

Abstract
Chronic stress impairs learning and memory in humans and rodents and disrupts long-term potentiation (LTP) in animal models. These effects are associated with structural changes in hippocampal neurons, including reduced dendritic arborization. Unlike the generally reversible effects of chronic stress on adult rat hippocampus, we have previously found that the effects of early-life stress endure and worsen during adulthood, yet the mechanisms for these clinically important sequelae are poorly understood. Stress promotes secretion of the neuropeptide corticotropin-releasing hormone (CRH) from hippocampal interneurons, activating receptors (CRF1) located on pyramidal cell dendrites. Additionally, chronic CRF1 occupancy negatively affects dendritic arborization in mouse organotypic slice cultures, similar to the pattern observed in middle-aged, early-stressed (CES) rats. Here we found that CRH expression is augmented in hippocampus of middle-aged CES rats, and then tested whether the morphological defects and poor memory performance in these animals involve excessive activation of CRF1 receptors. Central or peripheral administration of a CRF1 blocker following the stress period improved memory performance of CES rats in novel-object recognition tests and in the Morris water maze. Consonant with these effects, the antagonist also prevented dendritic atrophy and LTP attenuation in CA1 Schaffer collateral synapses. Together, these data suggest that persistently elevated hippocampal CRH–CRF1 interaction contributes importantly to the structural and cognitive impairments associated with early-life stress. Reducing CRF1 occupancy post hoc normalized hippocampal function during middle age, thus offering potential mechanism-based therapeutic interventions for children affected by chronic stress.
Introduction
Chronic early-life stress (CES), including neglect, loss of parent, or extreme poverty, affects the majority of children worldwide (UNICEF, 2005). Epidemiological studies have associated CES with enduring adverse consequences, including depression (Heim et al., 2008) and cognitive impairments (Nelson et al., 2007); these may emerge during adulthood and progress with age (Kaplan et al., 2001; Wilson, 2007). Elimination of global childhood stress is unlikely; therefore, effective therapies that can be used post hoc to prevent its long-term cognitive sequelae are needed to alleviate this significant problem.
The direct, causal role of early-life stress in subsequent memory dysfunction is difficult to study in humans. We therefore developed a rodent model of CES (during postnatal days 2–9) that uses fragmented maternal care to generate chronic stress in the pups (Avishai-Eliner et al., 2001b; Ivy et al., 2008). This model recapitulates the human condition in that it causes impaired memory later in life, and in addition, results in seriously impaired long-term potentiation (LTP), a variant of synaptic plasticity widely regarded as critical to the encoding of memory (Bliss and Collingridge, 1993). The LTP defects, which were selective to commissural/associational synapses in apical dendrites of fields CA1 and CA3 (Brunson et al., 2005), were accompanied by abnormal morphology of the corresponding neurons including impoverished dendritic arborization. However, the molecular mediators of these profound effects of early stress have remained unclear.
Glucocorticoids, the canonical stress hormones, mediate many actions of stress on hippocampus (Sapolsky, 1985; McEwen, 1999; de Kloet et al., 2008). More recently, the stress neuropeptide corticotropin-releasing hormone (CRH) has been implicated in stress-induced activation of neurons (Kovács and Sawchenko, 1996; Reyes et al., 2008), including hippocampal pyramidal cells (Chen et al., 2006; Joëls and Baram, 2009). CRH contributes to effects of acute stress on synaptic plasticity and memory (Lee and Davis, 1997; Blank et al., 2002; Chen et al., 2004b, 2010; Rainnie et al., 2004; Orozco-Cabal et al., 2006), and several lines of evidence support a role for CRH receptor occupancy in CES-induced hippocampal dysfunction: (1) interneurons expressing CRH are abundant in hippocampus (Chen et al., 2001); (2) CRH is released into hippocampal intercellular space during stress (Chen et al., 2004a, 2006); (3) CRH receptor type 1 (CRF1) resides on dendrites of CA1 cells, the same neurons sustaining dendritic atrophy after early-life stress (Chen et al., 2004b); (4) chronic exposure to CRH provokes dendritic atrophy in a pattern similar to that found after early stress; and (5) when administered into brains of immature rats, CRH led to persistent memory impairments reminiscent of those found after chronic early-life stress (Brunson et al., 2001b). Notably, CRH-induced impairments occurred even when glucocorticoid levels were “clamped” at low physiological levels (Akana et al., 1988), suggesting that they were independent of glucocorticoid receptor activation (Brunson et al., 2001b).
Based on these findings, the current study examined whether blocking CRF1 immediately following the early-life stress period would eliminate subsequent memory and morphological defects. We further tested whether CRF1 activation within the brain contributed to these effects of early-life stress.
Materials and Methods
Animals
All experiments conformed to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine (UCI).
Rats.
Time-pregnant Sprague Dawley rat dams gave birth in UCI vivaria, and dams and litters were maintained on 12 h light cycles with unlimited access to chow and water. Pups were mixed and adjusted to 12 per dam if necessary, to obviate the potential confounding effects of genetic variables and of litter size. Upon weaning on postnatal day 21 (P21), only male rats were kept and were housed 2–3 per cage.
Transgenic mice.
B6.Cg-TgN transgenic mice expressing yellow fluorescent protein (YFP) under the Thy1 promoter (Thy1-YFP; The Jackson Laboratory) were born in UCI vivaria and were used for organotypic hippocampal slice cultures on postnatal day 1, as described previously (Chen et al., 2008), and see below.
CES paradigm
Chronic stress lasting for 1 week was generated in rat pups using a previously established model (Gilles et al., 1996; Avishai-Eliner et al., 2001b; Brunson et al., 2005; Ivy et al., 2008). This stress is a result of erratic and fragmented nurturing behaviors of rat dams, which stem from limited nesting material in the cage. Briefly, on P2, dams and pups of the CES group were placed in cages fitted with plastic-coated aluminum mesh bottoms (2.5 cm above the floor, allowing collection of droppings), with no bedding material. Nesting material consisted of one paper towel that was shredded by the dam to construct a rudimentary nest. Control (CTL) dams and their litters resided in bedded cages, containing ∼0.33 cubic feet of sanitary chips. Control and experimental cages were undisturbed during P2–P9, and housed in a quiet room enclosed in a protective tent with strong laminar airflow, preventing ammonia accumulation. Maternal nursing behaviors were monitored during the week of CES as described previously (Ivy et al., 2008).
Administration of CRF1 antagonist
The small-molecule selective CRF1 antagonist (ANT) NBI30775 (3-[6-(dimethylamino)-4-methyl-pyrid-3-yl]-2,5-dimethyl-N,N-dipropyl-pyrazolo[2,3-a]pyrimidin-7-amine) or vehicle (VEH) solution was chronically infused via osmotic minipumps into control and CES male rats for 1 week, starting immediately after the stress period (P10–P17). The administration of the CRF1 blocker or vehicle was either subcutaneous [for systemic release (Fenoglio et al., 2005)] or intracerebroventricular. The latter administration mode was conducted to exclude the potential confounder of blocking pituitary CRF1 and downstream release of adrenal glucocorticoids. Briefly, NBI30775 was dissolved in warm distilled water and the pH was adjusted; vehicle solution was made using the same pH-adjusted distilled water without the compound. Drug solution (4 mg/ml) or vehicle was loaded into osmotic Alzet minipumps (model 1007D, Alzet), calibrated to release 0.5 μl/h (∼2.5 mg/kg/d) over 7 d. For systemic release of the CRF1 blocker, pups were anesthetized with vaporized isoflurane and pumps implanted subcutaneously between the shoulder blades through a small incision made at the nape of the neck. The implantation surgery took <5 min per animal, and, after recovery, pups returned to home cages. For intracerebroventricular infusions, pumps were implanted in the same manner and attached to a catheter connected to a 3.5 mm cannula placed into the lateral cerebral ventricle using the following coordinates (with bregma as landmark): AP −0.7, L 2.0, V 3.3. For both systemic and intracerebroventricular routes, pumps were implanted on the morning of P10, and CRF1 blocker or vehicle was continuously infused until P17. The pumps were removed upon weaning on P21 (Brunson et al., 2001b). Maternal care was monitored (Ivy et al., 2008; Rice et al., 2008), and there were no significant differences in dam interaction with implanted compared with nonimplanted pups during P10–P21. Implantation of pumps did not significantly alter body weight at weaning in either type of surgical procedure (p > 0.05): naive CTL = 42.3 ± 3.1, naive CES = 40.32 ± 1.8, subcutaneously implanted CTL = 41.3 ± 1.5, subcutaneously implanted CES = 42.6 ± 1.4, intracerebroventricularly implanted CTL = 47.4 ± 2.3, intracerebroventricularly implanted CES = 40.8 ± 2.7.
