformational contribution to inhibitor selectivity. The tiazofurin binding site is conserved among prokaryotic IMPDHs (Figure 2c), which predicts that CpIMPDH, HpIMPDH, BbIMPDH, SpIMPDH, EcIMPDH and TfIMPDH should all bind tiazofurin with similar affinity, yet the measured values of Ki vary from 1–69 mM. When the measured values of Ki are adjusted for competition from the mobile flap, that is, by dividing by a factor of 1 + Kc to normalize for the fraction of protein in the open conformation, the resulting “intrinsic" values are indeed nearly identical, ranging from 0.3–0.7 mM (Table 2). In contrast, the intrinsic values of Ki for ADP range from 0.2–9 mM, reflecting the structural divergence of the ADP binding sites among the six enzymes (Figure 2c). Therefore the intrinsic values reflect the structural similarity of the inhibitor binding sites.
The intrinsic values of IC50 for C are similar for CpIMPDH, HpIMPDH and BbIMPDH, indicating that the structures of inhibitor binding sites are also similar in these three enzymes. However, the intrinsic value of IC50 for C with SpIMPDH remains 40-fold greater than CpIMPDH. Figure 2 displays the conservation of the C64 binding site among these IMPDHs. While most of the residues within 5 Å of C64 are conserved among these enzymes, Ser164 and Met326 are variable; these residues are Thr and Leu, respectively in SpIMPDH. Perhaps these substitutions account for the lower potency of C with SpIMPDH. In contrast, the intrinsic values of IC50 for C91 are similar for CpIMPDH, HpIMPDH and SpIMPDH, indicating that the interactions that increase the potency of C91 in CpIMPDH are also found in the other enzymes.
Structural determinants of C susceptibility
To determine if the presence of Tyr358 is sufficient to install sensitivity to inhibitor C, we replaced the corresponding residue in EcIMPDH with Tyr. The resulting variant, L444Y, has similar kinetic properties to EcIMPDH, except that the value of Km for NAD+ is increased by 5-fold (Table S2). Unlike EcIMPDH, C inhibits L444Y (Figure S2); however, L444Y is still ~100-fold less sensitive than CpIMPDH.
Inspection of the residues within 5 Å of C64 revealed another potential structural determinant of C susceptibility: Ala165 (Figure 2a). The installation of Ala at the corresponding position of EcIMPDH also increased the sensitivity to C, although again the resulting variant, S250A, is ~100-fold less sensitive than CpIMPDH (Figure S2). However, the combination of these mutations created a variant S250A/L444Y with similar sensitivity to C as CpIMPDH (Table 1). The S250A/L444Y variant also displays similar sensitivity to C91 (Table 1). Therefore Ala250 and Tyr358 comprise a minimal structural motif that defines sensitivity to C.
All of the CpIMPDH inhibitors interact with Ala165 and Tyr358
The spectra of inhibition of compounds A, B and D–H are also very similar to C. These seven compounds inhibit HpIMPDH, BbIMPDH and SpIMPDH, but not EcIMPDH, TfIMPDH or LdIMPDH (Table 1). These compounds are noncompetitive (mixed) inhibitors of HpIMPDH with respect to NAD+ (data not shown), as observed with CpIMPDH (Umejiego, et al., 2008). Most importantly, the EcIMPDH variant S250A/L444Y is inhibited by A, B and D–H, indicating that together Ala165 and Tyr358 define the structural motif required for susceptibility to all the CpIMPDH inhibitors. Note that compound E has a single aromatic group, and therefore must interact exclusively with Tyr358 and not with the purine ring of IMP.
Two inhibitor binding modes exist
Although the above observations indicate that all of the compounds interact with Ala165 and Tyr358, inspection of the intrinsic values of IC50 for all of the CpIMPDH inhibitors reveals two distinct binding modes. The intrinsic values of IC50 of C range between 0.18–0.36 µM for CpIMPDH, HpIMPDH, BbIMPDH and S250A/L444Y (Table 2), reflecting the conservation of this binding site (Figure 2d) (MacPherson, et al.). Likewise, the intrinsic affinities of compounds A, B, D, E and F are within a factor of 2 for all four enzymes, indicating that the binding sites of these compounds are also conserved. In contrast, the intrinsic values of A–F for SpIMPDH are very different from CpIMDPH, indicating that this binding site is significantly different in SpIMPDH (Table 1). Only one substitution is present within 3.5 Å of C64: Met326 is a Leu in SpIMPDH. However, the Leu substitution is also present in HpIMPDH and BbIMPDH, and therefore cannot account for the different susceptibility. The next nearest substitution is Thr for Ser164; the side chain of Ser164 is 5 Å away from C64, but might be closer to A, B and D–F.
