

Abstract
Summary
Histone H2AX has a role in suppressing genomic instability and cancer. However, the mechanisms by which it performs these functions are poorly understood. After DNA breakage, H2AX is phosphorylated on serine 139 in chromatin near the break. We show here that H2AX serine 139 enforces efficient homologous recombinational repair of a chromosomal double-strand break (DSB) by using the sister chromatid as a template. BRCA1, Rad51, and CHK2 contribute to recombinational repair, in part independently of H2AX. H2AX−/− cells show increased use of single-strand annealing, an error-prone deletional mechanism of DSB repair. Therefore, the chromatin response around a chromosomal DSB, in which H2AX serine 139 phosphorylation plays a central role, “shapes” the repair process in favor of potentially error-free interchromatid homologous recombination at the expense of error-prone repair. H2AX phosphorylation may help set up a favorable disposition between sister chromatids.
Introduction
DSBs arising during replication may be repaired by sister chromatid recombination (SCR), a potentially error-free homologous recombination (HR) process in which the intact, neighboring sister chromatid is used as a template for repair (Johnson and Jasin, 2000; Kadyk and Hartwell, 1992). One trigger to SCR is thought to be the stalling of a DNA polymerase complex on a damaged DNA template (Kowalczykowski, 2000). SCR may be important for cancer predisposition because BRCA1, BRCA2, and the Bloom's syndrome gene have been implicated in SCR control (Scully and Livingston, 2000). However, the full spectrum of genes that regulate SCR is unknown. A remarkable feature of the DSB response is a modification of chromatin near the break, marked by phosphorylation of the histone H2A variant H2AX on C-terminal serine (S)139 (Rogakou et al., 1998). S. cerevisiae H2A proteins behave similarly (Downs et al., 2000). H2AX S139 phosphorylation is controlled by the DNA damage response kinases Atm, Atr, and DNA-PKCs (Burma et al., 2001; Stiff et al., 2004; Ward and Chen, 2001), and ScH2A S129 phosphorylation is controlled by MEC1 and TEL1 (Downs et al., 2000).
After exposure of mammalian cells to ionizing radiation (IR), phosphorylated H2AX (γ-H2AX) forms nuclear foci that colocalize with the Mre11/NBS1/Rad50 complex, Rad51, BRCA1, 53BP1, and MDC1 (Goldberg et al., 2003; Lou et al., 2003; Paull et al., 2000; Rappold et al., 2001; Stewart et al., 2003). The γ-H2AX tract near a DSB in mammalian cells may be megabases long (Rogakou et al., 1999); in S. cerevisiae, the equivalent γ-H2A tract at the MAT locus is tens of kilobases long (Shroff et al., 2004). γ-H2AX foci also form near DSBs induced in partial nuclear volumes in mammalian cells where they again colocalize with the above proteins (Paull et al., 2000; Rogakou et al., 1999). Early work with wortmannin to block H2AX phosphorylation suggested that γ-H2AX is essential for the accumulation of these repair/recombination proteins at a DSB (Paull et al., 2000). However, unlike Rad50−/−,Rad51−/−, and Brca1−/− mice, which die early in embryonic development, H2AX−/− mice are viable (Bassing et al., 2003; Celeste et al., 2002). Further, Rad50, Rad51, and BRCA1 can accumulate at defined regions of DSBs in H2AX−/− cells (Celeste et al., 2003b). Consequently, the role of the H2AX-dependent chromatin response is unclear at present.
H2AX−/− cells are hypersensitive to IR and show genomic instability, DSBR defects, and mild DNA damage checkpoint dysfunction (Bassing et al., 2002; Celeste et al., 2002; Fernandez-Capetillo et al., 2002). In thymocytes, γ-H2AX colocalizes with T cell receptor loci, suggesting a function in V(D)J recombination and nonhomologous end joining (NHEJ) (Chen et al., 2000). H2AX regulates class-switch recombination (CSR), a specialized form of NHEJ (Petersen et al., 2001). In p53−/− mice, H2AX is haploinsufficient for suppression of lymphoma (Bassing et al., 2003; Celeste et al., 2003a). B cell lymphomas in H2AX+/−p53−/− or H2AX−/−p53−/− mice show frequent IgH/c-myc translocations, with IgH breakpoints at V(D)J or CSR target sequences. In apparent support of a role in NHEJ, S. cerevisiae lacking H2A S129 show defective rejoining of an episomal plasmid (Downs et al., 2000).
In contrast, only circumstantial evidence implicates H2AX in HR. H2AX−/− mouse embryonic stem (ES) cells show modestly reduced gene targeting efficiency. H2AX−/− male mice are sterile (Celeste et al., 2002); however, this may be related to defective transcriptional silencing during pachytene (Fernandez-Capetillo et al., 2003). S. cerevisiae lacking H2A S129 are competent for mating-type switching, an intramolecular (intrachromatid) HR process (Downs et al., 2000). Interestingly, hydroxyurea treatment induces γ-H2AX at sites of replication arrest (Ward and Chen, 2001). This raises the possibility that H2AX regulates SCR, an intermolecular (interchromatid) HR process. To test this hypothesis, we developed a reporter specific for SCR and used it to analyze the role of H2AX in SCR.