Novel-object recognition
Tests of learning and memory were conducted once rats reached middle age (10–12 months); at this age, early-life stressed rats were previously found to have significant memory impairments and abnormal LTP (Brunson et al., 2005). For the object recognition (OR) procedure, transparent plastic cages were coated with opaque paper except the front panel, to allow for live experimenter observation (Broadbent et al., 2004; Brunson et al., 2005). After 3 d of habituation to the cage, rats explored two objects in a familiarization session (day 1). In the test session on day 2, rats were presented with a duplicate of a previously encountered object from day 1 and a novel object. Preliminary object exposure tests were used in a separate cohort of rats (n = 14) to select the three objects used for the OR procedure, yielding exploration times for objects 1 (light bulb), 2 (square Lego brick), and 3 (padlock) of 29.8 ± 8.4, 27.1 ± 9.7, and 33.7 ± 8.3 s, respectively). Counterbalancing of the objects was performed in an additional cohort of rats and did not influence the results (see Results). The duration of exploration of each object (sniffing with the animal's nose <2 cm from objects) as well as total object exploration time was recorded during the 5 min sessions on the familiarization and testing days.
Morris water maze
Rats were tested for hippocampus-dependent spatial memory using a variant of the Morris water maze (MWM) procedure with the least swim time [and thus minimal potential stress (Morris, 1984)], as described previously (Brunson et al., 2001b, 2005). Briefly, rats were trained to find a submerged platform in a pool of opaque water over 6 trials per day, for 2 d. During the 2 d of training, the platform remained in the same quadrant and all rats were trained to the same platform position. In an additional cohort of rats, the goal quadrant was counterbalanced, and this did not influence the time to find the platform (two-way repeated-measures ANOVA; effect of quadrant location F(1,23) = 0.48, p = 0.50; interaction of quadrant location and trial F(11,253) = 1.23, p = 0.19). For the reversal procedure, the platform was relocated to a new quadrant adjacent to the previous one. Rats entered the pool in one of three quadrants not containing the platform, and the start quadrant alternated between trials. At the start of each trial, rats were placed in the pool facing the wall and were allowed to swim for 60 s or until the platform was found. If the rat did not mount the platform during the trial, he was guided to the platform and remained on it for 15 s before removal from the pool. Rats were required to rely on spatial cues placed on the four walls of the room to learn the platform location during training and testing days, and short-term spatial memory was considered to have occurred if the latency time to finding the platform grew shorter upon repeated trials within the same day (acquisition of short-term memory does not exclude the possibility that long-term memory was being concurrently acquired). Long-term spatial memory was tested in a single, 60 s “probe” trial with no platform in the pool. This was conducted in separate cohorts of rats either 24 h after the second day of training or after the reversal procedure (fourth test day). A cued learning task was also conducted 2 weeks after water maze test. This cued procedure does not rely on hippocampus and thus controls for potential differences in motivation or sensorimotor skills that could influence performance in the MWM. The cued procedure consisted of six trials, using a proximal cue (spherical object attached to the platform, raised 10 cm above the water surface), while distal cues were removed. All trials were recorded, and data were analyzed for escape latency, mean swim speeds, and swim distances.
Tissue processing and immunocytochemistry
Rats were anesthetized with sodium pentobarbital and perfused with 0.9% saline, pH 7.4, for 5–8 min. Brains were removed and hemisected, and the left hemisphere was immersed in paraformaldehyde fixative for 24 h followed by cryoprotection in 25% sucrose for 36–48 h. The right hemisphere was used for Golgi impregnation studies (see below). Left hemispheres were sectioned 20 μm thick, and CRH immunocytochemistry (ICC) was performed on free-floating sections as previously described (Chen et al., 2001). Briefly, after washing (3 × 5 min) with 0.01 m PBS containing 0.3% Triton X-100 (PBS-T; pH 7.4), sections were treated for 30 min in 0.3% H2O2/PBS, followed by blockade of nonspecific sites with 5% normal goat serum in PBS for 30 min. After rinsing, sections were incubated for 2 d at 4°C with rabbit anti-CRH antiserum (1:20,000; a gift from Dr. W. W. Vale, Salk Institute) in PBS containing 1% bovine serum albumin, and washed in PBS-T. Sections were incubated in biotinylated goat-anti-rabbit IgG (1:200; Vector Laboratories) in PBS for 2 h at room temperature. After washing, sections were incubated in the avidin–biotin–peroxidase complex (ABC) solution (1:100; Vector Laboratories) for 2 h and rinsed (3 × 5 min PBS-T), and the reaction product was visualized by incubating the sections in 0.04% 3,3′-diaminobenzidine (DAB) containing 0.01% H2O2, with or without 0.3% nickel chloride. Sections were mounted on poly-l-lysine-coated slides and coverslipped with Permount (Fisher Scientific).
Golgi–Cox impregnation and quantification of CA1 pyramidal cell processes
Right hemispheres were immersed in Golgi–Cox solution (12–14 d), followed by 30% sucrose (3–5 d), both in the dark. Coronal sections (200 μm) were collected in 6% sucrose, developed in 14% NH4OH (30 min), and fixed (Kodak Fixative, Kodak). Individual, fully Golgi-impregnated CA1 neurons were imaged and reconstructed without knowledge of treatment group (n = 5–8 neurons per rat). Dendritic arborization was analyzed using the Sholl method (Sholl, 1953), which provides a quantitative description of the dendritic tree by counting the number of dendrites that cross virtual concentric circles drawn at fixed distances from the soma. The number of dendritic intersections at each concentric circle (20 μm, 40 μm, 60 μm, etc.), as well as total dendritic length, was measured and compared among groups.
Organotypic slice cultures
Hippocampal slice cultures were prepared from P1 YFP-expressing mice as described previously (Chen et al., 2008). Briefly, after decapitation, mouse brains were removed and placed in minimal essential medium (1× MEM; Invitrogen), and a block of tissue including entorhinal cortex and hippocampus was dissected from adhering tissue. Transverse slices (300 μm) were cut (McIlwain tissue chopper; Mickle Laboratory). Sections were placed onto Millicell-CM culture membranes (Millipore), and transferred to six-well plates filled with 1.2 ml of culture medium (0.5× MEM, 25% horse serum, 25% HBSS, 30 mm glucose, 7.5% NaHCO3, and 0.01% ascorbic acid, pH 7.2). Cultures were incubated in a humidified, 5% CO2 atmosphere at 36°C for 14 d in vitro (DIV), with medium changes every 2–3 d. To study the effects of chronic exposure to CRH on CA1 pyramidal cell dendrites, synthetic rat/human CRH (100 nm; Bachem) was applied to the culture medium on DIV 2 as well as to refreshed medium. After 13 d, cultures were fixed in fresh 4% paraformaldehyde for 30 min and processed for fluorescent ICC. Cultures were sectioned transversely (20 μm) using a cryostat, and ICC was performed on free-floating sections (Chen et al., 2004a) using mouse anti-GFP (1:8000; Sigma) followed by visualization with anti-mouse IgG conjugated to Alexa Fluor 488 (1:200; Invitrogen).
Long-term potentiation studies
Hippocampal slices were prepared as published previously (Brunson et al., 2005). Hippocampal slices (350 μm) were collected into artificial CSF maintained at 32°C for ∼1 h in an interface chamber (95% O2/5% CO2), superfused at 60–80 ml/h with artificial CSF of the following composition (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 2.5 MgSO4, 3.4 CaCl2, 26 NaHCO3). For LTP, a glass recording electrode (2 m NaCl; 2 MΩ), was positioned in CA1b stratum radiatum (s.r.) to record field EPSPs (fEPSPs). Stimulating test pulses were delivered (0.05 Hz) to the Schaffer collateral fibers in CA1c s.r. using a concentric bipolar stimulating electrode. Standard input–output curves were generated and test stimulation current was set to elicit a response = 50% of the maximum fEPSP amplitude. Baseline responses were recorded for 20 min, and unstable slices discarded. A train of high-frequency stimulation (HFS; 100 Hz, 1 s) was used to induce LTP. Analysis of fEPSP amplitudes and slopes (20–80% fall) was performed on digitized files (NacGather 2.0; Theta Burst). A postrecording correction factor for rising drift was applied to two CES+ANT slices before unblinding and did not affect statistical outcomes. Slope and amplitude analyses yielded identical results, and slope data are shown. Values from multiple slices were averaged for each rat, and statistical analysis performed by rat. One animal in the CES group failed to respond to the stress manipulations and was subsequently removed from analyses. Values are described in the text as percentage potentiation in means ± SD, whereas the figures demonstrate means ± SEM.