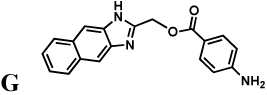
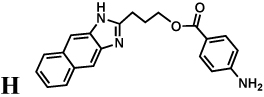
A different trend is observed in the intrinsic affinities of G and H. SpIMPDH is most similar to CpIMPDH, while HpIMPDH and BbIMPDH display lower affinities for these compounds (Table 2). These observations suggest that G/H bind in a region that is conserved in SpIMPDH and CpIMPDH, but different in the other enzymes. Therefore, at least a portion of the G/H binding site must be distinct from the site that binds A–F. Figure S3 reveals regions of conservation between CpIMPDH and SpIMPDH that are not shared with the other sensitive enzymes. These regions could serve as the binding site for G/H.
Inhibition of H. pylori growth
We chose to investigate the antibiotic potential of IMPDH inhibition in H. pylori because HpIMPDH is most similar to CpIMPDH. H. pylori causes gastrointestinal ulcers and stomach cancer; standard treatment involves triple therapy of a proton pump inhibitor, clarithromycin and amoxicillin or metronidazole (Selgrad and Malfertheiner, 2008). Moreover, resistance is developing to the standard triple therapy, and few new antibiotics are in the pipline. H. pylori has complex growth requirements that necessitate the use of rich media containing xanthine and guanine (Brucella broth) (Tomb, et al., 1997). H. pylori will be resistant to IMPDH inhibitors if its salvage pathways can provide sufficient guanine nucleotides to support proliferation. Therefore this bacteria provides a demanding test for the antibiotic potential of IMPDH-targeted inhibitors
Figure 3 shows that 20 µM C91 is sufficient to block the proliferation of a H. pylori culture exiting stationary phase. Higher concentrations of C91 display bactericidal effects, with only 23% of the colony forming units remaining after 24 hr treatment with 200 µM. Exponentially growing H. pylori cells are also sensitive to C91 (Figure S1); a concentration of 60 µM is sufficient to block growth while higher concentrations are bactericidal. Importantly, C91 failed to inhibit the growth of E. coli, suggesting that the antibacterial effects of C91 result from the inhibition of HpIMPDH rather than an off-target effect.
Figure 3. C91 inhibits H. pylori growth.

a. Compound C91 in DMSO was added to freshly diluted stationary cultures of H. pylori strain G27 in Brucella broth. Samples were removed at the indicated time points, diluted, and plated to determine bacterial proliferation/survival. Each point is the average of duplicate determinations; a representative of three experiments is shown. Black, DMSO alone. C91 concentrations: purple, 2 µM; blue, 7 µM; green, 20 µM; orange, 60 µM; red, 200 µM. b. Compound C91 was added to freshly diluted cultures of E. coli MG1655 in Luria broth. Each point is the average of three determinations; the standard deviations are smaller than the point. Black, DMSO alone. C91 concentrations: orange, 100 µM; red, 200 µM. Supported by Figure S4.
Implications for the design of antibiotics targeting IMPDH
The above findings indicate that Ala165 and Tyr358 comprise a structural motif that defines enzymes susceptible to CpIMPDH inhibitors. A BLAST search reveals that these critical residues are present in IMPDHs from a wide variety of pathogenic bacteria in addition to C. parvum, B. burgdorferi, H. pylori and S. pyogenes. Importantly, this list includes select agents and multi-drug resistant bacteria, where new antibiotics are urgently needed, but not E. coli (Table S1). As shown in Figure 2e, the inhibitor binding site is highly conserved among C. parvum and pathogenic bacteria, suggesting that IMPDH inhibition provides a promising strategy for the development of a broader spectrum antibiotic. Prokaryotic-specific inhibitors such as C91 will be invaluable in validating IMPDH as a target for antibiotic chemotherapy that will spare commensal bacteria.