Results
Recombination Reporters for Studying H2AX
To study recombination functions of H2AX, we used mouse H2AXfl/fl ES cells in which each allele of H2AX is flanked by loxP sites (Bassing et al., 2002). Cre treatment results in three alternative genotypes: H2AXfl/fl (H2AX+/+); H2AXfl/Δ (H2AX+/−), and H2AXΔ/Δ (H2AX−/−). We generated two HR reporters. HR-GFP (Figure 1A) has two tandem repeat mutant copies of the gene encoding the enhanced green fluorescent protein (GFP). The first, TR-GFP, carries a small 5′ deletion; the second, ISceI-GFP, is full length but is interrupted by an 18 bp restriction site for the rare-cutting restriction endonuclease, I-Sce I (Jasin, 1996) (Figure 1A). I-SceI-induced breakage triggers DSBR, whether through NHEJ, HR, or single-strand annealing (SSA). HR may occur between tandem copies of GFP when gene conversion generates wild-type (wt) GFP. Short tract gene conversion (STGC, Figure 1A) reflects either intramolecular/intrachromatid or intermolecular/interchromatid recombination (i.e., SCR). Some GFP+ SCR events entail long tract gene conversion (LTGC) between sister chromatids, causing “GFP triplication” (Johnson and Jasin, 2000; Puget et al., 2004) (Figure 1A). SCR followed by crossing over could cause the same outcome; GFP triplication is therefore diagnostic of SCR. Because crossing over is suppressed in somatic cells, including in SCR, we termed this outcome SCR/LTGC (Johnson and Jasin, 2000).
Figure 1. Impaired Gene Conversion of a Chromosomal DSB in H2AX−/− ES Cells.
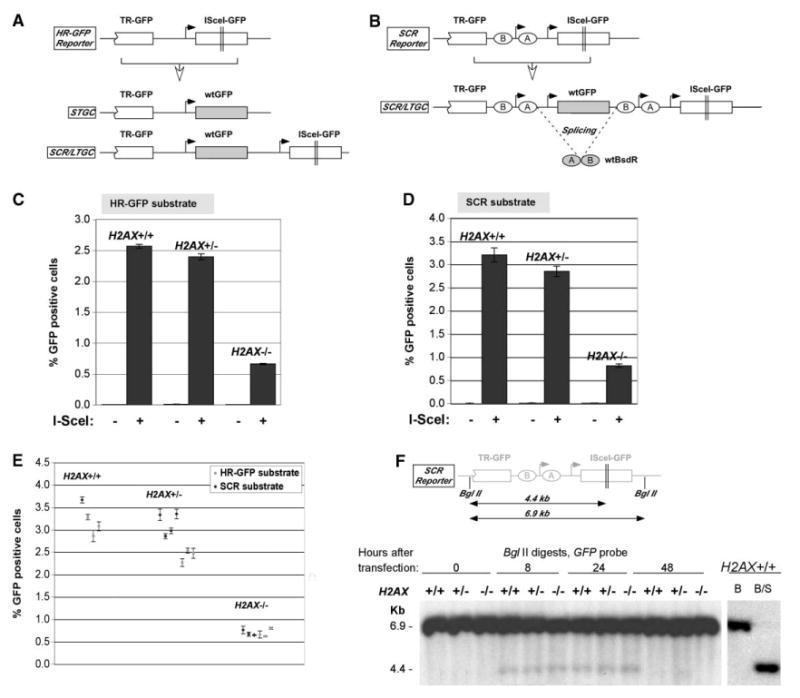
(A) HR-GFP reporter, filled arrows show promoters; open-headed arrow indicates I-SceI-induced HR, leading to alternative STGC or SCR/LTGC outcomes shown.
(B) SCR reporter, ellipses “A” and “B” indicate artificial BsdR exons.
(C) I-SceI induction of HR in HR-GFP reporter. Bars represent mean of four independent experiments, each with triplicates. Error bars indicate SEM. Paired t test H2AX−/− versus other groups, p < 0.001%.
(D) Similar experiments in SCR reporter. Paired t test H2AX−/− versus other samples, p < 0.001%.
(E) I-SceI-induced HR in isogenic ES cell clones containing HR-GFP or SCR reporters. Dots represent mean of triplicates in one experiment for each clone. Error bars indicate SEM. Unpaired t test (unknown variance) H2AX−/− versus other samples, p < 0.0001%.
(F) I-SceI-induced chromosomal DSBs in ES cell lines transfected with I-SceI plasmid or vector control and harvested at times shown after transfection. Southern blotting was performed on BglII-digested gDNA with a GFP probe. Schematic of the SCR reporter with BglII and I-SceI sites is shown in upper section. Right, control BglII (“B”) and BglII/I-SceI (“B/S”) digests of H2AX+/+ gDNA.
To positively select for the SCR/LTGC outcome, we devised a “nested intron” SCR reporter (Figure 1B) in which two artificial exons of the gene encoding blasticidin-S-deaminase (BsdR) are located “head to toe” as shown (Puget et al., 2004). Only in the context of SCR/LTGC (GFP triplication) is the BsdR cassette duplicated when splicing generates a BsdR+ cell. Hence, resistance to blasticidin is diagnostic of SCR. We validated this system previously in the human osteosarcoma cell line, U2OS (Puget et al., 2004).
We targeted HR-GFP or SCR reporters to the ROSA26 locus of H2AXfl/fl ES cells (Experimental Procedures and Supplemental Figure S1 available online at http://www.molecule.org/cgi/content/full/16/6/1017/DC1/) and transduced one clone of each reporter type with adenovirus encoding Cre (Bassing et al., 2002). We identified several clones of each H2AX genotype (Supplemental Figure S2). Although there were no loxP sites in the targeting vector, we rechecked the structure of the recombination reporter in each Cre-treated clone to confirm that Cre had not caused spurious rearrangement of the reporter (data not shown).