Image analysis of dendritic branching
Fixed organotypic cultures were imaged using a Zeiss LSM510 Meta or Bio-Rad Radiance 2000 confocal scanning system. Dendritic trees of individual YFP-expressing CA1 pyramidal cells were captured using “z-stack” three-dimensional projections (12–16 optical slices at 2 μm each; 10–14 neurons per group) and files were converted to Adobe Photoshop. For both slice culture and Golgi impregnation techniques, neurons were reconstructed and drawn using Zeiss LSM Image Browser and Adobe Photoshop software.
Statistical analyses
Analysis of CRH levels was accomplished using Student's t test and one-way ANOVA, as appropriate. Two-way repeated-measures (RM)-ANOVA was used for the LTP experiments and Sholl analyses of dendrites. Early-life treatment group was used as the “between-subjects” factor, and time after high-frequency stimulation (LTP) or distance away from soma (Sholl) was used for the “within-subjects” factor.
The analysis of learning and memory in the water maze used three-way RM-ANOVA, with two “between-subjects” factors (control vs CES, and vehicle vs CRF1 blocker treatment), and trial as the “within-subjects” factor. This was followed by pairwise comparisons among groups. To assess 24 h memory in the water maze, the percentage of total path length swum in the target quadrant was calculated, and these data were transformed (arcsine) to meet the assumptions of parametric statistics. One-sample t tests were then used to compare mean quadrant percentages to chance (25%), and Student's t tests were used to compare these percentages directly across groups (a priori planned comparisons; CES vs CTL, CES+ANT vs CTL, CES vs CES+ANT). For the novel-object recognition tests, simple one-way ANOVA was conducted.
All ANOVAs were followed by Bonferroni's post hoc multiple-comparisons test, with the exception of LTP experiments [that were followed by Tukey's honestly significantly different (HSD) post hoc test]. Significance levels were set at 0.05, and data are presented as mean ± SEM. Data were analyzed using the SPSS 16.0 (SPSS) and GraphPad Prism 5.0 Software.
Results
CRH immunoreactivity is augmented in hippocampus of early-life stressed rats
CRH was expressed in basket-cell type interneurons in the pyramidal cell layer of hippocampus (Fig. 1A,B, see also insets), consistent with previous findings (Yan et al., 1998; Chen et al., 2001). Comparing CRH expression in hippocampal areas CA1 and CA3 of control and early-stressed middle-aged rats, we found that immunoreactive cells were significantly more abundant in CA1, CA3a, and CA3c of the stressed rats (Fig. 1C) (by t tests; CA1: p = 0.049; CA3a: p = 0.028; CA3c: p = 0.012). Whereas ICC may not be fully quantitative, these findings suggested that early-life stress augmented the expression levels of CRH chronically, so that the number of detectable cells was higher than that in controls.
Figure 1.
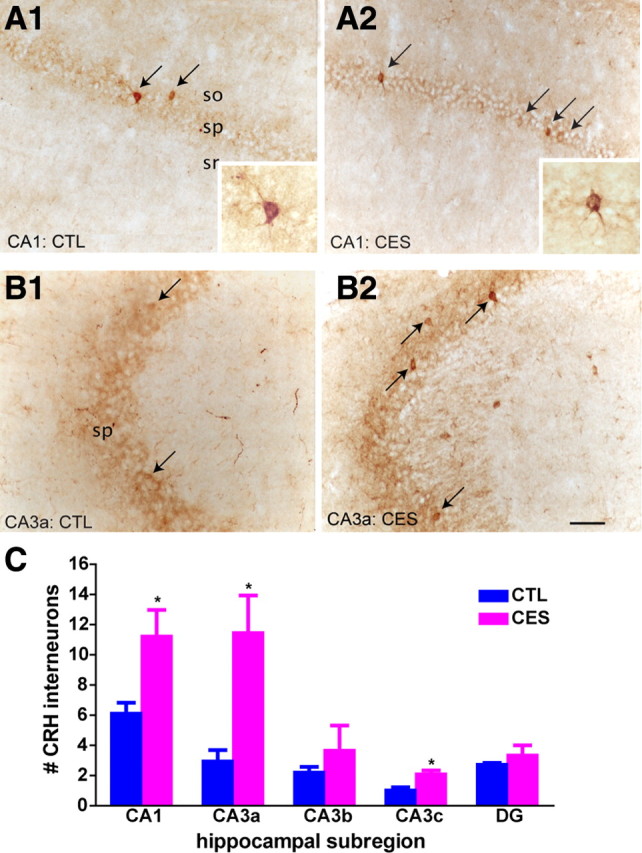
The number of CRH-immunoreactive cells was augmented in middle-aged rats that had experienced CES, compared to age-matched CTL rats. Representative sections from control (A1, B1) and CES (A2, B2) demonstrate a larger number of basket cell-like neurons within the pyramidal cell layers of area CA1 (A1, A2) and CA3 (B1, B2) in 12-month-old early-stressed rats (arrows) (n = 3 rats per group). Insets show individual CRH-immunoreactive interneurons. C, Quantitative analyses demonstrate significantly larger numbers of CRH-immunoreactive neurons detectable by ICC in the CA1 and CA3 regions of hippocampi from the early-stressed rats. ICCs were performed concurrently on all sections, and analyzed without knowledge of treatment group. (*p < 0.05, vs controls). Error bars indicate SEM. Scale bar, 100 μm.
Chronic exposure to nanomolar CRH levels reduces dendritic arborization in vitro
The ambient levels of CRH in extracellular space of control and early-life stressed rats are difficult to estimate. Available studies suggest that levels of peptide released within the hippocampus can reach ∼200 nm (Tringali et al., 2009), although lower stress-released CRH levels were measured in the amygdala (Merali et al., 2008). To evaluate the effects of similar levels of CRH on dendritic integrity, we used organotypic hippocampal slice cultures where the presence of other stress hormones can be controlled, exposed for 2 weeks to 100 nm CRH. We took advantage of transgenic mice expressing YFP in pyramidal cells to directly visualize dendritic branching of CA1 neurons (Fig. 2A). Chronic exposure to CRH (Fig. 2B) resulted in reduced dendritic arborization in a pattern similar to that observed after early-life stress (Brunson et al., 2005). Sholl analysis revealed that exposure to CRH affected dendritic branching most severely in the proximal apical dendritic regions that receive input from the commissural-associational fibers (Fig. 2C, left) (two-way RM-ANOVA, interaction: F(17,306) = 1.72, p = 0.04). The number of Sholl intersections was also significantly lower in the basal dendritic region of pyramidal cells exposed to CRH (Fig. 2C, right) (interaction: F(6,108) = 3.04, p = 0.008).
Figure 2.

CRH application onto hippocampal organotypic slice cultures reduced dendritic complexity. Cultures were prepared from postnatal day 1 mice and grown in the presence of CRH (100 nm) for 2 weeks. CA1 pyramidal neurons expressing YFP (n = 10 per group) are shown in control condition (A) and in cultures grown with CRH (B). C, Sholl analysis of both apical dendrites (left panel; F(1,16) = 26.99; p < 0.0001) and basal dendrites (right panel; F(1,6) = 18.35; p < 0.0001) revealed significant reductions in dendritic complexity induced by CRH exposure. Error bars indicate SEM. Scale bar, 70 μm.
Together, the results summarized in Figures 1 and 2 suggested that hippocampal pyramidal cells of rats stressed early in life remain exposed to high levels of CRH beyond the stress period and even during middle age, and that these levels might suffice to stunt dendritic arborization. Therefore, we queried whether interaction of hippocampal CRH with its principal hippocampal receptor, CRF1, contributed to the dendritic atrophy and associated spatial memory and LTP defects found after early-life stress.