Significance
The rising tide of antibiotic resistance creates an urgent need for new drugs to treat bacterial infections, but years of neglect have depleted the antibiotic pipeline. The re-purposing of other drug development programs for antibiotic discovery is a promising strategy to address this problem. Inosine 5’-monophosphate dehydrogenase (IMPDH), a key enzyme in the biosynthesis of the precursors for RNA and DNA, presents an intriguing opportunity for such re-purposing. IMPDH is a promising target for drugs against the protozoan parasite CryptoSporidium parvum, a major cause of diarrhea and malnutrition and a potential bioterrorism agent. Curiously, CpIMPDH is most closely related to prokaryotic IMPDHs, suggesting that the parasite obtained its IMPDH gene via horizontal transfer. We previously identified inhibitors of CpIMPDH that do not inhibit human IMPDHs. Here we show that selective inhibitors of CpIMPDH also inhibit IMPDHs from the pathogenic bacteria Helicobacter pylori, Borrelia burgdorferi, and Streptococcus pyogenes, but not Escherchia coli. Importantly, a second generation CpIMPDH inhibitor blocks H. pylori growth, demonstrating that these compounds have antibacterial activity. Susceptible enzymes are defined by a structural motif that is found in IMPDHs from a wide variety of pathogenic bacteria, suggesting that IMPDH-targeted inhibitors can be developed into a new class of broader spectrum antibiotics that will spare some commensal bacteria.
Experimental Procedures
Materials
Compounds D, E, F, G, and H were purchased from ChemDiv Inc. (San Diego, CA), Compounds A, B and C were synthesized as described previously (Umejiego, et al., 2008). Compound C91 was synthesized as described in Supplementary Material. Full SAR of the C series will be described in (Kirubakaran, et al.). All other chemicals were obtained from Fisher Scientific, unless mentioned otherwise. Plasmid containing the guaB gene of S. pyogenes was a generous gift of Dr. Cameron Ashbaugh (Ashbaugh and Wessels, 1995). H. pylori total genomic DNA was obtained from American Type Culture Collection (ATCC). L. donovani IMPDH coding sequence was the gift of Dr. Buddy Ullman (Wilson, et al., 1991).
Enzyme Cloning and Purification
Recombinant T. foetus, B. burgdorferi, E. coli and C. parvum IMPDH were expressed in guaB strains of E. coli (which lack endogenous IMPDH) and purified as described previously (Digits and Hedstrom, 1999; Kerr and Hedstrom, 1997; Umejiego, et al., 2004; Zhou, et al., 1997). The S250A, L444Y and S250A/L444Y mutants of E. coli IMPDH were constructed using Quikchange (Stratagene, La Jolla, CA). Enzymes were expressed and purified as previously described (Kerr and Hedstrom, 1997).
To express L. donovani IMPDH, a NcoI site was created at the beginning of the LdIMPDH coding sequence and the NcoI-PstI fragment was cloned into pKK233-2 to create the plasmid pLDI, which expresses LdIMPDH under control of the trc promoter. Cultures were induced with 0.5 mM IPTG and grown overnight. Cells were harvested by centrifugation, resuspended in 50 mM Tris-HCl, pH 8.0, 1 mM dithiothreitol and 10 % glycerol (Buffer A), lysed by sonication and clarified by centrifugation followed by filtration through a 45 µm cellulose acetate filter. Protein was applied to a Poros HS strong cation exchange resin (PerSeptive Biosystems) pre-equilibrated with 20 mM sodium phosphate buffer, pH 7.5, 1 mM dithiothreitol (Buffer B). LdIMPDH was eluted with a gradient of 0–0.9 M NaCl. Fractions containing IMPDH activity were pooled and applied to IMP affinity resin. The column was washed with Buffer B and enzyme was eluted with Buffer B containing 0.5 M KCl, 1 mM IMP. The specific activity of the final preparation was 2.6 µmoles/min-mg.
