

Abstract
An improved strategy for the effective synthesis of enantiomerically pure (2R,3aS,7aS)-octahydroindole-2-carboxylic acid (Oic), based on the formation of a trichloromethyloxazolidinone derivative, has been developed. Additionally, the completely diastereoselective α-alkylation of such oxazolidinone provides a very convenient and concise route to enantiopure α-tetrasubstituted derivatives of this Oic stereoisomer.
Keywords: quaternary amino acid, bicyclic proline analogue, perhydroindole-2-carboxylic acid, trichloromethyloxazolidinone, α-alkylation, self-reproduction of chirality, crystal structure
1. Introduction
The development of practical synthetic routes to render enantiopure non-proteinogenic α-amino acids is receiving considerable attention due to their wide applicability in engineering peptide drugs.1 The incorporation of non-coded α-amino acids into the sequence of bioactive peptides is considered a very useful approach to tailor their properties in order to make therapeutic applications feasible. Such applications are very limited due to poor metabolic stability, low bioavailability and lack of selectivity for a specific receptor. The incorporation of non-natural α-amino acids generates modifications in the secondary structure of these peptides and, as a result, improvements in selectivity and stability can often be achieved.2
Octahydroindole-2-carboxylic acid (Oic) is a non-coded α-amino acid with a bicyclic structure that is able to introduce certain backbone rigidity when incorporated into peptides, similar to that induced by proline,3 and, at the same time, is endowed with greater lipophilicity. This latter property is very relevant for peptides and peptidomimetics because it is related to their rate of absorption and distribution through biological membranes and, hence, to their bioactivity.4 For this reason, the incorporation of residues of increased lipophilicity into bioactive peptides is a convenient way to improve their bioavailability.5
Interestingly, octahydroindole-2-carboxylic acid has been used as a surrogate of proline and phenylalanine in bradykinin, a linear nonapeptide hormone involved in pain and inflammation. In this way, Oic has contributed to improve the resistance to degrading enzymes of a family of orally available bradykinin B2 receptor antagonists with attractive anti-cancer properties.6 It has also been used to stabilize a type-II′ β-turn in small cyclic peptides in order to find a starting point for the design of alternative non-peptidic bradykinin B2 antagonists.7 In addition, Oic is a key intermediate in the elaboration of medicinally relevant drugs like potent ACE inhibitors for the treatment of hypertension (perindopril).8
Moreover, the octahydroindole-2-carboxylic acid motif with a diverse substitution profile has been found to be part of the structure of natural products like marine aeruginosins with potent antithrombotic properties.9 Besides, quaternary derivatives of Oic are the core structures of investigational compounds with potential applications in diseases characterized by joint cartilage damage.10
All of the aforementioned applications involve octahydroindole-2-carboxylic acid with the absolute configuration (2S,3aS,7aS) (Fig. 1). This particular stereoisomer is usually denoted as L-Oic, and will be referred to as (S,S,S)-1 in the following text – a nomenclature without numerical locants is adopted for the sake of simplicity.
Figure 1.

Structure of (2S,3aS,7aS)-octahydroindole-2-carboxylic acid and its epimer at the α carbon.
The significant occurrence of this bicyclic α-amino acid has stimulated our interest in developing efficient synthetic routes to access the different stereoisomers of octahydroindole-2-carboxylic acid in enantiomerically pure form. The so called L-Oic, (S,S,S)-1, is the only commercially available stereoisomer and has therefore been the most widely used in peptidomimetic and medicinal chemistry research. Various strategies for the preparation of enantiopure (S,S,S)-1 have been reported in the literature and include stereoselective processes,11 chemical resolutions12 of diastereoisomeric salts of (S,S,S)-1 derivatives or their indoline precursors, and, more recently, enzymatic13 and chromatographic14 resolutions. In contrast, there is only one reported synthesis12a of enantiomerically pure (R,S,S)-1 (Fig. 1), which involves an epimerization reaction of (S,S,S)-1 followed by laborious crystallizations, and suffers from a poor overall yield.
In this paper we report a more efficient method for the convenient synthesis of enantiomerically pure (R,S,S)-1, which will also allow access to α-alkyl derivatives in a concise manner.
2. Results and discussion
The preparation of enantiomerically pure (R,S,S)-1 has only been described in the literature by Vincent et al.,12a who used an epimerization reaction of enantiopure (S,S,S)-1 under acidic conditions, according to a general procedure reported by Yamada.15 This approach affords a 50:50 mixture of epimers from which the desired compound is isolated after consecutive preferential crystallizations. As a result, the synthesis becomes laborious and suffers from a low overall isolated yield. Previously, Henning described16 a racemic synthesis of this compound through a base-promoted Favorskii-type ring contraction of an α-chlorinated lactam, a process that proceeds in a quite diastereoselective fashion. However, this approach involves more synthetic steps and details were not given concerning the isolation of the desired major stereoisomer (as a racemic mixture).
In principle, it seems that the easiest procedure for the synthesis of (R,S,S)-1 would be an epimerization reaction of enantiopure (S,S,S)-1, obtained from commercial (S)-indoline-2-carboxylic acid, since other retrosynthetic possibilities from the disconnection approach do not seem to lead to more appropriate, or easily available, starting materials. Indeed, an improved epimerization reaction followed by a more competitive procedure for the separation of diastereoisomers other than preferential crystallizations, would be desirable for Vincent’s strategy12a to become efficient.