Chronic early-life stress leads to late-onset impairment in hippocampus-dependent cognitive functions that are prevented by post hoc systemic administration of a CRF1 antagonist
To test whether a CRF1 antagonist could prevent early-stress-provoked cognitive defects, hippocampus-dependent learning and memory were assessed in naive, vehicle-treated, or CRF1 blocker-treated middle-aged CES rats, in comparison with controls. We used the Morris water maze test because learning in this task requires the integrity of dorsal hippocampus (Morris, 1984; Hollup et al., 2001; Broadbent et al., 2010). During the first 2 training days, all groups successfully learned to find the hidden platform (Fig. 3A) (effect of trial: F(11,539) = 7.73, p < 0.001). Early-life stress impaired acquisition of the water maze (effect of stress condition: F(1,49) = 11.09, p < 0.005), and CRF1 blocker treatment improved performance regardless of stress exposure (F(1,49) = 5.7, p < 0.05). Furthermore, planned comparisons revealed that by the end of each training day, CES rats did not find the platform as fast as controls (trial 6: t(40) = 2.13, p = 0.04; trial 12: t(40) = 2.4, p = 0.02). Antagonist treatment of CES rats improved their ability to find the platform on the final training trial (trial 12) (CES+ANT vs CES; t(23) = 2.43, p = 0.02) such that their performance was comparable to controls (CTL vs CES+ANT; t(31) = 0.98, p = 0.34). On the third day (consisting of the reversal procedure), the location of the platform was changed, and rats were forced to rely on spatial cues to navigate to this new location (Fig. 3B). Although all groups reduced their escape latencies across subsequent trials (F(5,245) = 5.61, p < 0.001), early-life stressed rats took significantly longer to reach the platform relative to controls (effect of stress condition: F(1,49) = 6.9, p < 0.012). Again, escape latencies were significantly shorter in CRF1 blocker-treated rats regardless of stress exposure (F(1,49) = 11.12, p < 0.005). Analysis of the last trial of the reversal procedure (trial 6) indicated that by the end of testing, CES rats had not learned the new platform location as well as controls (t(40) = 3.26, p < 0.05), and CRF1 antagonist treatment reversed this deficit (CES vs CES+ANT: t(23) = 3.34, p < 0.005; CES+ANT vs CTL: t(31) = 1.33, p = 0.19).
Figure 3.

Hippocampus-dependent learning and memory were preserved in early-life stress rats treated systemically with a blocker of CRF1, NBI30775, during P10–P17. A, During the first 2 training days, all groups successfully learned to find the hidden platform (effect of trial: F(11,539) = 7.73, p < 0.001). Early-life stress impaired acquisition of the water maze (effect of stress condition: F(1,49) = 11.09, p < 0.005), and CRF1 blocker treatment improved performance regardless of stress exposure (F(1,49) = 5.7, p < 0.05). Compared to CTL middle-aged rats (n = 20), age-matched CES rats (n = 13) required more time on the last trial of each water maze training day (trials 6 and 12) to find a hidden platform (*p < 0.05, CES vs CTL). At the end of training (trial 12), this deficit was completely abolished in the stressed group that received CRF1 antagonist (CES+ANT; n = 8; #p < 0.05, vs CES; p > 0.05, vs CTL). B, On day 3 of the water maze, the platform was moved to a new quadrant and rats relied on strategically placed spatial cues to learn the new platform location. Untreated control rats, as well as control (CTL+ANT; n = 4) and early-stressed groups treated with a CRF1 blocker, successfully learned the task, whereas untreated stressed rats had significantly higher escape latencies (*p < 0.05, CES vs all other groups). Analysis of the last trial of the reversal procedure indicated that by the end of testing, CES rats had not learned the new platform location as well as controls (t(40) = 3.26, p < 0.05), and CRF1 antagonist treatment reversed this deficit (CES vs CES+ANT: t(23) = 3.34, p < 0.005; CES+ANT vs CTL: t(31) = 1.33, p = 0.19). C, On day 1 of the object recognition task, exploration times of both objects were similar among the four experimental groups. D, On day 2, preferential exploration of the novel object (apparent from the increased ratio of time spent with novel versus the familiar objects) was observed in untreated control rats (ratio: 2.14), as well as CRF1 blocker-treated controls (ratio: 1.75). The cohort of middle-aged CES rats given the CRF1 receptor blocker also discriminated between the novel and familiar objects (ratio: 1.84), evidenced by a significantly higher mean exploration ratio compared to untreated CES rats (ratio: 0.99; p < 0.0001) (*p < 0.05, CES vs all other groups). Error bars indicate SEM.
The increased latency to finding the platform in the stressed group was not a result of differences in motivation or physical ability, because hippocampus-independent cued platform learning (measured by escape latency), and distance traveled to platform did not differ among the four experimental groups (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material) (p > 0.05). Notably, the memory of the early-stress group for platform location from the previous day was impaired, as tested in a probe trial on the post reversal day. In fact, untreated CES rats swam only 28% of their total path length within the target quadrant during the probe test. This performance was significantly worse than controls (43%; t(6) = 2.76, p < 0.05) and not statistically better than chance (one-sample t test, t(3) = 0.97, p > 0.05) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Similar impairments in early-stressed rats were noted when the probe test was performed immediately after the 2 d of training (CES vs CTL, t(13) = 2.02, p = 0.06). Furthermore, treatment with the CRF1 blocker prevented the early stress-provoked difficulties in the probe test (CES+ANT vs CTL, t(4) = 0.86, p > 0.05) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Note that vehicle treatment did not influence spatial memory in control rats (p = 0.22) or CES rats (p = 0.23), so that data from naive and vehicle-treated groups were combined in Figure 3, A and B.
Together, these data demonstrate a profound impairment in hippocampus-dependent spatial memory of the stressed rats, apparent from their failure to remember the location of the platform found in the previous trial as well as from their poor performance in the probe test. Notably, CRF1 blocker treatment improved the affected memory functions in early-stress rats.
Although the version of the MWM used in this study was chosen to minimize stress (Morris, 1984), the results might be confounded by group-specific differences in “coping” with averse, potentially stressful components of the task (forced swimming in room temperature water). To exclude this possibility, we tested the ability of the CRF1 antagonist to mitigate the effects of early stress on recognition memory using the OR task. This relatively stress-free test requires an intact limbic circuit, including the hippocampus and related cortices (Broadbent et al., 2004, 2010; Winters et al., 2004; Squire et al., 2007). The OR task examined whether rats could differentiate between a novel object and one encountered 24 h previously, and the ratio of novel:familiar object exploration time was used as an index of memory. There was no significant difference in exploration times on day 1 of the test between naive and vehicle-treated groups, for either the controls or the CES rats (object 1: CTL vs CTL+VEH: p = 0.29; CES vs CES+VEH: p = 0.30; object 2: CTL vs CTL+VEH: p = 0.47; CES vs CES+VEH: p = 0.41). Therefore, data for naive and vehicle-treated groups were combined. As shown in Figure 3C, exploration durations of each object on day 1 did not differ significantly among any of the experimental groups (F(7,100) = 0.18, p = 0.986). On the second (testing) day, control rats spent approximately twice as long exploring the novel object compared with the familiar one (Fig. 3D) (mean ratio ± SEM = 2.14 ± 0.18). In contrast, middle-aged CES rats failed to distinguish a novel object from one they had previously encountered (ratio: 0.99 ± 0.04; p < 0.0001 compared to the control group). The cohort of middle-aged CES rats given the CRF1 receptor blocker discriminated between the novel and familiar objects, spending significantly more time than untreated CES rats in exploring the novel object (ratio: 1.84 ± 0.16; p < 0.0001), and this ratio was not significantly different from that of controls (p = 0.26). Total exploration times did not vary among groups on day 2 (F(3,50) = 0.62, p = 0.61). In summary, the adverse effects of early-life stress on learning and memory, demonstrated in two independent tests, were abrogated by the systemic administration of a selective antagonist of CRF1 during the week immediately after the stressful period.
CRF1 antagonist treatment prevents LTP deficits in hippocampal CA3–CA1 synapses of middle-aged CES rats
To examine the cellular mechanisms responsible for the protective effects of the CRF1 blocker on cognitive function in CES rats, electrophysiological studies evaluated LTP, generally considered to underlie memory functions in the hippocampal circuit (Roman et al., 1987; Bliss and Collingridge, 1993). Focusing on synaptic plasticity in CA3–CA1 apical dendritic synapses that is disrupted by early-life stress (Brunson et al., 2005), we found that CES had no significant effect on normal synaptic transmission within a range of stimulus currents that do not elicit a population spike; additionally, treatment with the CRF1 antagonist during the week that followed the CES had no effect on this measure (Fig. 4A) (p = 0.64; one-way ANOVA; n = 3–6 slices from 3 to 6 rats per group). However, HFS demonstrated profound differences in synaptic plasticity among the four tested groups: In middle-aged control rats, HFS induced robust, enduring potentiation of field EPSPs that was stable for the 40 min duration of the recording (Fig. 4C) (41 ± 6% above baseline, n = 6 slices from 6 rats), and this potentiation was not significantly influenced by the administration of the CRF1 blocker on postnatal days 10–17 (47 ± 10%, n = 3 slices from 3 rats). In contrast, hippocampal slices from middle-aged CES rats demonstrated attenuated LTP (14 ± 6%, n = 4 slices from 4 rats; p = 0.02 vs controls, Tukey's HSD following two-way RM-ANOVA for minutes 30–40 after HFS). In marked contrast to slices from vehicle-treated CES rats, potentiation in slices from CRF1 antagonist-treated CES rats resembled that from controls (44 ± 11%; n = 5 slices from 4 rats) or blocker-treated controls, and differed significantly from nontreated CES slices (p = 0.03). These findings indicate that LTP in CA3–CA1 synapses was disrupted by chronic early-life stress in middle-aged hippocampus, concordant with the disruption of learning and memory. Notably, limiting CRF1 occupancy during the week subsequent to the stress period abrogated these derangements of synaptic potentiation, in parallel with, and perhaps as a mechanism for, the prevention of cognitive deficits.