The H. pylori and S. pyogenes guaB genes were cloned into pET28a with 6x His-tags. Bacteria were grown at 30°C in LB medium containing 25 µg/mL kanamycin until the OD600 reached approximately 0.6. Expression was initiated by the addition of 0.5 mM IPTG and the temperature was changed to 25°C. Bacteria were harvested after 16 hours. The cell pellet was rinsed (3x) with 50 mM phosphate buffer, 500 mM NaCl, 5 mM imidazole, pH 8.0, 1 mM IMP and 5 mM β-mercaptoethanol, and lysed by sonication. The lysate was clarified by centrifugation and loaded on a Ni-NTA column (Qiagen). The purified protein were eluted in 50 mM phosphate buffer, 500 mM NaCl, 250 mM imidazole, pH 8.0, 1 mM IMP and 5 mM β-mercaptoethanol, concentrated and dialyzed against Buffer A. The protein concentration was determined by using Bradford dye procedure (BioRad).
Steady State Enzyme Kinetics
IMPDH assays were performed in 50 mM Tris-HCl, pH 8.0, 100 mM KCl, 3 mM EDTA and 1 mM DTT. Activity was routinely assayed in the presence of 50 nM IMPDH at 25 °C. NADH production was monitored either by following absorbance change at 340 nm using a Hitachi U-2000 spectrophotometer (ε = 6.2 mM−1 cm−1). IMPDHs are prone to NAD+ substrate inhibition (Hedstrom, 2009). Therefore the steady state kinetics for HpIMPDH were initially analyzed by varying NAD+ at saturating IMP concentrations to determine the value of Km for NAD+, then by varying IMP at the fixed NAD+ concentration as close to saturating as practical. Using the SigmaPlot program (SPSS, Inc.), initial velocity data were fit to the Michaelis-Menten equation (Equation 1) and/or the uncompetitive substrate inhibition equation (Equation 2), as follows,
| (1) |
| (2) |
where v represents the velocity, Vm is the maximal velocity, S is the substrate concentration, Km is the Michaelis constant, Kii is the intercept inhibition constant. The values of kcat determined under both conditions are in good agreement.
Inhibitor Kinetics
Enzyme was incubated with inhibitor (50 pM – 100 µM) for 10 min at room temperature prior to addition of substrates. The conditions used for each enzyme is listed in Supplemental Material. IC50 values were calculated for each inhibitor according to Equation 3 using the SigmaPlot program (SPSS, Inc.):
| (3) |
where υi is initial velocity in the presence of inhibitor (I) and υ0 is the initial velocity in the absence of inhibitor.
Assays were carried out in assay buffer at 25 °C with ~50 nM IMPDH and NADH production was monitored by following fluorescence. The values of Ki with respect to NAD+ were determined by using fixed concentrations of IMP and varied NAD+ concentrations. Data were fitted according to Equation 4 (noncompetitive inhibition) using SigmaPlot program (SPSS, Inc.):
| (4) |
where Kii and Kis represent the intercept and slope inhibition constants, respectively. The best fits were determined by the relative fit error. The Morrison equation was used to evaluate tight-binding inhibitors (Morrison, 1969).