With this aim in mind, our synthesis began with the hydrogenation of commercially available (S)-indoline-2-carboxylic acid to afford diastereoisomeric (S,S,S)-1 and (S,R,R)-1 in a 90:10 ratio (Scheme 1), in a similar way to that described by Vincent.12a In our case, the use of platinum oxide as a catalyst, instead of palladium on charcoal, allowed the hydrogenation to proceed at atmospheric pressure. The desired compound (S,S,S)-1 was isolated as a pure material, in 85% yield, after crystallization from ethanol.
Scheme 1.

Synthesis of enantiomerically pure (S,S,S)-1. Reagents and conditions: (a) H2, PtO2, AcOH, 60 °C. Recrystallization of the mixture from EtOH furnishes pure (S,S,S)-1.
Enantiomerically pure (S,S,S)-1 was then submitted to Yamada’s epimerization procedure,15 by heating in acetic acid in the presence of a catalytic amount of salicylaldehyde to promote the formation of a Schiff base (Scheme 2). According to the literature,12a the epimerization progresses to give a 50:50 mixture of diastereoisomers after 1 hour. However, we found that a prolonged reaction time leads to a considerable enrichment of the diastereoisomeric mixture in the desired epimer, with the final ratio (S,S,S)-1/(R,S,S)-1 = 23:77 after 24 hours of reaction. Undoubtedly, this represents a substantial improvement and is expected to have a very positive effect on the overall efficiency of the method. In comparison, in the synthesis reported by Vincent,12a a 50:50 mixture of diastereoisomers was obtained and used for the isolation of (R,S,S)-1. This leads to a significant reduction in the final yield and also sets hurdles for its isolation by recrystallization techniques.
Scheme 2.

Epimerization of (S,S,S)-1. Reagents and conditions: (a) salicylaldehyde, AcOH, reflux.
At this point, our efforts focused on finding a more competitive way to isolate the desired compound from the 23:77 mixture of epimers, other than laborious recrystallizations. In doing so, we turned our attention to reported investigations dealing with the highly stereoselective synthesis of bicyclic oxazolidinones derived from L-proline, such as those depicted in Scheme 3.17 This type of oxazolidinone, prepared by condensation of L-proline with a bulky alkyl aldehyde, has proven to be very valuable for the synthesis of α-tetrasubstituted prolines in enantiomerically pure form.17a,c
Scheme 3.

The most commonly used bicyclic oxazolidinones derived from L-proline.
The condensation reaction gives a single diastereoisomer with the bulky alkyl group in a cis disposition with respect to the bridgehead hydrogen. A perspective view of the structure reveals a “roof” shaped bicyclic framework, with the bulky alkyl moiety pointing towards the less crowded convex face (exo).17a,b
According to this observation, it seems reasonable to consider that the ease of such a stereoselective condensation, when applied to octahydroindole-2-carboxylic acids, will be conditioned to the structural constraint of the resulting tricyclic system. Based on this premise, we envisaged the possibility of using this transformation as a tool for discrimination between (R,S,S)-1 and (S,S,S)-1, where the two theoretical tricyclic systems, (S,S,S,R)-2 and (R,S,S,S)-2 respectively,18 should show a priori remarkably different spatial constraints (Fig. 2). In fact, molecular models of these structures reflect a more sterically disfavoured situation for oxazolidinone (R,S,S,S)-2, due to partially overlapping hydrogens at the concave face defined by the tricyclic scaffold. To the best of our knowledge, the use of this reaction for selective discrimination between substituted epimeric prolines has not been explored, with all previous investigations focused on the reactivity of the oxazolidinones derived from L-proline.17,19
Figure 2.
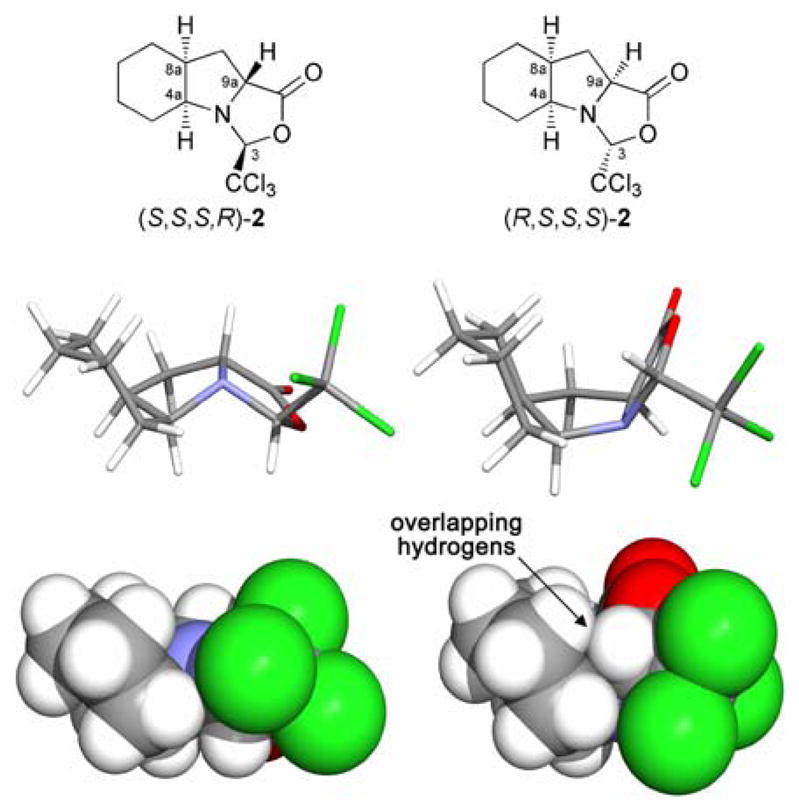
Stick and space filling models for the theoretical trichloromethyloxazolidinones derived from (R,S,S)-1 (left) and (S,S,S)-1 (right). The partially overlapping hydrogens in (R,S,S,S)-2 are indicated.