Figure 4.

Hippocampal LTP was attenuated by chronic early-life stress and rescued by a CRF1 blocker given during the week that followed the stress. Acute hippocampal slices were prepared from middle-aged rats killed without any early-life manipulation (CTL) or exposed to CES without or with CRF1 blocker administration (ANT). A, Field EPSP amplitudes (±SEM) plotted as a function of input stimulus duration within a range subthreshold for eliciting a population spike revealed no differences between the three groups. B, Inset: Representative fEPSP traces collected during baseline (black, labeled “1”) or 40 min after HFS (gray overlay, labeled “2”) from controls (left), CES (middle), and CES+ANT (right) rats illustrate the greater potentiation of the fEPSP in the control and CES+ANT rats as compared to the stressed rats. Calibration: 0.5 mV, 5 ms. Plot, fEPSP slopes measured from 10 min before to 1 h after HFS (arrow). LTP in the stressed rats (open triangles) stabilized at significantly lower levels than did potentiation in controls (open circles; p < 0.05 vs control; repeated measure ANOVA) or CES+ANT rats (gray triangles; p < 0.05); values from control and CES+ANT rats were not significantly different. C, Bar graph shows group mean slope values for the final 10 min of recording (*p = 0.02 vs CTL).
CRF1 antagonist administration directly into the lateral ventricles of CES rats preserves their cognitive function
During stress, CRH is released from the paraventricular nucleus of the hypothalamus and binds to pituitary CRF1, thus initiating a cascade of events ultimately leading to glucocorticoid release from the adrenal gland. Glucocorticoids cross the blood–brain barrier freely, and may reach the hippocampus to influence learning and memory through the mineralocorticoid or glucocorticoid receptors (Joëls, 2007; Joëls and Baram, 2009; Lupien et al., 2009). In the experiments described above, the small-molecule CRF1 antagonist NBI30775, which crosses the blood–brain barrier (Heinrichs et al., 1995), was administered via subcutaneous osmotic minipump. Thus, the compound reached the systemic circulation and blocked peripheral (pituitary) CRF1 receptors, and, after crossing the blood–brain barrier, also central CRF1 receptors, including those on hippocampal pyramidal cells (Chen et al., 2000, 2004b). In view of these facts, it might be argued that systemic CRF1 antagonist administration prevented the effects of CES on learning and memory by altering signaling through peripheral CRF1 receptors and consequently modifying physiologic stress responses and levels of plasma glucocorticoids. To examine whether blocking CRH receptor within the brain or in the pituitary was responsible for the improved learning and memory, we administered the CRF1 blocker directly into the lateral ventricles (intracerebroventricularly) of control and early-stressed rats during P10–P17, at doses found previously not to influence peripheral glucocorticoid release (Chen et al., 2006). We then tested for the effects of this treatment on hippocampus-dependent learning and memory using the water maze.
During both the training (Fig. 5A) and reversal tests (Fig. 5B), all rats successfully learned to find the hidden platform (training: F(11,473) = 6.31, p < 0.001; reversal: F(5,215) = 3.19, p = 0.008). There were no overall differences in escape latencies between stress or drug treatment conditions (p > 0.05). However, when we compared untreated early-stressed rats directly to control rats, we confirmed our previously observed impairments in short-term spatial memory as a result of early-life stress in both tests (two-way ANOVA, main effect of group, training: F(1,36) = 5.230, p < 0.05; reversal: F(1,36) = 13.986, p < 0.05). The effects of intracerebroventricular administration of the CRF1 blocker surfaced during the reversal testing (Fig. 5B). Indeed, planned comparisons revealed that by the last trial of reversal testing (trial 18), untreated CES rats did not find the platform as fast as controls (t(36) = 3.94, p < 0.001). Importantly, intracerebroventricular antagonist administration significantly improved escape latencies of early-stressed rats (CES+ANT vs CES; t(18) = 2.52, p = 0.02), such that their performance was comparable to controls (CTL vs CES+ANT; t(26) = 0.49, p = 0.63). Note that in this analysis (similar to Fig. 3), mean escape latencies were comparable among naive and vehicle-treated control rats as well as between naive and vehicle-treated stressed rats (p = 0.12 and p = 0.31, respectively); therefore, data from naive and vehicle-treated groups were collapsed in Figure 5A. Together, these results are in line with the results of the systemic CRF1 blocker administration, showing the effects of early-stress on specific memory measures, and the amelioration of these deficits by blocking CRH-CRH receptor signaling immediately after the early-life stress.
Figure 5.

Administration of the CRF1 antagonist directly into the cerebral ventricles (intracerebroventricularly) sufficed to prevent late-onset cognitive impairments after chronic early-life stress. A, Deficits in spatial learning were found in early-life stressed (CES; n = 15) rats during the 2 d of Morris water maze training, evidenced by higher escape latencies when compared to control (p < 0.001 vs CTL; n = 23). B, The effects of intracerebroventricular CRF1 antagonist treatment surfaced during the water maze tests in which the platform was moved to a new quadrant (reversal). By the last trial of reversal testing (trial 18), the untreated CES rats took significantly longer to find the platform compared to control rats or early-stressed rats given intracerebroventricular CRF1 blocker treatment (CES+ANT; n = 5) (*p < 0.05, CES vs CTL, CES vs CES+ANT). Antagonist treatment of control rats (CTL+ANT; n = 4) had no effect on escape latencies (p > 0.05, vs CTL). C, In the novel-object recognition task, rats from all groups explored two separate objects equally on day 1 of the task, eliminating the possibility of differences in motivation to explore. D, The ratio of time spent with a novel versus a familiar object, a measure of 24 h recognition memory, was significantly lower in early-stressed rats compared to controls (*p < 0.05). CRF1-blocker treatment significantly increased this ratio (*p < 0.05, vs CES), and had little effect on recognition memory in control rats (p > 0.05, vs untreated controls). E, A similar pattern of results was found when the novel objects were counterbalanced across groups. The ratio of time spent exploring a novel versus a familiar object was again lower in untreated early-stressed rats, and CRF1-blocker treatment significantly increased this ratio (*p < 0.05, CES vs all other groups). Data indicate mean ± SEM.
The novel-object recognition task described above was also used to assess the efficacy of intracerebral administration of the CRF1 blocker. On day 1 of the task, total exploration times of objects 1 and 2 were similar among control and stressed groups that were either naive or vehicle-treated (p > 0.05; t tests), and the groups were collapsed. On this first day, exploration time for each of the two objects was not significantly different among the four major experimental groups (Fig. 5C) (F(7,22) = 2.42, p > 0.05). On the second, testing day, early-stressed rats failed to recognize a novel object, as apparent from the ratio of novel:familiar object exploration (Fig. 5D) (ratio: 1.2 ± 0.44), and there was a significant difference in these ratios across groups (F(3,9) = 4.59, p = 0.03). Specifically, NBI30775-treated early-stressed rats had significantly higher novel:familiar exploration ratios than untreated early-stressed rats (Bonferroni's post hoc tests, p < 0.05), and were comparable to both antagonist-treated and vehicle-treated controls (p > 0.05), indicating that CRF1 blocker treatment prevented the early-stress-induced deficits in recognition memory. The longer exploration of the novel object on day 2 in the vehicle-treated controls and CRF1-blocker-treated control and stressed groups was not due to differences in motivation for exploring objects, because total exploration times were indistinguishable among groups (p = 0.39), and potential bias toward one of the objects was excluded using counterbalancing (Fig. 5E). Together with the water maze data, these findings demonstrate that selectively blocking central CRF1 receptors during the week that followed the early-life stress period sufficed to abolish deficits in hippocampus-dependent memory.