Determination of Kc
The values of Kc were determined by measuring the interaction between tiazofurin and ADP (Digits and Hedstrom, 2000; Hedstrom and Wang, 1990). Tiazofurin binds in the nicotinamide subsite of the NAD site while ADP binds in the adenosine subsite. If the closed conformation is favored, tiazofurin and ADP will interact synergistically-one inhibitor shifts the enzyme into the open conformation and promotes the association of the second inhibitor (Guillén Schlippe, et al., 2004). If the open conformation is favored, the two inhibitors bind independently. Tiazofurin and ADP concentrations are varied at constant IMP and NAD+. Initial velocities were fit to equation 5 using SigmaPlot:
| (5) |
where v is the initial velocity, v0 is the initial velocity in the absence of inhibitor, Ki and Kj are the inhibition constants for the inhibitors I and J, respectively and α is the interaction constant. The value of α approximates the fraction of enzyme in the open conformation, so the value of Kc is obtained (Guillén Schlippe, et al., 2004):
| (6) |
Bacterial growth assays
H. pylori strain G27 (Baltrus, et al., 2009) was stored at −80°C in Brucella broth (Becton, Dickinson) supplemented with 10% fetal bovine serum (FBS) and 20% glycerol. Bacteria were plated on TSA/sheep blood agar plates (Remel) or grown in growth media (Brucella broth supplemented with 10% FBS and 4µg/mL fungizone/amphotericin B) at 37°C in a 10% CO2 incubator. For growth inhibition assays, a frozen stock of H. pylori strain G27 was plated on blood agar plates and allowed to grow for 48 hours. The bacteria were re-suspended in growth media (30 ml) and incubated for ~18–20 hours. The resulting stationary phase bacteria were diluted into assay media (Brucella broth with 10% FBS) to an OD600 = 0.025 (~104 colony forming units/µl) and aliquots (200µl) were added to 96 well tissue-culture grade polystyrene plates (Costar, Corning Inc.). A DMSO solution of C91 (2 µl) was added to each well and the plates were incubated with shaking. Colony forming units (CFUs) were measured by diluting aliquots at 0, 2, 4, 8, 12, and 24 hours, and plating on blood agar plates. Alternatively, logarithmic phase bacteria were obtained by diluting stationary phase bacteria 5x into fresh growth media (25 ml) and incubating with shaking for 4 hours prior to the growth inhibition assay.
An exponentially growing culture of E. coli MG1655 was diluted into fresh Luria broth containing C91 (100 and 200 µM) or DMSO. The triplicate cultures were incubated at 37°C in a plate reader and optical density was recorded every minute over 3.5 hours.
Supplementary Material
Acknowledgements
This work was supported by NIH-NIAID U01 AI075466 (L.H.). GWL was supported in part by the University of Virginia Cancer Training Grant (5T32 CA 009109).
Abbreviations
- IMP
inosine 5'-monophosphate
- NAD+
nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleotide
- IPTG
isopropyl-β-D-thiogalactoside
- DTT
dithiothreitol
- IMPDH
inosine monophosphate dehydrogenase
- CpIMPDH
IMPDH from C. parvum
- HpIMPDH
IMPDH from H. pylori
- BbIMPDH
IMPDH from B. burgdorferi
- SpIMPDH
IMPDH from S. pyogenes
- EcIMPDH
IMPDH from E. coli
- TfIMPDH
IMPDH from T. foetus
- LdIMPDH
IMPDH from L. donovani
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- Ashbaugh CD, Wessels MR. Cloning, sequence analysis and expression of the group A streptococcal guaB gene encoding inosine monophosphate dehydrogenase. Gene. 1995;165:57–60. doi: 10.1016/0378-1119(95)00422-3. [DOI] [PubMed] [Google Scholar]
- Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, Stein M, Salama NR, Guillemin K. The complete genome sequence of Helicobacter pylori strain G27. J Bacteriol. 2009;191:447–448. doi: 10.1128/JB.01416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, Philippe H. The potential value of indels as phylogenetic markers: position of trichomonads as a case study. Mol Biol Evol. 2002;19:972–977. doi: 10.1093/oxfordjournals.molbev.a004156. [DOI] [PubMed] [Google Scholar]