To test our hypothesis, the 23:77 mixture of epimers (S,S,S)-1 and (R,S,S)-1 was treated with trichloroacetaldehyde in acetonitrile at room temperature.20 We were delighted to observe that, while (R,S,S)-1 reacted quantitatively with the aldehyde, (S,S,S)-1 remained unreacted (Scheme 4). The condensation reaction, as in the case of L-proline, furnishes a single compound with a new chiral center at C3 as a result of the trichloromethyl group being exclusively in a cis relationship with the hydrogen in the 9a position. This experimental result suggests that, indeed, the oxazolidinone derived from (S,S,S)-1 (Fig. 2, right) is not formed because it would result in a tricyclic system with substantial steric hindrance at the inner concave face defined by the rings. In particular, the hydrogen at C3 is oriented towards the cyclohexane ring and would therefore lie in close proximity to one of the hydrogens at C6, thus producing a steric clash.
Scheme 4.

Synthesis of (S,S,S,R)-2. Reagents and conditions: (a) CCl3CHO, CH3CN, r.t., 12 h.
The resulting oxazolidinone was easily isolated as a solid, after filtration through a short pad of silica eluting with dichloromethane.21 Single crystals were obtained from an ethyl acetate/hexanes solution and X-ray diffraction analysis corroborated the expected stereochemistry (Fig. 3). Also in this case, the two five-membered rings define a “roof” shaped structure, with the bulky trichloromethyl group pointing towards the exo face. The pyrrolidine ring assumes an envelope conformation with carbon 8a deviating by 0.63 Å from the plane defined by the other four atoms, which corresponds to a Cγ-exo conformation22 for the proline subunit. The junction to the chair of the six-membered ring is sitting at the flap of the envelope.
Figure 3.
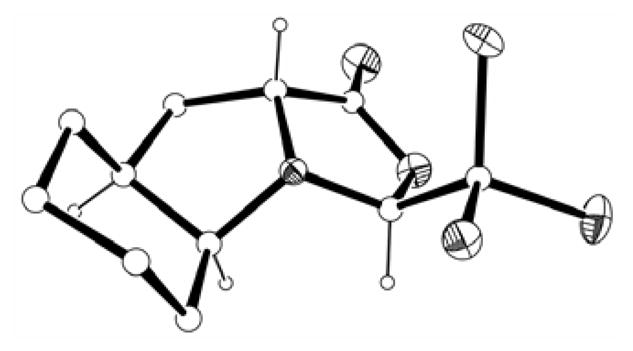
X-ray crystal structure of (S,S,S,R)-2. Heteroatoms are shown as thermal ellipsoids. Most hydrogens have been omitted for clarity.
The trichlomethyloxazolidinone obtained was then converted to the corresponding free amino acid through hydrolysis in acidic conditions (Scheme 5). Thus, treatment of (S,S,S,R)-2 with a saturated solution of hydrogen chloride in ethyl acetate furnished (R,S,S)-1 in enantiomerically pure form, as the hydrochloride salt. It is worth noting that the overall isolated yield of the final amino acid resulting from the epimerization-condensation-hydrolysis sequence was 56%, which represents a tremendous improvement when compared with the 30% described for direct crystallizations.12a
Scheme 5.

Isolation of the enantiomerically pure amino acid (R,S,S)-1.
Thus, the trichlomethyloxazolidinone (S,S,S,R)-2 has proven to be a key intermediate to allow a more efficient synthesis of (R,S,S)-1. At this point, we considered that this oxazolidinone could also be a valuable precursor to afford α-tetrasubstituted derivatives of this Oic stereoisomer. We decided to explore this possibility driven by the attractiveness of α-alkylated Oic derivatives for peptide-engineering purposes and the potential therapeutic value shown by structurally related compounds for the treatment of osteoarthritis.10
Treatment of oxazolidinone (S,S,S,R)-2 with lithium diisopropylamide in THF at −78 °C generated a lithium enolate that successfully reacted with several alkyl electrophiles in high yields (Scheme 6). As in the pioneering investigations by Seebach et al.,17a we observed that both the trichloromethyl group and the newly introduced substituent are cis to each other on the exo side of the bicycle constituted by the two five-membered rings. This indicates that the enolate underwent alkylation exclusively at the Si face. The bridgehead nitrogen of (S,S,S,R)-2 is strongly pyramidalised and its inversion does not take place, even when the enolate is generated, because of the high steric constraints that such a change would have on the system. For this reason, the enolate attacks the electrophile only from the face that was previously occupied by the α hydrogen, thus leading to the formation of a single stereoisomer with retention of configuration at C9a. This behaviour was named by Seebach as self-reproduction of chirality.17a The complete stereocontrol of this process becomes particularly remarkable in comparison with the only example of the α-alkylation of an L-Oic derivative reported to date.10 It has been shown that an N-protected methyl ester derivative of L-Oic undergoes methylation, upon treatment with LiHMDS, to afford the stereoisomer with retention of configuration as the major compound, together with a non-negligible percentage of the α-methylated epimer. In contrast, in our case, not even traces of the other possible stereoisomer could be detected. Single crystal X-ray diffraction analysis of the resulting α-tetrasubstituted compounds 3a–c provided evidence for the alkylation reaction taking place with retention of configuration23 (Fig. 4).