Impaired HR in H2AX−/− Cells
We transfected H2AX+/+,H2AX+/−, or H2AX−/− ES reporter clones in parallel with I-SceI plasmid or empty vector (Figures 1C and 1D) and quantified gene conversion as induction of GFP+ cells (see Experimental Procedures). Cultures not exposed to I-SceI were always < 0.015% GFP+ (Figures 1C and 1D); this negative control is not shown subsequently. All data were corrected for transfection efficiency, which was typically between 55% and 80%, and did not vary with H2AX status. After transfection of I-SceI plasmid into H2AX+/+ or H2AX+/− cells, 2.5%–3.5% of transfected cells became GFP+ (Figures 1C and 1D). In contrast, H2AX−/− cells showed a highly significant ∼4-fold reduction, with I-SceI converting 0.5%–1.0% of transfected cells to GFP+ (Figures 1C and 1D). To exclude errors caused by clone-to-clone variation, we examined, in parallel, four H2AX+/+, seven H2AX+/−, and six H2AX−/− independent clones carrying either of the two reporters (Figure 1E). All six H2AX−/− clones were defective for HR. Interestingly, we noted a slight reduction of HR efficiency in H2AX+/− cells, the mean value for H2AX+/+ being 3.21% GFP+ and for H2AX+/−, 2.85% GFP+.
Conceivably, access of I-SceI to its target site might vary with H2AX status. To assess this, we measured breakage in the reporter at discrete times after I-SceI transfection (Figure 1F). At 8 and 24 hr after I-SceI transfection, the broken locus is seen as a 4.4 kb GFP-hybridizing band. Importantly, H2AX+/+, H2AX+/−, and H2AX−/− cells reveal identical degrees of breakage. (I-SceI transfection efficiencies in the experiment shown were 80.4%, 79.1%, and 76.9%, respectively.) This shows that the inefficient induction of GFP+ cells by I-SceI in H2AX−/− cells is caused by a true defect in HR, rather than by inefficient breakage at the I-SceI site.
H2AX Serine 139 Is Essential for H2AX-Mediated Homologous Recombination
We asked whether the HR defect in H2AX−/− ES cells could be reversed by expression of wt H2AX. To determine this, we selected pools of H2AX−/− ES cells stably transfected with control vector or expression vector encoding wt human H2AX bearing an N-terminal influenza hemagglutinin (HA) tag. Consistent with its integration into chromatin, HA-H2AX could be extracted efficiently only by acid extraction or by other histone preparation methods (data not shown). Immunostaining for HA-H2AX with anti-HA monoclonal antibody 12CA5 revealed a strong signal throughout the nucleus, with relative nucleolar exclusion (data not shown). Unphosphorylated endogenous H2AX behaves similarly in all H2AX+/+ cells we have examined (A.X. and R.S., unpublished data). H2AX−/− ES cells stably expressing wt H2AX showed a ∼4-fold increase in recombination compared to control transfectants (Figures 2A and 2B). The N-terminal HA tag appears not to compromise H2AX function. Indeed, the histone H2A N and C termini occupy distinct surfaces of the nucleosome (Luger et al., 1997).
Figure 2. Serine 139 Is Essential for H2AX HR Function.
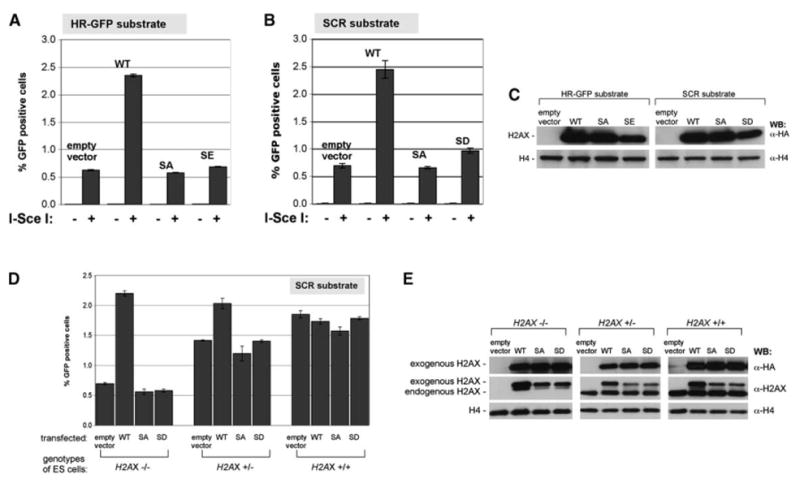
(A and B) I-SceI-induced GFP+ frequencies in H2AX−/− reporter cell lines HR-GFP (A) or SCR (B), stably expressing H2AX alleles indicated. Bars represent mean of four independent experiments, each with triplicates. Error bars indicate SEM. Paired t test between wt and other samples, p < 0.0001%; between SD and SA in (B): p < 0.001%.
(C) Steady state levels of HA-H2AX protein in ES cell lines shown in (A) and (B).
(D) I-SceI-induced GFP+ frequencies in ES cells transiently transfected with H2AX expression plasmids. Bars represent mean of two independent experiments, each with triplicates. Error bars indicate SEM. Paired t test between wt and any other samples in H2AX−/−, p < 0.0001%; in H2AX+/−, p < 0.005%; in H2AX+/+, not significant.
(E) H2AX protein levels after transient transfection of H2AX alleles in ES cells containing SCR reporter—same experiment as (D). HA-H2AX migrates slower than endogenous H2AX. Antibody specific to wt unphosphorylated H2AX C-terminal tail binds poorly to S139 mutant proteins (middle row and data not shown); this accounts for the discrepancy between anti-HA and anti-H2AX signal seen here.
To assess the role of H2AX S139 in HR, we generated mutants of HA-tagged human H2AX in which S139 was changed to alanine (SA), aspartate (SD), or glutamate (SE). We generated parallel pools of H2AX−/− ES cells stably expressing empty vector, wt H2AX or the SA, SD, or SE mutants. Steady-state levels of different H2AX proteins were similar, although SD and SE abundance was slightly reduced (Figure 2C). Replacement of S139 with an acidic residue (which could potentially mimic phosphoserine) or by SA abolished the HR function of H2AX (Figures 2A and 2B). Taken together, these results show that S139 is essential for this H2AX function.