Dendritic atrophy of CA1 pyramidal neurons after chronic early-life stress is abrogated by post hoc treatment with a CRF1 antagonist
The cognitive deficits provoked by chronic early-life stress were associated with reduced arborization and total length (atrophy) of apical dendrites of CA1 pyramidal cells (Brunson et al., 2005). This finding raised the possibility that the loss of dendritic spines and excitatory synapses that would normally reside on the missing dendrites might be the foundation of memory and LTP impairments. Therefore we tested whether CRF1-blocker administration that preserved hippocampal function in CES rats also protected the complexity of apical dendrites in CA1. Sholl analysis revealed a significant effect of group (F(3,62) = 6.79, p < 0.001) and distance from soma (F(12,744) = 55.79, p < 0.0001). Figure 6B illustrates that the early-stress related atrophy was most pronounced in proximal stratum radiatum, a region carrying the postsynaptic elements of Schaffer collateral axons synapses that were tested in the LTP experiments (between 60 and 120 μm away from the soma; Bonferroni's post hoc tests: p < 0.05, comparing CES to CTL, CTL+ANT, and CES+ANT). In addition, there was a reduction in total length of apical dendrites in the early-stress cohort compared to all other treatment groups (Fig. 6C) (F(3,22) = 4.6, p = 0.01). In contrast, in basal dendrites, total length did not distinguish among groups (effect of group: F(3,42) = 1.28, p = 0.3). In summary, the location of dendritic abnormalities resulting from CES was congruent with the loss of LTP, and these abnormalities were rescued by CRF1 antagonist treatment. These findings suggest that prevention of excessive CRF1 occupancy abrogated dendritic atrophy in selective regions of hippocampal CA1, which in turn preserved LTP and memory function.
Figure 6.

CA1 dendritic atrophy in middle-aged, early-life stressed rats was prevented by treatment with a blocker of CRF1 during the week following the stress. A, Golgi-impregnated CA1 pyramidal cells from control, CES, CES+ANT, and CTL+ANT groups were reconstructed using camera lucida. Representative neurons from each group illustrate the reduction in dendritic branching in neuron from the stress group, and the normalization of dendritic arborization of neurons from CES+ANT rats (n = 5–8 neurons per animal, 3–5 animals per group; scale bar, 60 μm). B, Complexity of dendritic arbors was quantified using Sholl analyses, counting the number of dendritic intersections in apical (left) and basal (right) dendritic regions. Significantly reduced dendritic complexity was found in the proximal stratum radiatum of neurons from early-stress rats when compared to either controls (p < 0.05) or CES+ANT (p < 0.05). C, Total apical dendritic length was significantly reduced in the early stress group, but not in stressed rats given the CRH receptor blocker NBI30775. Error bars indicate SEM. *p < 0.05 versus CTL, ‡p < 0.05 versus CES+ANT.
Post hoc CRF1 blocker treatment of early-life stressed rats blunts the augmented expression of hippocampal CRH
If persistent and excessive CRF1 activation resulted from the enduringly increased levels of CRH in hippocampus and contributed to the dendritic atrophy in early-stressed rats, then did the CRF1 blocker given after the stress prevent dendritic atrophy by abrogating the increase in CRH expression? Quantification of CRH-immunoreactive interneurons in anatomically matched sections from all groups revealed that vehicle-treated early-stressed rats had a significantly higher number of CRH-positive cells in areas CA1 and CA3, as shown in Figure 7A–C using a cohort different from that of Figure 1 [p = 0.04 (CA1) and p < 0.0005 (CA3), t tests control vs stressed]. Treatment of stressed rats with NBI30775 during the week following the stress period attenuated this effect. Thus, as shown in Figure 7C, numbers of CRH-immunoreactive neurons in treated stressed rats were not significantly different from those of naive and blocker-treated controls (p > 0.05, Bonferroni's post hoc tests; CA1: F(3,13) = 5.20, p = 0.01; CA3: F(3,13) = 13.58, p = 0.0003, one-way ANOVA). Together, these data suggest that prevention of the increased numbers of ICC-detectable CRH cells, and of the presumed augmented release of CRH throughout life, might contribute to the mechanisms by which administration of the CRF1 blocker during P10–P17 prevented dendritic atrophy and loss of hippocampus-dependent memory.
Figure 7.
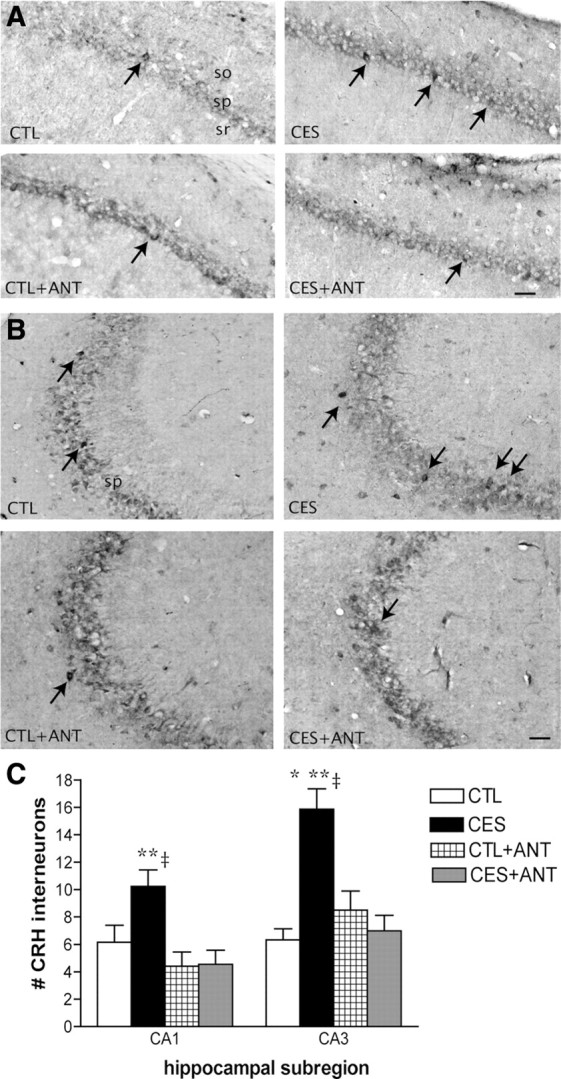
CRF1 blocker treatment during P10–P17 prevented augmentation of CRH-immunoreactive (IR) cells in middle-aged rats that had experienced CES. A, CRH-IR neurons in hippocampal CA1 of CTL, early-stressed, antagonist-treated CTL (CTL+ANT), and antagonist-treated early-stressed (CES+ANT) rats. Arrows point to CRH-positive interneurons in the pyramidal cell layer. B, Representative images from hippocampal CA3 region of the same groups. C, Numbers of CRH-IR neurons in areas CA1 and CA3 of NBI30775-treated early-stressed rats were significantly lower than in untreated stressed littermates, and were comparable to controls. *p < 0.05 versus CTL, **p < 0.05 versus CTL+ANT, ‡p < 0.05 versus CES+ANT.
Discussion
The current studies demonstrate that a selective blocker of the CRH receptor type 1, given during the week after early-life chronic stress, abolishes the deficits in hippocampus-dependent cognitive function provoked by this stress in middle-aged rats. The receptor antagonist was efficacious when given either systemically or directly into the brain, suggesting that blocking central CRH receptors contributed to the ameliorating effect of the blocker on hippocampal function. The cellular basis of the protective effects of the CRF1 antagonist involved rescue of LTP in hippocampal synapses and preservation of dendritic branches of hippocampal pyramidal cells innervated by the projections tested in the electrophysiology experiments. These novel findings directly implicate CRF1 activation in the enduring effects of early-life stress on hippocampal synaptic plasticity and learning and memory, and offer promising translational therapeutic opportunities.
Alternative mechanisms for the effects of CRF1 blocker on early-life stress-induced defects of cognitive function and hippocampal structure
Chronic stress is associated with increased levels of plasma glucocorticoids. These may penetrate the blood brain barrier and act on receptors found in hippocampal neurons, resulting in dendritic atrophy (Magariños and McEwen, 1995) and attenuated LTP (Pavlides et al., 2002; Joëls et al., 2004). Involvement of glucocorticoids in the mechanisms by which early-life stress causes hippocampal defects could not be excluded by blocking these defects via systemic administration of a CRF1 antagonist, because the antagonist blocked both pituitary and central CRH receptors (Brunson et al., 2002; Rivier et al., 2003). Blocking pituitary CRF1 would attenuate ACTH and glucocorticoid release and interaction with hippocampal neurons. Therefore, it is conceivable that systemic administration of the CRF1 blocker ameliorated the effects of early-life stress by reducing glucocorticoid levels and their subsequent effects on hippocampal neurons. To address this possibility, we also administered the blocker directly into the brain (Figs. 5–7), and found that doses of the blocker that did not affect plasma glucocorticoids (Chen et al., 2006) were still effective in preventing cognitive defects and dendritic atrophy in middle-aged rats that had experienced early-life stress. These data support an alternative mechanism of action of the blocker, namely, preventing directly the effects of CRH—presumably originating from within the hippocampus (Chen et al., 2001)—on hippocampal CRF1 receptors (Chen et al., 2004a,b, 2010). Whereas it is quite likely that glucocorticoids significantly contribute to any effects of stress, the efficacy of centrally administered CRF1 blocker is consistent with a critical role for local CRH receptors in the deleterious effects of early-life stress on hippocampal structure and function (Chen et al., 2008, 2010).