Scheme 6.

Synthesis of α-quaternary derivatives.
Figure 4.
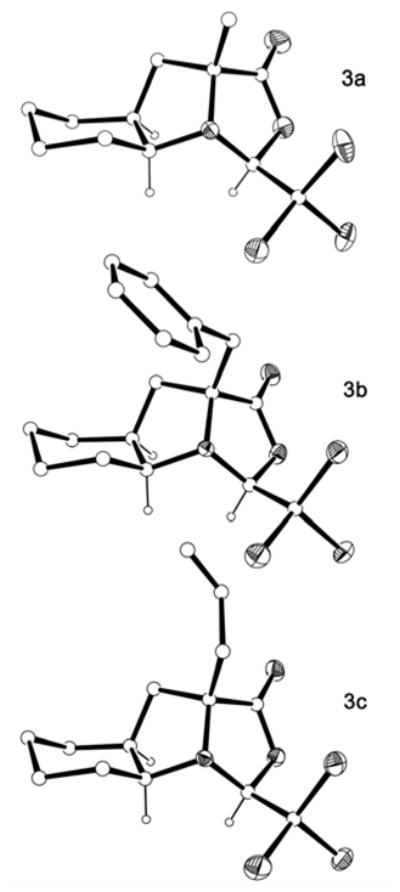
X-ray crystal structures of the α-tetrasubstituted oxazolidinones (S,S,S,R)-3a, (S,S,S,S)-3b and (S,S,S,R)-3c. Heteroatoms are shown as thermal ellipsoids. Most hydrogens have been omitted for clarity.
In the three crystalline structures, the pyrrolidine ring assumes an envelope conformation and, as already observed for (S,S,S,R)-2 (Fig. 3), it is the atom at the 8a position (Cγ considering the proline system) that deviates from planarity (0.58–0.61 Å). However, in contrast to (S,S,S,R)-2, in all three tetrasubstituted derivatives 3a–c a Cγ-endo conformation22 is observed, i.e. the flap of the envelope points in the opposite direction. This probably helps to accommodate the newly introduced bridgehead substituent.
These α-tetrasubstituted oxazolidinones can be hydrolyzed in an acidic medium, although stronger conditions are required in comparison to the non-substituted analogue, as exemplified for the α-methyl derivative in Scheme 7. Thus, compound (S,S,S,R)-3a was heated under reflux in a mixture of hydrochloric acid and acetic acid to give the free α-methylated amino acid (R,S,S)-4 as the hydrochloride salt in 92% yield.
Scheme 7.

Isolation of the (R,S,S) isomer of (αMe)Oic in enantiomerically pure form.
3. Conclusion
We have established an improved methodology for the efficient synthesis of enantiomerically pure (2R,3aS,7aS)-octahydroindole-2-carboxylic acid. This stereoisomer can be efficiently separated from its epimer by means of a selective condensation reaction with trichloroacetaldehyde. In addition, the resulting oxazolidinone constitutes a versatile intermediate that undergoes completely diastereoselective α-alkylation reactions, thus providing a very convenient and concise route to access enantiomerically pure α-alkyl derivatives of (2R,3aS,7aS)-octahydroindole-2-carboxylic acid.
4. Experimental
4.1. General
All reagents were used as received from commercial suppliers without further purification. Thin-layer chromatography (TLC) was performed on Macherey-Nagel Polygram syl G/UV precoated silica gel polyester plates. The products were visualized by exposure to UV light (254 nm), iodine vapour or submersion in cerium molybdate stain [aqueous solution of phosphomolybdic acid (2%), CeSO4·4H2O (1%) and H2SO4 (6%)]. Column chromatography was performed using 60Å silica gel purchased from SDS. Melting points were determined on a Gallenkamp apparatus and are uncorrected. IR spectra were registered on a Mattson Genesis or a Nicolet Avatar 360/370 FTIR spectrophotometer; νmax is given for the main absorption bands. 1H and 13C NMR spectra were recorded on a Bruker AV-400 instrument at room temperature using the residual solvent signal as the internal standard; chemical shifts (δ) are expressed in ppm and coupling constants (J) in Hertz. Optical rotations were measured at room temperature using a JASCO P-1020 polarimeter. High-resolution mass spectra were obtained on a Bruker Microtof-Q spectrometer.