We asked whether transient expression of wt H2AX could reverse the HR defect in H2AX−/− ES cells. We cotransfected, in parallel, mutant or wt H2AX expression vectors, together with either empty vector or I-SceI expression vector. A similar pattern of rescue was seen, with wt H2AX causing a ∼4-fold increase in HR efficiency and H2AX S139 mutants failing to rescue (Figure 2D). In H2AX+/+ ES cells, forced overexpression of either wt or mutant H2AX did not affect HR, despite a 2-fold increase in the abundance of wt H2AX over the endogenous protein level (Figure 2E). In H2AX+/− ES cells, forced overexpression of wt, but not S139 mutant, H2AX improved recombination efficiency (Figure 2D). This raises the possibility, hinted at in previous experiments (Figure 1E), that H2AX+/− ES cells may be slightly impaired for HR. However, we noted variability of this effect between different H2AX+/− clones (Figure 1E and data not shown).
H2AX Serine 139 Regulates Recombination between Sister Chromatids
To determine whether H2AX controls SCR, we compared H2AX+/+, H2AX+/−, and H2AX−/− ES cells carrying the SCR reporter for the efficiency of SCR/LTGC, as measured by induction of BsdR+ clones by I-SceI (Puget et al., 2004). All data were corrected for both I-SceI transfection efficiency and plating efficiency of ES cells on gelatinized plates—the latter being typically ∼35% for H2AX+/+ or H2AX+/− ES cells and ∼25% for H2AX−/− ES cells (see Experimental Procedures). H2AX−/− ES cells showed a ∼4-fold reduction in I-SceI-induced BsdR+ (Figure 3B) outcomes, compared to equivalent H2AX+/+ or H2AX+/− ES cells. The ratio of I-SceI-induced BsdR+ to GFP+ frequency was 6%–7% (Figure 3C) and did not vary as a function of H2AX status. Stable expression of wt, but not S139 mutated, H2AX restored efficient SCR/LTGC to H2AX−/− ES cells (Figure 3E) but did not affect the ratio of I-SceI-induced BsdR+ to GFP+ frequency, irrespective of the form of H2AX expressed (Figure 3F). These results show that H2AX regulates SCR and that H2AX S139 plays a central role in this process. However, the fixed ratio of BsdR+ to GFP+ frequency implies that the “choice” between STGC and LTGC is not affected by H2AX status.
Figure 3. H2AX Regulates SCR.
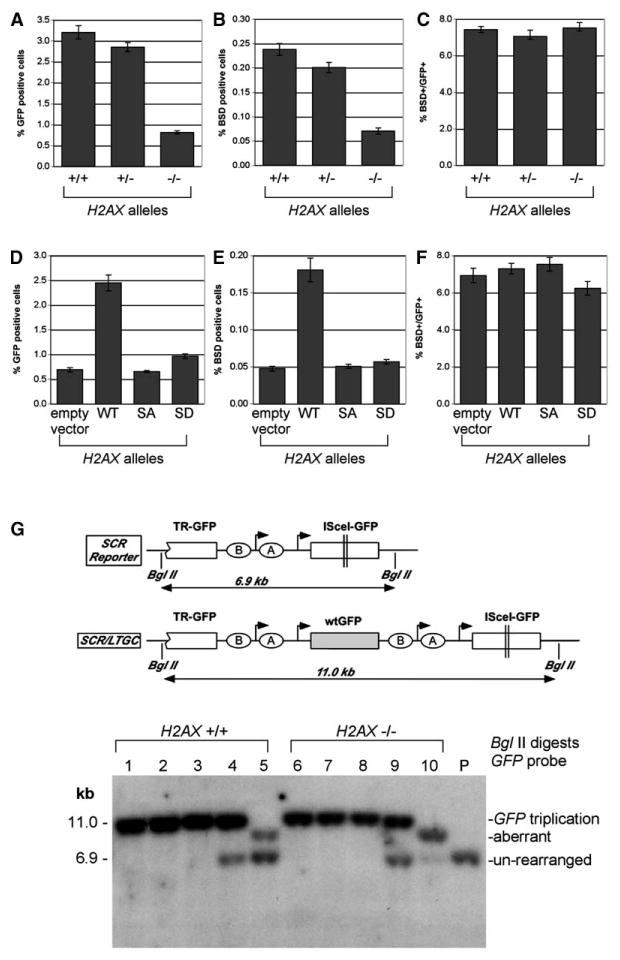
(A and D) I-SceI-induced GFP+ frequencies in ES cell lines containing the SCR reporter (A) or H2AX−/− ES cell lines containing the SCR reporter, stably reconstituted with H2AX alleles (D). These figures reproduce the data shown in Figures 1D and 2B. Bars represent mean of four independent experiments, each with triplicates. Error bars indicate SEM.
(B and E) I-SceI-induced BsdR+ frequencies in ES cell lines in the same experiments depicted in (A) and (D). Bars represent mean of four independent experiments, each with triplicates. Error bars indicate SEM. Paired t test between H2AX−/− cells and others in (B), p < 0.001%. Paired t test between wt and any other samples in (D), p < 0.001%. Frequency of spontaneously arising BsdR+ cells was ∼0.001% in all cases and is not shown.
(C and F) Ratios of I-SceI-induced BsdR+ to GFP+ frequency from the same experiments shown in (A) and (B) or (D) and (E). Bars represent mean of ratios in four independent experiments, each with triplicates. Error bars indicate SEM. All differences, not significant. (G) Southern blot analysis of I-SceI-induced BsdR+ colonies (BglII digests of gDNA, GFP probe). Cartoon shows schematic of unre-arranged reporter and of GFP triplication outcome. Lanes 1–3 and 6–8 are representative of GFP triplication events (H2AX status as indicated). Lanes 4 and 9 are representative of mixed clones with GFP triplication and parental reporter (compare Puget et al., 2004). Lanes 5 and 10 are representative of aberrant SCR/LTGC events. P, parental (unrearranged) reporter.