- Colby TD, Vanderveen K, Strickler MD, Markham GD, Goldstein BM. Crystal structure of human type II inosine monophosphate dehydrogenase: implications for ligand binding and drug design. Proc. Natl. Acad. Sci., USA. 1999;96:3531–3536. doi: 10.1073/pnas.96.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digits JA, Hedstrom L. Drug selectivity is determined by coupling across the NAD+ site of IMP dehydrogenase. Biochemistry. 2000;39:1771–1777. doi: 10.1021/bi992288e. [DOI] [PubMed] [Google Scholar]
- Digits JA, Hedstrom L. Kinetic mechanism of Tritrichomonas foetus inosine-5'-monophosphate dehydrogenase. Biochemistry. 1999;38:2295–2306. doi: 10.1021/bi982305k. [DOI] [PubMed] [Google Scholar]
- Dobie F, Berg A, Boitz JM, Jardim A. Kinetic characterization of inosine monophosphate dehydrogenase of Leishmania donovani. Mol Biochem Parasitol. 2007;152:11–21. doi: 10.1016/j.molbiopara.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Fayer R. Cryptosporidium: a water-borne zoonotic parasite. Veterinary Parasitology. Veterinary Parasitology. 2004;126:37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Petsko GA, Hedstrom L. Crystal structure of a ternary complex of Tritrichomonas foetus inosine 5'-monophosphate dehydrogenase: NAD+ orients the active site loop for catalysis. Biochemistry. 2002;41:13309–13317. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- Gan L, Seyedsayamdost MR, Shuto S, Matsuda A, Petsko GA, Hedstrom L. The immunosuppressive agent mizoribine monophosphate forms a transition state analog complex with IMP dehydrogenase. Biochemistry. 2003;42:857–863. doi: 10.1021/bi0271401. [DOI] [PubMed] [Google Scholar]
- Guillén Schlippe YV, Hedstrom L. Guanidine derivatives rescue the Arg418Ala mutation of Tritrichomonas foetus IMP dehydrogenase. Biochemistry. 2005;44:16695–16700. doi: 10.1021/bi051603w. [DOI] [PubMed] [Google Scholar]
- Guillén Schlippe YV, Hedstrom L. Is Arg418 the Catalytic Base Required for the Hydrolysis Step of the IMP Dehydrogenase Reaction? Biochemistry. 2005;44:11700–11707. doi: 10.1021/bi048342v. [DOI] [PubMed] [Google Scholar]
- Guillén Schlippe YV, Riera TV, Seyedsayamdost MR, Hedstrom L. Substitution of the Conserved Arg-Tyr Dyad Selectively Disrupts the Hydrolysis Phase of the IMP Dehydrogenase Reaction. Biochemistry. 2004;43:4511–4521. doi: 10.1021/bi035823q. [DOI] [PubMed] [Google Scholar]
- Hedstrom L. IMP Dehydrogenase: structure, mechanism and inhibition. Chem. Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L, Wang CC. Mycophenolic acid and thiazole adenine dinucleotide inhibition of Tritrichomonas foetus inosine 5'-monophosphate dehydrogenase: implications on enzyme mechanism. Biochemistry. 1990 doi: 10.1021/bi00456a001. 849-554. [DOI] [PubMed] [Google Scholar]
- Kerr KM, Hedstrom L. The roles of conserved carboxylate residues in IMP dehydrogenase and identification of a transition state analog. Biochemistry. 1997;36:13365–13373. doi: 10.1021/bi9714161. [DOI] [PubMed] [Google Scholar]
- Kirubakaran S, Gorla SureshKumar, Johnson CR, Zhang M, Golapalli DR, Hedstrom L, Cuny GD. Benzimidazole based inhibitors for Cryptosporidium parvum IMPDH. manuscript in preparation. [Google Scholar]
- Kohler GA, Gong X, Bentink S, Theiss S, Pagani GM, Agabian N, Hedstrom L. The functional basis of mycophenolic acid resistance in Candida albicans IMP dehydrogenase. J Biol Chem. 2005;280:11295–11302. doi: 10.1074/jbc.M409847200. [DOI] [PubMed] [Google Scholar]
- MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D'Aquino JA, Zhang M, Lu J, Cuny GD, Hedstrom L. The Structural Basis of Cryptosporidium-Specific IMP Dehydrogenase Inhibitor Selectivity. J. Am. Chem. Soc. 2010 doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. Triazole inhibitors of Cryptosporidium parvum inosine 5'-monophosphate dehydrogenase. J Med Chem. 2009;52:4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan FM, Cahoon M, White A, Hedstrom L, Petsko GA, Ringe D. Crystal structure at 2.4 Å resolution of Borrelia burgdorferi Inosine 5'-monophosphate dehydrogenase: evidence of a substrate-induced hinged-lid motion by loop 6. Biochemistry. 2000;39:4533–4542. doi: 10.1021/bi992645l. [DOI] [PubMed] [Google Scholar]