How might a CRF1 blocker prevent disruption of synaptic plasticity by chronic early-life stress?
Synaptic plasticity, measured by LTP, is generally believed to underlie hippocampus-mediated learning and memory (Bliss and Collingridge, 1993). In the current studies, administration of the CRF1 blocker during the week subsequent to the early-life stress restored both LTP and memory during middle age. LTP requires the functional integrity of dendritic spines (Segal, 2005; Chen et al., 2007; Harvey et al., 2008). For example, reduced spine number decreases total postsynaptic area of excitatory synapses, influencing receptor density, synaptic signaling (Harris et al., 2003; Kennedy et al., 2005), and the number of functional excitatory synapses (Segal, 2005). These changes, in turn, will affect synaptic plasticity via several routes, including a reduction in local density of NMDA receptor-gated synaptic currents (Engert and Bonhoeffer, 1999; Toni et al., 1999; Nägerl et al., 2004; Zhou et al., 2004). Early-life stress attenuated dendritic length and arborization with the associated loss of spines and excitatory synapses. The efficacy of the CRF1 blocker in reversing these morphological defects suggests that endogenous hippocampal CRH contributes to their occurrence. In accord with this, early-life stress caused a persistent increase in the number of neurons expressing CRH, as assessed with ICC (Figs. 1, 7), and the peptide is known to promote spine and dendrite loss (Chen et al., 2008). While the mechanisms by which early-life stress enhances CRH expression in hippocampus are not fully understood, CRH mRNA expression in hippocampal neurons of immature rats is augmented by glucocorticoids (Brunson et al., 2001a), and plasma levels of the latter were chronically elevated during the period of early-life stress (Gilles et al., 1996; Avishai-Eliner et al., 2001a).
How does a transient administration of a CRF1 blocker result in enduring amelioration of hippocampal function in early-stressed rats?
Treatment with the CRF1 blocker lasted for only one poststress week (P10–P17) in our studies, yet resulted in enduring rescue of hippocampal LTP and memory function. This suggests that both the effects of early-life stress and the therapeutic actions of the CRF1 blocker interact with developmental events within hippocampus. How might these two factors (i.e., CES and CRF1 blockade) influence hippocampal development permanently? Stress causes release of CRH within immature hippocampus (Chen et al., 2004a, 2006). Therefore, persistent release of CRH during the chronic early-life stress and the consequent activation of CRF1 receptors located on dendritic spines might promote dendritic spine loss, as found previously (Chen et al., 2008). Loss of CA1 and CA3 pyramidal cell spines would eliminate the postsynaptic targets of incoming axons, and interfere with the establishment and maturation of synaptic connections onto hippocampal pyramidal cells (Gall and Lynch, 1978; Avishai-Eliner et al., 2002). Whereas the axons of the granule cells (“mossy fibers”) can overcome such disruption via a strong sprouting response that persists throughout adulthood (Pitkänen et al., 2000; Brunson et al., 2001b, 2005), injured commissural/associational axons sprout poorly beyond a critical period terminating during the second postnatal week (Gall and Lynch, 1978).
Previous studies obtained evidence for the above argument: mossy fiber sprouting was found in middle-aged, early-stressed rats (Brunson et al., 2005), together with sparing of the electrophysiology of mossy fiber–CA3 pyramidal cell synapses. However, functional and structural disturbances were evident in the commissural-associational innervations of CA3, and in CA3–CA1 Schaffer collateral pathways (Brunson et al., 2005). Together, these data suggest that the efficacy of a transient administration of CRH receptor antagonist results from blocking the excessive activation of CRH receptors during a critical developmental period in which excess CRH irreversibly injures dendrites and spines. Protecting these elements allows for the anchoring of incoming axons and creation of a normal mature hippocampal network.
Similar critical windows for vulnerability and therapy in the developing hippocampus have been described for a number of other insults. For example, early-life seizures can disrupt hippocampus-mediated learning, and these deficits can be restored via environmental enrichment that follows the seizures (Faverjon et al., 2002). Similarly, the consequences of suboptimal maternal care can be reversed by post hoc enrichment of the environment (Bredy et al., 2003). The enduring efficacy of CRF1-antagonist treatment that was initiated already during the second postnatal week indicates that the developmental window of opportunity for reversing early-stress-induced hippocampal defects extends beyond the stress period itself, and likely involves a number of age-specific aspects of hippocampal maturation (Swann et al., 1991).
A remaining conundrum involves the basis of the persisting effect of the CRF1-antagonist treatment in abrogating the augmented expression of CRH in hippocampus of CES rats. The mechanisms by which CES leads to persistent upregulation of CRH expression are not yet known. Epigenetic modulation of this gene in response to early-life experience has recently been demonstrated in hypothalamus (Korosi et al., 2010), and might take place in hippocampus as well. In this scenario, blocking excessive CRF1 occupancy might interfere with the mechanisms of CRH upregulation, consistent with previously demonstrated cross-regulation of CRH and its receptors (e.g., Bale et al., 2002; Brunson et al., 2002)
Translational, therapeutic relevance of the results of the current experiments
In summary, the long-lasting deleterious effects of chronic early-life stress on hippocampal anatomy, synaptic plasticity, and learning were prevented by post hoc administration of a CRF1 blocker. Although the precise mechanisms underlying the rescue of hippocampal structure and function will require further investigation, the present results point to a clinically plausible molecular target for preventing the cognitive consequences of early life stress. It has now been established that the majority of the world's children grow up under chronic stress (UNICEF, 2005), and this cannot be prevented at present. Therefore, interventions with translational and clinical potential will need to be effective when applied post hoc, as is the case for the treatments described here.
Footnotes
This work was supported by National Institutes of Health Grants MH73136 (T.B.Z.), NS28912 (T.B.Z.), and NS045260 (T.Z.B., C.M.G.); NS051823 and NS045260 (G.L.); MH087467 (A.S.I.); and AG00358 (C.S.R.). We thank Kimberly Fok and Albert Cho for excellent technical help.