To study SCR at a molecular level, we examined the SCR reporter of individual I-SceI-induced BsdR+ colonies by Southern blotting (Figure 3G). We studied 32 H2AX+/+ and 41 H2AX−/− colonies in this way. We found that 27/32 (84%) BsdR+ H2AX+/+ colonies showed GFP triplication (Figure 3G, lanes 1–4, 11 kb band), whereas 5/32 (16%) were “aberrant” events; for example, those in which LTGC had resolved prior to full GFP triplication (e.g., lane 5). Similarly, 34/41 (83%) BsdR+ H2AX−/− colonies showed GFP triplication (lanes 6–9), whereas 7/41 (17%) were aberrant (e.g., lane 10). These proportions are virtually identical to those measured previously in U2OS cells (Puget et al., 2004). In U2OS cells, we noted additional rare (2%–3% of BsdR+ colonies) clones in which multiple rounds of gene amplification had occurred within the reporter (Puget et al., 2004). In a total of 99 I-SceI-induced BsdR+ ES cell colonies of various H2AX genotypes, we found no examples of this rearrangement (data not shown). Taken together, the results do not indicate a role for H2AX in controlling termination of the gene conversion tract during SCR.
Suppression of Single-Strand Annealing by H2AX
Reduced efficiency of SCR in H2AX−/− cells might allow more error-prone DSBR mechanisms to come into play. We assessed mutagenic NHEJ and SSA in SCR reporter ES cell lines of different H2AX status. Each of these mechanisms should produce a colony that has no GFP+ cells and no cells containing an I-SceI site in the reporter. SSA involves a homologous deletion between the two GFP copies in the reporter (Figure 4A). To measure these outcomes, we transfected H2AX+/+ or H2AX−/− cells with I-SceI and grew them without selection. We assessed colonies for GFP status and for the presence of the I-SceI site by the ability of retransfected I-SceI to induce GFP+ in a fraction of cells of the colony in question. A number of colonies had the characteristics we sought, which were those containing no GFP+ cells and with no induction of GFP+ cells after I-SceI retransfection. We analyzed these colonies by Southern blotting. Strikingly, all (100%) such colonies revealed a deletion within the reporter suggestive of SSA (Figures 4A and 4B). To confirm that the break point was homologous (an obligatory feature of SSA), we PCR amplified and sequenced the GFP copy and found it to be identical in sequence to the known TR-GFP sequence. These events were therefore SSA events. We did not identify any mutagenic NHEJ events in this screen.
Figure 4. H2AX Suppresses SSA.
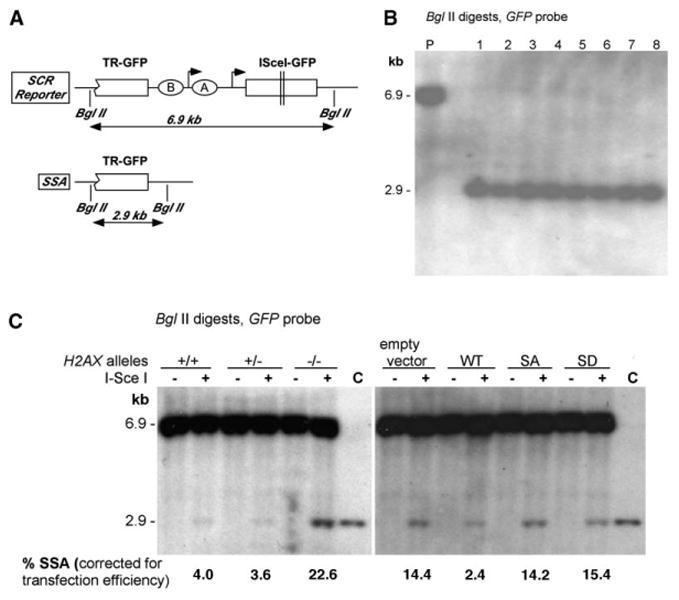
(A) The SCR reporter and its SSA product after DSBR.
(B) Southern blot analysis of I-SceI-trans-fected ES cell/SCR reporter clones that have lost the I-SceI site (Lanes 1–8, BglII digests of gDNA; GFP probe). P, parental (unrearranged) reporter.
(C) Southern blot analysis of unselected bulk cultures 5 days after transfection of I-SceI (+) or control (−) plasmid (BglII digested gDNA; GFP probe). “C” shows migration of sequence-defined SSA product. Numbers below gel indicate proportion of 2.9 kb SSA product in lane, quantified by densitometry, corrected for transfection efficiency and GFP copy number.
After correction for I-SceI transfection efficiency, we calculated the percentage of I-SceI-induced SSA events in H2AX+/+ and H2AX−/− cells over four independent experiments (Table 1). The mean SSA frequencies in H2AX+/+ and H2AX−/− cells were 5.4% and 28.8%, respectively, a highly significant difference (χ2 test, p < 0.001). Stable expression of wt H2AX, but not S139 mutants, appeared to suppress SSA in H2AX−/− cells (Table 1), although the differences by χ2 test did not reach statistical significance.
Table 1. I-SceI-Induced SSA Events in ES Cell Clones of Different H2AX Status.
| Measurement | H2AX Statusa | H2AX−/− Cells Stably Expressing H2AXb | ||||
|---|---|---|---|---|---|---|
| +/+ | −/− | Empty Vector | Wt | SA | SD | |
| SSA events/total screened (corrected for transfection efficiency) | 7/129* | 24/83* | 6/49 | 2/95 | 5/64 | 4/72 |
| Percentage of SSA (corrected for transfection efficiency) | 5.4% | 28.8% | 12.2% | 2.1% | 7.8% | 5.6% |
SSA events were identified and quantified as described in the text and in the Experimental Procedures.