4.2. X-ray diffraction
Colourless single crystals of (S,S,S,R)-2 and (S,S,S,R)-3a were obtained by slow evaporation from ethyl acetate solutions. Compounds (S,S,S,S)-3b and (S,S,S,R)-3c, which are oils, furnished single crystals upon standing. The X-ray diffraction data were collected at room temperature on an Oxford Diffraction Xcalibur diffractometer provided with a Sapphire CCD detector, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods using SHELXS-9724a and refinement was performed using SHELXL-9724b by the full-matrix least-squares technique with anisotropic thermal factors for heavy atoms. Hydrogen atoms were located by calculation and affected by an isotropic thermal factor fixed to 1.2 times the Ueq of the carrier atom (1.5 for the methyl protons). Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 658042–658045. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or e-mail: deposit@ccdc.cam.ac.uk].
Crystallographic data for (S,S,S,R)-2: orthorhombic, space group P212121; a = 8.6296(2) Å, b = 9.9887(3) Å, c = 15.4787(4) Å; Z = 4; dcalcd = 1.486 g·cm−3; 7764 reflections collected, 2570 unique (Rint = 0.024); data/parameters: 2570/155; final R indices (I > 2σI): R1 = 0.028, wR2 = 0.062; final R indices (all data): R1 = 0.042, wR2 = 0.065. Highest residual electron density: 0.18 e Å−3.
Crystallographic data for (S,S,S,R)-3a: orthorhombic, space group P212121; a = 8.0709(4) Å, b = 10.0848(5) Å, c = 17.7838(8) Å; Z = 4; dcalcd = 1.434 g·cm−3; 8697 reflections collected, 2868 unique (Rint = 0.026); data/parameters: 2868/165; final R indices (I > 2σI): R1 = 0.030, wR2 = 0.061; final R indices (all data): R1 = 0.048, wR2 = 0.065. Highest residual electron density: 0.16 e Å−3.
Crystallographic data for (S,S,S,S)-3b: orthorhombic, space group P212121; a = 8.6469(1) Å, b = 9.5584(1) Å, c = 22.3424(4) Å; Z = 4; dcalcd = 1.398 g·cm−3; 24447 reflections collected, 4017 unique (Rint = 0.038); data/parameters: 4017/218; final R indices (I > 2σI): R1 = 0.027, wR2 = 0.050; final R indices (all data): R1 = 0.052, wR2 = 0.053. Highest residual electron density: 0.15 e Å−3.
Crystallographic data for (S,S,S,R)-3c: orthorhombic, space group P212121; a = 8.7355(4) Å, b = 13.1329(5) Å, c = 14.1367(5) Å; Z = 4; dcalcd = 1.387 g·cm−3; 8804 reflections collected, 2851 unique (Rint = 0.031); data/parameters: 2851/181; final R indices (I > 2σI): R1 = 0.030, wR2 = 0.058; final R indices (all data): R1 = 0.058, wR2 = 0.063. Highest residual electron density: 0.17 e Å−3.
4.3. Synthesis of (2S,3aS,7aS)-octahydroindole-2-carboxylic acid, (S,S,S)-1
A solution of (S)-indoline-2-carboxylic acid (3.0 g, 18.38 mmol) in acetic acid (60 mL) was hydrogenated at 60 °C in the presence of PtO2 (300 mg). After 24 h, the catalyst was filtered off and washed with acetic acid. The solvent was evaporated to dryness and the resulting residue was crystallized from ethanol to afford pure (S,S,S)-1 as a white solid (2.64 g, 15.60 mmol, 85% yield). Mp 267–269 °C. [α]D −45.6 (c 0.46, MeOH). IR (KBr) ν 3600–2200, 1623 cm−1. 1H NMR (D2O, 400 MHz) δ 1.23–1.64 (m, 7H), 1.73–1.82 (m, 1H), 1.91–2.01 (m, 1H), 2.25–2.36 (m, 2H), 3.65 (m, 1H), 4.06 (m, 1H). 13C NMR (D2O, 100 MHz) δ 20.88, 21.36, 24.38, 25.06, 32.36, 36.91, 59.34, 59.70, 175.42. HRMS (ESI) C9H16NO2 [M + H]+: calcd. 170.1176, found 170.1174.
4.4. Synthesis of (3S,4aS,8aS,9aR)-3-trichloromethyloctahydrooxazolo[3,4-a]indol-1-one, (S,S,S,R)-2
A solution of (S,S,S)-1 (2.0 g, 11.82 mmol) in acetic acid (20 mL) was treated with salicyl aldehyde (75 μL, 0.71 mmol) and heated under reflux for 24 h. The solvent was evaporated and the residue lyophilized to furnish a 23:77 mixture of epimers (S,S,S)-1 and (R,S,S)-1. This mixture was suspended in dry acetonitrile (40 mL) and treated with anhydrous trichloroacetaldehyde (2.0 mL, 20.50 mmol). The resulting suspension was stirred under an argon atmosphere at room temperature for 24 h. The mixture was filtered through a pad of Celite® and the filtrate was concentrated. The residue was purified through a short pad of silica gel eluting with dichloromethane to afford pure (S,S,S,R)-2 as a white solid (2.11 g, 7.07 mmol, 60% yield). Mp 144–146 °C (dec). [α]D +59.4 (c 0.47, CHCl3). IR (Nujol) ν 1739 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.20–1.48 (m, 3H), 1.54–1.71 (m, 4H), 1.87 (m, 1H), 1.96–2.20 (m, 3H), 3.18 (m, 1H), 4.27 (dd, 1H, J = 8.8, 7.2 Hz), 5.18 (s, 1H). 13C NMR (CDCl3, 100 MHz) δ 20.26, 23.62, 26.80, 27.67, 34.46, 38.49, 61.27, 66.42, 100.48, 102.14, 176.88. HRMS (ESI) C11H15Cl3NO2 [M + H]+: calcd. 298.0163, found 298.0157.