- Miller JR, Dunham S, Mochalkin I, Banotai C, Bowman M, Buist S, Dunkle B, Hanna D, Harwood HJ, Huband MD, et al. A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore. Proc Natl Acad Sci U S A. 2009;106:1737–1742. doi: 10.1073/pnas.0811275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D, Josephine HR, Li H, Lakner C, MacPherson IS, Naylor GJ, Swofford D, Hedstrom L, Yang W. An enzymatic atavist revealed in dual pathways for water activation. PLoS Biol. 2008;6:e206. doi: 10.1371/journal.pbio.0060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JF. Kinetics of reversible inhibition of enzyme catalyzed reactions by tight binding inhibitors. Biochim. Biophys. Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera- a visualization system for exploratory research and analysis. J. Comp. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Prosise GL, Luecke H. Crystal Structures of Tritrichomonas foetus Inosine Monophosphate Dehydrogenase in Complex with Substrate, Cofactor and Analogs: A Structural Basis for the Random-in Ordered-out Kinetic Mechanism. J Mol Biol. 2003;326:517–527. doi: 10.1016/s0022-2836(02)01383-9. [DOI] [PubMed] [Google Scholar]
- Prosise GL, Wu JZ, Luecke H. Crystal Structure of Tritrichomonas foetus Inosine Monophosphate Dehydrogenase in Complex with the Inhibitor Ribavirin Monophosphate Reveals a Catalysis-dependent Ion-binding Site. J Biol Chem. 2002;277:50654–50659. doi: 10.1074/jbc.M208330200. [DOI] [PubMed] [Google Scholar]
- Riera TV, Wang W, Josephine HR, Hedstrom L. A kinetic alignment of orthologous inosine-5'-monophosphate dehydrogenases. Biochemistry. 2008;47:8689–8696. doi: 10.1021/bi800674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selgrad M, Malfertheiner P. New strategies for Helicobacter pylori eradication. Curr Opin Pharmacol. 2008;8:593–597. doi: 10.1016/j.coph.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko M, Wilson KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- Sintchak MD, Nimmesgern E. The structure of inosine 5'-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology. 2000;47:163–184. doi: 10.1016/s0162-3109(00)00193-4. [DOI] [PubMed] [Google Scholar]
- Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci U S A. 2004;101:3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr, Liu C, Abrahamsen MS. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci U S A. 2002;99:6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem Biol. 2008;15:70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: Identification of functional, structural and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- Walsh CT, Fischbach MA. Repurposing libraries of eukaryotic protein kinase inhibitors for antibiotic discovery. Proc Natl Acad Sci U S A. 2009;106:1689–1690. doi: 10.1073/pnas.0813405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby FG, Luecke H, Kuhn P, Somoza JR, Huete-Perez JA, Philips JD, Hill CP, Fletterick RJ, Wang CC. Crystal structure of Tritrichomonas foetus inosine-5'-monophosphate dehydrogenase and the enzyme-product complex. Biochemistry. 1997;36:10666–10674. doi: 10.1021/bi9708850. [DOI] [PubMed] [Google Scholar]
- Wilson K, Collart F, Huberman E, Stringer J, Ullman B. Amplification and molecular cloning of the IMP dehydrogenase gene of Leishmania donovani. J. Biol. Chem. 1991;266:1665–1671. [PubMed] [Google Scholar]
- Zhang R, G E, Rotella F, Westbrook E, Huberman E, Joachimiak A, Collart FR. Differential signatures of bacterial and mammalian IMP dehydrogenase enzymes. Curr. Med. Chem. 1999;6:537–543. [PubMed] [Google Scholar]
- Zhang R-G, Evans G, Rotella FJ, Westbrook EM, Beno D, Huberman E, Joachimiak A, Collart FR. Characteristics and crystal structure of bacterial inosine-5'-monophosphate dehydrogenase. Biochemistry. 1999;38:4691–4700. doi: 10.1021/bi982858v. [DOI] [PubMed] [Google Scholar]
- Zhou X, Cahoon M, Rosa P, Hedstrom L. Expression, purification and charcterization of inosine-5'-monophosphate dehydrogenase from Borrelia burgdorferi. J. Biol. Chem. 1997;272:21977–21981. doi: 10.1074/jbc.272.35.21977. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.