Results of four independent experiments.
Results of two independent experiments.
χ2 test, p < 0.001
The high frequency of SSA events in H2AX−/− cells suggested that we might detect the 2.9 kb SSA product by Southern blotting in bulk I-SceI transfected ES cell cultures. We therefore harvested gDNA from H2AX+/−, H2AX+/−,or H2AX−/− cells, without selection or cloning, 5 days after transfection with I-SceI or control vector—i.e., at a time when all repair events should be complete. Southern blotting for GFP revealed a prominent 2.9 kb SSA band in H2AX−/− cultures and a much weaker one in H2AX+/+ or H2AX+/− cultures (Figure 4C). Transfection efficiencies in the experiment shown were 60.4% (H2AX+/+ cells), 50.1% (H2AX+/− cells), and 50.2% (H2AX−/− cells). We used phosphoimaging and densitometry to quantify the SSA band as a percentage of the parental 6.9 kb band, correcting for transfection efficiency and for copy number (one GFP copy in the SSA band, two copies in the parental band). Results of densitometry and colony screening experiments were similar. In particular, expression of wt, but not S139 mutant, H2AX in H2AX−/− ES cells suppressed appearance of the SSA band, reinforcing the conclusions of the above-noted colony formation experiments. Therefore, by two independent criteria, H2AX suppresses SSA.
BRCA1 and Rad51 Act Independently of H2AX to Regulate HR
As noted above, there is paradoxical information regarding the relationship between H2AX and BRCA1 (Bassing et al., 2002; Celeste et al., 2002, 2003b; Paull et al., 2000). We asked whether Rad51, BRCA1, and CHK2 recombination functions are independent of H2AX (Moynahan et al., 1999; Zhang et al., 2004). We used RNA interference (RNAi) to deplete proteins in either H2AX+/+ or H2AX−/− cells and determined the effect on HR (measured as I-SceI induction of GFP+ cells, Figure 5). RNAi-mediated depletion of BRCA1, Rad51, or CHK2 caused reductions in HR efficiency of similar proportions in H2AX+/+ and H2AX−/− cells (Figure 5). This suggests that these proteins contribute HR functions that are, at least in part, independent of H2AX.
Figure 5. BRCA1, Rad51, and CHK2 Act Independently of H2AX to Control HR.
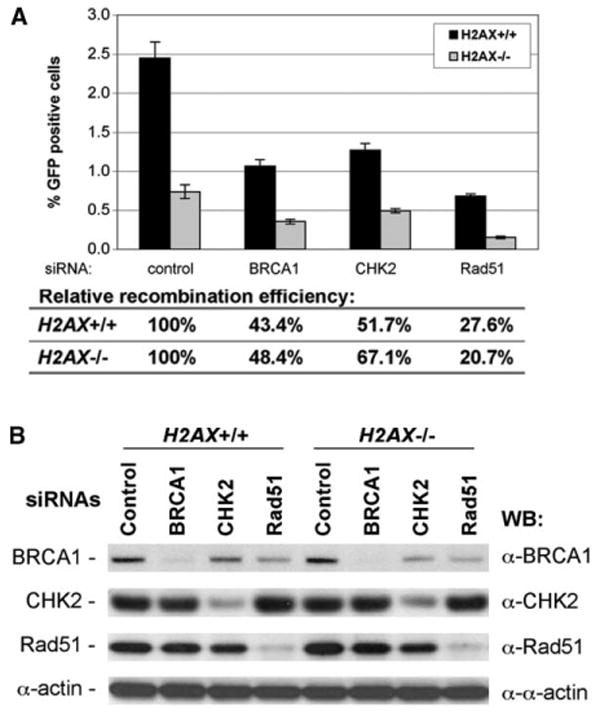
(A) The percentage of I-SceI-induced GFP+ cells from ES cell lines transfected with RNAi duplexes. H2AX+/+ or H2AX−/− ES cell lines, as indicated, were transiently transfected with control RNAi or RNAi against BRCA1, CHK2, or Rad51, together with I-SceI plasmid. GFP+ cells were quantified by FACS analysis 3 days posttransfection. Bars represent the average from three independent experiments, each with triplicates. Error bars indicate SEM.
(B) Depletion of proteins in ES cells by corresponding RNAi. ES cells, as indicated, were transfected with RNAi and lysed 3 days posttransfection. Whole-cell extracts were resolved by SDS-PAGE and analyzed by Western blotting by using antibody against target proteins (BRCA1, CHK2, and Rad51) or α-actin as a loading control.
Discussion
The “generic” role of phosphorylated H2AX at DSBs has been unclear. Work described here shows that H2AX regulates homologous recombination between sister chromatids and suppresses single-strand annealing. These functions are therefore regulated by the chromatin response around a DSB. The ∼4-fold effects of H2AX on HR, SCR, and SSA require serine 139, showing the importance of this residue as a regulator of recombination. H2AX status appears not to influence resolution of the gene conversion tract once it is established. Somewhat surprisingly, these experiments provided no information regarding the role of H2AX in NHEJ. We cannot explain why we captured no mutagenic NHEJ events in this screen. We believe that NHEJ is operative in fixing a substantial proportion of I-SceI-induced DSBs in these experiments, but presumably these are almost all simple, nonmutagenic religations. Perhaps the combination of cell type and locus of integration, in the experiments described here, is relatively nonpermissive of mutagenic NHEJ.