4.5. Synthesis of (2R,3aS,7aS)-octahydroindole-2-carboxylic acid hydrochloride, (R,S,S)-1·HCl
A 3N solution of HCl in anhydrous ethyl acetate (8 mL) was added to (S,S,S,R)-2 (300 mg, 1.01 mmol) and the resulting mixture was stirred at room temperature for 24 h. The solvent was concentrated in vacuo and the resulting solid was washed with small portions of ethyl acetate to afford pure (R,S,S)-1 as the hydrochloride salt (193 mg, 0.94 mmol, 93% yield). Mp 176–178 °C. [α]D +29.6 (c 0.47, MeOH). IR (KBr) ν 3507, 3200–2300, 1732, 1578 cm−1. 1H NMR (D2O, 400 MHz) δ 1.20–1.51 (m, 5H), 1.54–1.70 (m, 2H), 1.72–1.82 (m, 1H), 2.02–2.15 (m, 1H), 2.23–2.36 (m, 2H), 3.72 (m, 1H), 4.38 (m, 1H). 13C NMR (D2O, 100 MHz) δ 20.51, 21.01, 23.95, 24.72, 32.44, 36.33, 57.61, 59.94, 172.76. HRMS (ESI) C9H16NO2 [M − Cl]+: calcd. 170.1176, found 170.1177.
4.6. Synthesis of (3S,4aS,8aS,9aR)-9a-methyl-3-trichloromethyloctahydrooxazolo[3,4-a]indol-1-one, (S,S,S,R)-3a
A 1.8 M solution of LDA in hexanes (720 μL, 1.29 mmol) was added dropwise by syringe to a stirred solution of (S,S,S,R)-2 (200 mg, 0.67 mmol) in anhydrous THF (3.5 mL) at −78 °C. After stirring at this temperature for 30 min, methyl iodide (169 μL, 2.71 mmol) was added dropwise. The resulting solution was then warmed to −50 °C and stirred at this temperature overnight. The reaction mixture was treated with water (4 mL) and extracted with dichloromethane several times. The combined organic layers were dried over anhydrous MgSO4 and filtered. The solvent was concentrated in vacuo and the residue was purified by column chromatography (eluent: hexanes/ethyl acetate 10/1) to afford pure (S,S,S,R)-3a as a white solid (200 mg, 0.64 mmol, 95% yield). Mp 134–136 °C. [α]D +9.4 (c 0.45, CHCl3). IR (Nujol) ν 1787 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.09–1.22 (m, 1H), 1.23–1.39 (m, 2H), 1.47–1.66 (m, 2H) overlapped with 1.62 (s, 3H), 1.68–1.85 (m, 2H), 1.88–1.97 (m, 1H), 2.02 (m, 1H), 2.15–2.30 (m, 2H), 3.15 (m, 1H), 5.05 (s, 1H). 13C NMR (CDCl3, 100 MHz) δ 19.95, 24.50, 24.96, 27.43, 28.79, 36.98, 40.88, 68.77, 70.45, 100.41, 102.35, 177.84. HRMS (ESI) C12H17Cl3NO2 [M + H]+: calcd. 312.0319, found 312.0320.
4.7. Synthesis of (3S,4aS,8aS,9aS)-9a-benzyl-3-trichloromethyloctahydrooxazolo[3,4-a]indol-1-one, (S,S,S,S)-3b
Using benzyl bromide (326 μL, 2.74 mmol) as the alkylating agent, an identical procedure to that described above was applied to the transformation of (S,S,S,R)-2 (200 mg, 0.67 mmol) into (S,S,S,S)-3b (230 mg, 0.59 mmol, 88% yield). The product was isolated as an oil that crystallized upon standing. Mp 150–151 °C. [α]D −26.6 (c 0.48, CHCl3). IR (Nujol) ν 1794 cm−1. 1H NMR (CDCl3, 400 MHz) δ 0.80–0.97 (m, 2H), 1.23–1.59 (m, 5H), 1.71 (m, 1H), 1.97–2.22 (m, 3H), 2.91–3.09 (m, 2H), 3.29 (d, 1H, J = 13.8 Hz), 5.12 (s, 1H), 7.21–7.36 (m, 5H). 13C NMR (CDCl3, 100 MHz) δ 19.91, 24.43, 25.00, 27.67, 35.84, 36.26, 42.32, 71.45, 72.58, 100.04, 103.48, 127.13, 127.93, 131.63, 135.28, 177.42. HRMS (ESI) C18H20Cl3NNaO2 [M + Na]+: calcd. 410.0452, found 410.0443.