References
- Akana SF, Jacobson L, Cascio CS, Shinsako J, Dallman MF. Constant corticosterone replacement normalizes basal adrenocorticotropin (ACTH) but permits sustained ACTH hypersecretion after stress in adrenalectomized rats. Endocrinology. 1988;122:1337–1342. doi: 10.1210/endo-122-4-1337. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Gilles EE, Eghbal-Ahmadi M, Bar-El Y, Baram TZ. Altered regulation of gene and protein expression of hypothalamic-pituitary-adrenal axis components in an immature rat model of chronic stress. J Neuroendocrinol. 2001a;13:799–807. doi: 10.1046/j.1365-2826.2001.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avishai-Eliner S, Eghbal-Ahmadi M, Tabachnik E, Brunson KL, Baram TZ. Down-regulation of hypothalamic corticotropin-releasing hormone messenger ribonucleic acid (mRNA) precedes early-life experience-induced changes in hippocampal glucocorticoid receptor mRNA. Endocrinology. 2001b;142:89–97. doi: 10.1210/endo.142.1.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Picetti R, Contarino A, Koob GF, Vale WW, Lee KF. Mice deficient for both corticotropin-releasing factor receptor 1 (CRFR1) and CRFR2 have an impaired stress response and display sexually dichotomous anxiety-like behavior. J Neurosci. 2002;22:193–199. doi: 10.1523/JNEUROSCI.22-01-00193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Eckart K, Spiess J. Priming of long-term potentiation in mouse hippocampus by corticotropin-releasing factor and acute stress: implications for hippocampus-dependent learning. J Neurosci. 2002;22:3788–3794. doi: 10.1523/JNEUROSCI.22-09-03788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Humpartzoomian RA, Cain DP, Meaney MJ. Partial reversal of the effect of maternal care on cognitive function through environmental enrichment. Neuroscience. 2003;118:571–576. doi: 10.1016/s0306-4522(02)00918-1. [DOI] [PubMed] [Google Scholar]
- Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci U S A. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Gaskin S, Squire LR, Clark RE. Object recognition memory and the rodent hippocampus. Learn Mem. 2010;17:794–800. doi: 10.1101/lm.1650110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Khan N, Eghbal-Ahmadi M, Baram TZ. Corticotropin (ACTH) acts directly on amygdala neurons to down-regulate corticotropin-releasing hormone gene expression. Ann Neurol. 2001a;49:304–312. [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, Bender R, Chen Y, Baram TZ. Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci U S A. 2001b;98:8856–8861. doi: 10.1073/pnas.151224898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Grigoriadis DE, Lorang MT, Baram TZ. Corticotropin-releasing hormone (CRH) downregulates the function of its receptor (CRF1) and induces CRF1 expression in hippocampal and cortical regions of the immature rat brain. Exp Neurol. 2002;176:75–86. doi: 10.1006/exnr.2002.7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Kramár E, Lin B, Chen Y, Colgin LL, Yanagihara TK, Lynch G, Baram TZ. Mechanisms of late-onset cognitive decline after early-life stress. J Neurosci. 2005;25:9328–9338. doi: 10.1523/JNEUROSCI.2281-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Müller MB, Cariaga W, Baram TZ. Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF(1))-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J Comp Neurol. 2000;420:305–323. doi: 10.1002/(sici)1096-9861(20000508)420:3<305::aid-cne3>3.0.co;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004a;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W, Baram TZ. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci U S A. 2004b;101:15782–15787. doi: 10.1073/pnas.0403975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fenoglio KA, Dubé CM, Grigoriadis DE, Baram TZ. Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol Psychiatry. 2006;11:992–1002. doi: 10.1038/sj.mp.4001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dubé CM, Rice CJ, Baram TZ. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J Neurosci. 2008;28:2903–2911. doi: 10.1523/JNEUROSCI.0225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Rex CS, Rice CJ, Dubé CM, Gall CM, Lynch G, Baram TZ. Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc Natl Acad Sci U S A. 2010;107:13123–13128. doi: 10.1073/pnas.1003825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Karst H, Joëls M. Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrinol. 2008;29:268–272. doi: 10.1016/j.yfrne.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Faverjon S, Silveira DC, Fu DD, Cha BH, Akman C, Hu Y, Holmes GL. Beneficial effects of enriched environment following status epilepticus in immature rats. Neurology. 2002;59:1356–1364. doi: 10.1212/01.wnl.0000033588.59005.55. [DOI] [PubMed] [Google Scholar]
- Fenoglio KA, Brunson KL, Avishai-Eliner S, Stone BA, Kapadia BJ, Baram TZ. Enduring, handling-evoked enhancement of hippocampal memory function and glucocorticoid receptor expression involves activation of the corticotropin-releasing factor type 1 receptor. Endocrinology. 2005;146:4090–4096. doi: 10.1210/en.2004-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall C, Lynch G. Rapid axon sprouting in the neonatal rat hippocampus. Brain Res. 1978;153:357–362. doi: 10.1016/0006-8993(78)90415-8. [DOI] [PubMed] [Google Scholar]
- Gilles EE, Schultz L, Baram TZ. Abnormal corticosterone regulation in an immature rat model of continuous chronic stress. Pediatr Neurol. 1996;15:114–119. doi: 10.1016/0887-8994(96)00153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Fiala JC, Ostroff L. Structural changes at dendritic spine synapses during long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:745–748. doi: 10.1098/rstb.2002.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33:693–710. doi: 10.1016/j.psyneuen.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Menzaghi F, Merlo Pich E, Britton KT, Koob GF. The role of CRF in behavioral aspects of stress. Ann N Y Acad Sci. 1995;771:92–104. doi: 10.1111/j.1749-6632.1995.tb44673.x. [DOI] [PubMed] [Google Scholar]
- Hollup SA, Kjelstrup KG, Hoff J, Moser MB, Moser EI. Impaired recognition of the goal location during spatial navigation in rats with hippocampal lesions. J Neurosci. 2001;21:4505–4513. doi: 10.1523/JNEUROSCI.21-12-04505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy AS, Brunson KL, Sandman C, Baram TZ. Dysfunctional nurturing behavior in rat dams with limited access to nesting material: a clinically relevant model for early-life stress. Neuroscience. 2008;154:1132–1142. doi: 10.1016/j.neuroscience.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M. Role of corticosteroid hormones in the dentate gyrus. Prog Brain Res. 2007;163:355–370. doi: 10.1016/S0079-6123(07)63021-0. [DOI] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Karst H, Alfarez D, Heine VM, Qin Y, van Riel E, Verkuyl M, Lucassen PJ, Krugers HJ. Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress. 2004;7:221–231. doi: 10.1080/10253890500070005. [DOI] [PubMed] [Google Scholar]
- Kaplan GA, Turrell G, Lynch JW, Everson SA, Helkala EL, Salonen JT. Childhood socioeconomic position and cognitive function in adulthood. Int J Epidemiol. 2001;30:256–263. doi: 10.1093/ije/30.2.256. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Korosi A, Shanabrough M, McClelland S, Liu ZW, Borok E, Gao XB, Horvath TL, Baram TZ. Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J Neurosci. 2010;30:703–713. doi: 10.1523/JNEUROSCI.4214-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács KJ, Sawchenko PE. Regulation of stress-induced transcriptional changes in the hypothalamic neurosecretory neurons. J Mol Neurosci. 1996;7:125–133. doi: 10.1007/BF02736792. [DOI] [PubMed] [Google Scholar]
- Lee Y, Davis M. Role of the hippocampus, the bed nucleus of the stria terminalis, and the amygdala in the excitatory effect of corticotropin-releasing hormone on the acoustic startle reflex. J Neurosci. 1997;17:6434–6446. doi: 10.1523/JNEUROSCI.17-16-06434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69:83–88. doi: 10.1016/0306-4522(95)00256-i. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- Merali Z, Anisman H, James JS, Kent P, Schulkin J. Effects of corticosterone on corticotropin-releasing hormone and gastrin-releasing peptide release in response to an aversive stimulus in two regions of the forebrain (central nucleus of the amygdala and prefrontal cortex) Eur J Neurosci. 2008;28:165–172. doi: 10.1111/j.1460-9568.2008.06281.x. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nelson CA, 3rd, Zeanah CH, Fox NA, Marshall PJ, Smyke AT, Guthrie D. Cognitive recovery in socially deprived young children: the Bucharest Early Intervention Project. Science. 2007;318:1937–1940. doi: 10.1126/science.1143921. [DOI] [PubMed] [Google Scholar]
- Orozco-Cabal L, Pollandt S, Liu J, Shinnick-Gallagher P, Gallagher JP. Regulation of synaptic transmission by CRF receptors. Rev Neurosci. 2006;17:279–307. doi: 10.1515/revneuro.2006.17.3.279. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Nivón LG, McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12:245–257. doi: 10.1002/hipo.1116. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Nissinen J, Lukasiuk K, Jutila L, Paljärvi L, Salmenperä T, Karkola K, Vapalahti M, Ylinen A. Association between the density of mossy fiber sprouting and seizure frequency in experimental and human temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S24–S29. doi: 10.1111/j.1528-1157.2000.tb01552.x. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Valentino RJ, Van Bockstaele EJ. Stress-induced intracellular trafficking of corticotropin-releasing factor receptors in rat locus coeruleus neurons. Endocrinology. 2008;149:122–130. doi: 10.1210/en.2007-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology. 2008;149:4892–4900. doi: 10.1210/en.2008-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivier CL, Grigoriadis DE, Rivier JE. Role of corticotropin-releasing factor receptors type 1 and 2 in modulating the rat adrenocorticotropin response to stressors. Endocrinology. 2003;144:2396–2403. doi: 10.1210/en.2002-0117. [DOI] [PubMed] [Google Scholar]
- Roman F, Staubli U, Lynch G. Evidence for synaptic potentiation in a cortical network during learning. Brain Res. 1987;418:221–226. doi: 10.1016/0006-8993(87)90089-8. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoid toxicity in the hippocampus: temporal aspects of neuronal vulnerability. Brain Res. 1985;359:300–305. doi: 10.1016/0006-8993(85)91440-4. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Wixted JT, Clark RE. Recognition memory and the medial temporal lobe: a new perspective. Nat Rev Neurosci. 2007;8:872–883. doi: 10.1038/nrn2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JW, Smith KL, Brady RJ. Age-dependent alterations in the operations of hippocampal neural networks. Ann N Y Acad Sci. 1991;627:264–276. doi: 10.1111/j.1749-6632.1991.tb25930.x. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Tringali G, Lisi L, De Simone ML, Aubry JM, Preziosi P, Pozzoli G, Navarra P. Effects of olanzapine and quetiapine on corticotropin-releasing hormone release in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1017–1021. doi: 10.1016/j.pnpbp.2009.05.012. [DOI] [PubMed] [Google Scholar]
- UNICEF. State of the world's children 2006. New York: UNICEF; 2005. [Google Scholar]
- Wilson P. Recognising childhood neuropsychiatric disorders. Practitioner. 2007;251:26–27. 29–30. [PubMed] [Google Scholar]
- Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ. Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J Neurosci. 2004;24:5901–5908. doi: 10.1523/JNEUROSCI.1346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]