In vertebrate cells, recombination genes such as Rad54 and BRCA2 are known to facilitate HR and suppress SSA (Dronkert et al., 2000; Tutt et al., 2001). H2AX may therefore work in a concerted fashion with these genes. In S. cerevisiae, the SSA:HR ratio is increased in the absence of RAD51, RAD54, RAD55, or RAD57 (Ivanov et al., 1996). The apparent reciprocal relationship between HR and SSA revealed by these studies could indicate a role for each of these genes in facilitating Rad51 function. Indeed, the mechanisms of action of Rad54, Rad55, Rad57, and BRCA2 in Rad51 regulation are increasingly well understood. H2AX might therefore also regulate Rad51 function, perhaps through an intermediary such as BRCA1. Alternatively, H2AX might promote SCR and suppress SSA by a quite different mechanism. For example, it is unknown whether γ-H2AX marks both sister chromatids at the site of damage or whether it is restricted to the damaged sister. If the latter were the case, H2AX might conceivably play a role in discriminating between repair templates, favoring intermolecular SCR at the expense of intramolecular SSA, by means of a differential marking of sister chromatids.
Because H2AX is part of the nucleosome, its function in recombination is likely executed in situ from within the chromatin fiber. This function is exerted up to several kilobases (or perhaps megabases) away from the DSB (Rogakou et al., 1999). Indeed, mapping of the γ-H2A chromatin tract in S. cerevisiae shows a relative dearth of γ-H2A signal in the immediate ∼1000 bp vicinity of an HO endonuclease-induced DSB (Shroff et al., 2004). Although this could represent epitope masking by a γ-H2A interacting protein binding at 100% occupancy, it may alternatively indicate that γ-H2A acts only in chromatin at a certain distance from the DSB. Taken together, these findings appear to run counter to the hypothesis that γ-H2AX helps to concentrate repair factors at the site of a DSB. An alternative hypothesis would propose that H2AX functions in processes parallel to the enzymology of the recombination reaction. Barring epistasis analysis, the direct proof of this is missing. However, consistent with this notion, we found that BRCA1, Rad51, and CHK2 all contribute recombination functions that are, at least in part, independent of H2AX. This is also consistent with the relatively mild genomic instability of H2AX−/− cells (Bassing et al., 2002; Celeste et al., 2002), the viability of H2AX−/− mice, and the H2AX independence of recruitment of BRCA1 and Rad51 to DSBs induced in partial nuclear volumes (Celeste et al., 2003b).
These results suggest ways to clarify understanding of BRCA1, which acts with BRCA2, Rad51, and other HR components to prevent catastrophic chromosome breakage and cell lethality (Scully and Livingston, 2000). Paradoxically, IR-induced BRCA1 focus formation is greatly diminished in H2AX−/− cells (Bassing et al., 2002; Celeste et al., 2002). This suggests that BRCA1 performs at least two separate functions in the DSB response: first, an H2AX-independent function with BRCA2/Rad51 at the break; second, an H2AX-dependent function on chromatin at a distance from the break.
One model of H2AX function proposes that the chromatin response around DSBs is part of a signal amplification mechanism involved in checkpoint function (Fernandez-Capetillo et al., 2002). Our work suggests a different model in which γ-H2AX—and the specialized chromatin structure established upon it—acts directly at the site of H2AX phosphorylation to set up a favorable disposition between sister chromatids. Both enzymatic and structural elements might collaborate to promote error-free SCR over illegitimate recombination (Kadyk and Hartwell, 1992). In this regard, proper sister chromatid cohesion and alignment might be prerequisites of efficient SCR (Sjögren and Nasmyth, 2001). One possible player in this process is cohesin, which is recruited to sites of DNA damage and phosphorylated by Atm in the DNA damage response (Kim et al., 2002). Recent work in S. cerevisiae directly supports this idea. Cohesin is recruited to chromatin near an HO endonuclease-induced DSB (Ström et al., 2004 [this issue of Molecular Cell]) in an H2A S129-dependent fashion (Ünal et al., 2004 [this issue of Molecular Cell]). Intriguingly, pulsed field gel measurement of postreplicative repair of IR-induced DSBs revealed a 3- to 4-fold defect in this process in yeast H2A S129 mutants as well as in cohesin mutants (Ström et al., 2004; Ünal et al., 2004). If these gel-based assays reflect true SCR, then yeast and mammalian H2A(X) may have strikingly similar generic functions, each contributing a ∼4-fold degree of efficiency to SCR. It will be of interest to study the functional interplay between H2AX and cohesin in SCR control in mammalian cells. At the same time, genes such as BRCA1, 53BP1, and MDC1 may not have direct orthologs in S. cerevisiae, indicating that the full spectrum of activities controlled by or controlling H2A(X) may vary between yeast and mammals.
A slight but systematic dysfunction of SCR may make the genome moderately plastic—sufficient, in the lifetime of a mammal, to promote cancer. If so, mild genetic or epigenetic disturbances in SCR regulation might have an impact on cancer risk in the human population. An analogy to this is suggested by the increased cancer predisposition of p53−/−H2AX+/− mice in comparison to p53−/−H2AX+/+ littermates (Bassing et al., 2003; Celeste et al., 2003a). It will be interesting to examine whether H2AX+/− cells are, under some circumstances, haploin-sufficient for HR and SCR. This would support the idea of a quantitative relationship between SCR control and cancer susceptibility.