4.8. Synthesis of (3S,4aS,8aS,9aR)-9a-allyl-3-trichloromethyloctahydrooxazolo[3,4-a]indol-1-one, (S,S,S,R)-3c
Using allyl bromide (237 μL, 2.74 mmol) as the alkylating agent, an analogous procedure to that described above, allowed the transformation of (S,S,S,R)-2 (200 mg, 0.67 mmol) into (S,S,S,R)-3c (185 mg, 0.55 mmol, 82% yield), which was isolated as an oil that crystallized upon standing. Mp 114–115 °C. [α]D −3.3 (c 0.48, CHCl3). IR (Nujol) ν 1791 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.04–1.18 (m, 1H), 1.19–1.39 (m, 2H), 1.43–1.64 (m, 2H), 1.65–1.75 (m, 1H), 1.76–1.90 (m, 2H), 1.98–2.23 (m, 3H), 2.54 (ddd, 1H, J = 14.0, 7.1, 0.9 Hz), 2.72 (dd, 1H, J = 14.0, 7.6 Hz), 3.12 (m, 1H), 5.08 (s, 1H), 5.11–5.23 (m, 2H), 5.97 (m, 1H). 13C NMR (CDCl3, 100 MHz) δ 20.09, 24.57, 25.07, 28.57, 36.46, 36.79, 43.56, 70.75, 71.17, 100.08, 102.88, 119.24, 132.67, 176.86. HRMS (ESI) C14H19Cl3NO2 [M + H]+: calcd. 338.0476, found 338.0463.
4.9. Synthesis of (2R,3aS,7aS)-2-methyl-octahydroindole-2-carboxylic acid hydrochloride, (R,S,S)-4·HCl
Acetic acid (4 mL) and 6N HCl (4 mL) were added to (S,S,S,R)-3a (200 mg, 0.64 mmol) and the reaction mixture was heated under reflux for 12 h. The solvent was concentrated in vacuo and the resulting solid was washed with small portions of ethyl acetate to afford pure (R,S,S)-4 as the hydrochloride salt (130 mg, 0.59 mmol, 92% yield). Mp 206–208 °C. [α]D +28.2 (c 0.47, MeOH). IR (KBr) ν 3434, 3200–2200, 1731, 1591 cm−1. 1H NMR (D2O, 400 MHz) δ 1.18–1.71 (m, 7H) overlapped with 1.65 (s, 3H), 1.78–1.89 (m, 1H), 2.07 (dd, 1H, J = 17.1, 12.9 Hz), 2.29 (m, 1H), 2.39 (dd, 1H, J = 17.1, 8.9 Hz), 3.74 (m, 1H). 13C NMR (D2O, 100 MHz) δ 20.19, 21.73, 23.37, 24.85, 24.93, 36.09, 39.02, 59.85, 68.59, 175.45. HRMS (ESI) C10H18NO2 [M − Cl]+: calcd. 184.1332, found 184.1341.
Acknowledgments
Financial support from the Ministerio de Educación y Ciencia–FEDER (project CTQ2004-5358) and Gobierno de Aragón (project PIP206/2005 and research group E40) is gratefully acknowledged. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Dedicated to Miguel Yus on the occasion of his 60th birthday
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Adessi C, Soto C. Curr Med Chem. 2002;9:963–978. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 2.(a) Cowell SM, Lee YS, Cain JP, Hruby VJ. Curr Med Chem. 2004;11:2785–2798. doi: 10.2174/0929867043364270. [DOI] [PubMed] [Google Scholar]; (b) Toniolo C, Crisma M, Formaggio F, Peggion C. Biopolymers (Pept Sci) 2001;60:396–419. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; (c) Hruby VJ, Li G, Haskell-Luevano C, Shenderovich M. Biopolymers (Pept Sci) 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Karoyan P, Sagan S, Lequin O, Quancard J, Lavielle S, Chassaing G. Substituted Prolines: Syntheses and Applications in Structure-Activity Relationship Studies of Biologically Active Peptides. In: Attanasi OA, Spinelli D, editors. Targets in Heterocyclic Systems. Chemistry and Properties. Vol. 8. Royal Society of Chemistry; Cambridge: 2005. pp. 216–273. [Google Scholar]
- 4.van de Waterbeemd H, Karajiannis H, El Tayar N. Amino Acids. 1994;7:129–145. doi: 10.1007/BF00814156. [DOI] [PubMed] [Google Scholar]
- 5.El Tayar N, Karajiannis H, van de Waterbeemd H. Amino Acids. 1995;8:125–139. doi: 10.1007/BF00806487. [DOI] [PubMed] [Google Scholar]
- 6.(a) Reissmann S, Imhof D. Curr Med Chem. 2004;11:2823–2844. doi: 10.2174/0929867043364135. [DOI] [PubMed] [Google Scholar]; (b) Stewart JM. Peptides. 2004;25:527–532. doi: 10.1016/j.peptides.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 7.Monteagudo ES, Calvani F, Catrambone F, Fincham CI, Madami A, Meini S, Terracciano R. J Pept Sci. 2001;7:270–283. doi: 10.1002/psc.321. [DOI] [PubMed] [Google Scholar]
- 8.(a) Alfakih K, Hall AS. Expert Opin Pharmacother. 2006;7:63–71. doi: 10.1517/14656566.7.1.63. [DOI] [PubMed] [Google Scholar]; (b) Hurst M, Jarvis B. Drugs. 2001;61:867–896. doi: 10.2165/00003495-200161060-00020. [DOI] [PubMed] [Google Scholar]; (c) Todd PA, Fitton A. Drugs. 1991;42:90–114. doi: 10.2165/00003495-199142010-00006. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hanessian S, Del Valle JR, Xue Y, Blomberg N. J Am Chem Soc. 2006;128:10491–10495. doi: 10.1021/ja0625834. [DOI] [PubMed] [Google Scholar]; (b) Hanessian S, Tremblay M, Petersen JFW. J Am Chem Soc. 2004;126:6064–6071. doi: 10.1021/ja030669g. [DOI] [PubMed] [Google Scholar]; (c) Carroll AR, Pierens GK, Fechner G, de Almeida Leone P, Ngo A, Simpson M, Hyde E, Hooper JNA, Boström S-L, Musil D, Quinn RJ. J Am Chem Soc. 2002;124:13340–13341. doi: 10.1021/ja020814a. [DOI] [PubMed] [Google Scholar]
- 10.Barvian NC, Connolly CJC, Guzzo PR, Hamby JM, Hicks JL, Johnson MR, Le V-D, Mitchell LH, Roark WH. 2004092132 PCT Patent WO.