Experimental Procedures
Plasmid and RNAi
HR-GFP and SCR reporters were constructed as previously described (Puget et al., 2004), with replacement of the CMV promoter driving ISceI-GFP with a pgk promoter. Reporters were assembled in a modified form of vector pBigT (Srinivas et al., 2001) from which we removed loxP sites and introduced an upstream splice acceptor site to allow expression of the neo gene from the endogenous ROSA26 promoter. Reporters were inserted into the PacI-AscI site of the ROSA26-PA targeting vector (Srinivas et al., 2001) to generate final targeting vectors (Supplemental Figure S1). The expression vector for HA-wt H2AX was constructed by standard cloning procedures, and the mutants SA, SD, and SE were generated by PCR and confirmed correct by sequencing. H2AX cDNAs were transferred to pcDNA3.1-Hyg (Invitrogen). Control RNAi duplex (5-AAUAACAGUGACCUUUAUGGAdTdT-3), RNAi duplex against BRCA1 (5′-AACCAGAAGAAAGGGCCUUCAdTdT-3′) and CHK2 (5′-AACTCTGTTCTATTCCTGAGGdTdT-3′), and RNAi smart-pool against Rad51 were purchased from Chemicon.
Antibodies and Western Blotting
To analyze histones, cells were lysed for 30 min by cyto-lysis buffer (10 mM Hepes [pH 7.9], 50 mM NaCl, 0.25 M sucrose, 0.1 mM EDTA, and 0.5% Triton X100), and pellets were dissolved in 0.25M HCl and 5% glycerol buffer. HCl extracts were resolved by SDS-PAGE electrophoresis and analyzed by Western blotting by using either anti-HA mAb 12CA5 or rabbit polyclonal antibody to unphosphorylated H2AX (Bassing et al., 2002). To analyze nonhistone proteins, cells were lysed with RIPA buffer, and Western blotting was performed by using antibody against Rad51, mouse BRCA1, or mouse CHK2 (Santa Cruz).
Cell Lines and Cell Culture
H2AXfl/fl ES cells were grown in ES medium on either MEF feeder cells or gelatinized plates, and adeno-Cre infection was performed as described previously (Bassing et al., 2002). To construct the ES reporter cell lines, 20 × 106 ES cells were electroporated with 20 μg of linearized targeting vector and seeded in four 6 cm plates with neo+ feeders, and G418 (400 μg/ml) was added to the medium 24 hr later. After ∼7 days selection, G418R colonies were isolated and expanded. To generate ES cell lines stably expressing H2AX, 6 × 106 H2AX−/− ES cells were electroporated with 8 μg of HygR H2AX expression vector and seeded on a 10 cm plate. Hygromycin (400 μg/ml) was added 48 hr later, and clones were pooled after a further ∼7 days.
Southern Analysis
gDNA was extracted from 5–10 × 106 ES cells by using the Puregene DNA Isolation Kit (Gentra Systems). Southern blotting for GFP was performed as described previously (Puget et al., 2004). The 5′ probe for ROSA26 targeting was a fragment used previously (Mao et al., 1999), and the probe for H2AX status was as described previously (Bassing et al., 2002).
Recombination Assays
2–4 × 105 trypsinized ES cells were transfected with 1 μg pcDNA3β-mycNLS-I-SceI (Puget et al., 2004) or control pcDNA3β by using Lipofectamine 2000 (Invitrogen). Transfection efficiency was measured by parallel transfection of wt GFP expression vector. GFP+ frequencies were measured 3–5 days posttransfection (Puget et al., 2004). To assay SCR events, cells were counted and replated at 20,000–50,000 cells per gelatinized 10 cm dish (triplicates). Blastici-din (5 μg/ml, Invitrogen) was added 2 days later. 2 weeks later, BsdR+ colonies were stained and counted or expanded for molecular analysis. Plating efficiency was also determined accordingly. Statistical comparisons between two unpaired populations were analyzed by two-tailed t test (unknown variance) or for paired samples by two-tailed paired t test.
PCR Analysis and Sequencing
For PCR analysis of SSA events we used 200 ng of gDNA from SSA clones and primers #1 (5′-CAGGTTACTCGGATCTCGAC-3′) and #2 (5′-GACTCCCGCCCATCTTCTAG-3′) to amplify a ∼1.1 kb GFP fragment. PCR products were sequenced with primers #3 (5′-GAGCG CACCATCTTCTTCAAG-3′) and #4 (5′-CTTTACTTGTACAGCTCG TCC-3′).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Scully lab members for stimulating discussions and Katerina Vlasakova for technical assistance. Drs. Phil Soriano, Frank Costatini, and Stuart Orkin provided ROSA26 sequences, plasmids, and protocols. We thank Drs. James Haber, Douglas Koshland, and Camilla Sjögren for communicating work “in press” to us during preparation of this manuscript. This work was supported by grants CA95175 and RSG-0419801MGO (to R.S.), by a Pew Scholars Award and a Howard Temin Award (to R.S.), by grants CA92625 and CA109901 (to F.W.A.), and by a Lymphoma Research Foundation Fellowship (to C.H.B.).
References
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003a;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2 AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003b;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, Nussenzweig A. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science. 2000;290:1962–1965. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- Dronkert ML, Beverloo HB, Johnson RD, Hoeijmakers JH, Jasin M, Kanaar R. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol Cell Biol. 2000;20:3147–3156. doi: 10.1128/mcb.20.9.3147-3156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyk LC, Hartwell LH. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics. 1992;132:387–402. doi: 10.1093/genetics/132.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem Sci. 2000;25:156–165. doi: 10.1016/s0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci USA. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2 AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puget N, Knowlton M, Scully R. Molecular analysis of sister chromatid recombination in mammalian cells. DNA Repair (Amst) 2004 doi: 10.1016/j.dnarep.2004.08.010. Published online November 25, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–620. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Mega-base chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren C, Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol. 2001;11:991–995. doi: 10.1016/s0960-9822(01)00271-8. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus BMC. Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Ström L, Lindroos HB, Shirahige K, Sjögren C. Post-replicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16(this issue):1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ünal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 16(this issue):991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. CHK2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.