- 11.(a) Belvisi L, Colombo L, Colombo M, Di Giacomo M, Manzoni L, Vodopivec B, Scolastico C. Tetrahedron. 2001;57:6463–6473. [Google Scholar]; (b) Pyne SG, Javidan A, Skelton BW, White AH. Tetrahedron. 1995;51:5157–5168. [Google Scholar]
- 12.(a) Vincent M, Marchand B, Remond G, Jaguelin-Guinamant S, Damien G, Portevin B, Baumal JY, Volland JP, Bouchet JP, Lambert PH, Serkiz B, Luitjen W, Laubie M, Schiavi P. Drug Des Discovery. 1992;9:11–28. [PubMed] [Google Scholar]; (b) Blankley CJ, Kaltenbronn JS, DeJohn DE, Werner A, Bennett LR, Bobowski G, Krolls U, Johnson DR, Pearlman WM, Hoefle ML, Essenburg AD, Cohen DM, Kaplan HR. J Med Chem. 1987;30:992–998. doi: 10.1021/jm00389a006. [DOI] [PubMed] [Google Scholar]; (c) Vincent M, Remond G, Portevin B, Serkiz B, Laubie M. Tetrahedron Lett. 1982;23:1677–1680. [Google Scholar]
- 13.Hirata N. 20050106690 US Patent.
- 14.Sayago FJ, Jiménez AI, Cativiela C. Tetrahedron: Asymmetry. 2007;18:2358–2364. doi: 10.1016/j.tetasy.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada S, Hongo C, Yoshioka R, Chibata I. J Org Chem. 1983;48:843–846. [Google Scholar]
- 16.Henning R, Urbach H. Tetrahedron Lett. 1983;24:5339–5342. [Google Scholar]
- 17.(a) Seebach D, Boes M, Naef R, Schweizer WB. J Am Chem Soc. 1983;105:5390–5398. [Google Scholar]; (b) Orsini F, Pelizzoni F, Forte M, Sisti M, Bombieri G, Benetollo F. J Heterocyclic Chem. 1989;26:837–841. [Google Scholar]; (c) Germanas JP, Wang H. Synlett. 1999:33–36. [Google Scholar]
-
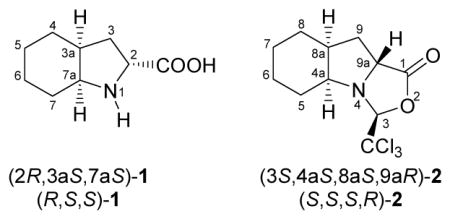
18.Note that the order of stereochemical descriptors R/S obey different numeration profiles in compounds 1 and 2. In particular, the α carbon occupies position 2 in the former and position 9a in the latter. Abreviated names, with and without numerical locants, are exemplified below for clarity.
- 19.Amedjkouh M, Ahlberg P. Tetrahedron: Asymmetry. 2002;13:2229–2234. [Google Scholar]
- 20.The preparation of trichloromethyloxazolidinones was preferred to tert-butyloxazolidinones because of their greater stability. In the case of proline, the corresponding trichloromethyloxazolidinone is a crystalline solid, while the tert-butyl derivative is highly sensitive to hydrolysis and unstable in air.
- 21.Unreacted (S,S,S)-1 can be isolated, if desired, by washing the silica pad with MeOH.
- 22.(a) DeTar DF, Luthra NP. J Am Chem Soc. 1977;99:1232–1244. doi: 10.1021/ja00446a040. [DOI] [PubMed] [Google Scholar]; (b) Ashida T, Kakudo M. Bull Chem Soc Jpn. 1974;47:1129–1133. [Google Scholar]
- 23.Although the relative spatial disposition of substituents at carbon 9a is identical for all three tetrasubstituted compounds 3a–c, the stereochemical descriptors R/S giving the absolute configuration of the carbon centre differ. Thus, the compound with a benzyl substituent (3b) has an S configuration at position 9a due to the different priority of the substituents in comparison with the methyl or allyl derivatives.
- 24.(a) Sheldrick GM. SHELXS-97, Program for the Solution of Crystal Structures. University of Göttingen; Göttingen: 1997. [Google Scholar]; (b) Sheldrick GM. SHELXL-97. Program for the Refinement of Crystal Structures. University of Göttingen; Göttingen: 1997. [Google Scholar]