

Abstract
Adenosine A1 receptors are inhibitory G-protein coupled receptors that presynaptically regulate neurotransmitter release, but their role in self-regulating adenosine release is not known. In this study, we examined the modulation of evoked adenosine and dopamine efflux by A1 receptors and studied whether D1 receptors mediate these effects. Fast-scan cyclic voltammetry at carbon-fiber microelectrodes was used for the simultaneous detection of adenosine and dopamine efflux on a subsecond time scale. Short electrical stimulation trains delivered to the substantia nigra (60 pulses, 60 Hz) were used to evoke dopamine and adenosine release in the striatum. The adenosine A1 receptor agonist N6-cyclopentyladenosine (CPA, 1 mg/kg i.p.) decreased both adenosine and dopamine efflux, although the effect for adenosine occurred more quickly than for dopamine. The A1 antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 6 mg/kg i.p.) increased stimulated adenosine release. The effects of CPA were partially attenuated by the dopamine D1 receptor antagonist SCH-23390. Thus, A1 and D1 receptors have a synergistic interaction that modulates both stimulated adenosine and dopamine. The decrease in adenosine is not a downstream effect of lowered dopamine release, as decreasing dopamine synthesis and release with α-methyl-p-tyrosine or increasing release with haloperidol had no effect on adenosine release. This study shows that A1 receptors have some characteristics of an autoreceptor, including self-regulation of adenosine release.
Keywords: adenosine, dopamine, A1 receptor, stimulated release, voltammetry, microelectrode
1. Introduction
In the central nervous system, the purine nucleoside adenosine is a metabolic byproduct that is also a ubiquitous neuromodulator. Much research has focused on the neuroprotective effects of adenosine and the role that it plays in protecting tissue from damage following ischemia (Masino and Dulla, 2005). However, adenosine also has general properties of modulating neuronal responses and neurotransmission that are relevant in non-pathological situations. Many of the neuroprotective and modulatory effects of adenosine are regulated by A1 receptors (Latini and Pedata, 2001), which are inhibitory and act by reducing intracellular calcium and increasing K+ and Cl− channel conductances (Yawo and Chuhma, 1993). A1 receptors are also linked to Gi proteins and decrease adenyl cyclase activity, further reducing neural excitability. In neurons, A1 receptors are expressed presynaptically and postsynaptically where they modulate both neurotransmitter release and signaling. The extent to which A1 receptors modulate adenosine release has not been elucidated.
In the striatum, a brain region involved in behavioral responses to motivational stimuli and locomotion, A1 adenosine receptors are highly expressed (Ferre et al., 1997). The two main striatal inputs are dopaminergic afferents from the substantia nigra and glutamatergic afferents from the cortical, limbic, and thalamic areas. A1 receptors are expressed presynaptically in the striatum on both glutamatergic neurons and dopaminergic neurons, where they can modulate neurotransmitter release (Ferre et al., 1997). The main outputs from the striatum are GABAergic medium spiny neurons, which can be divided into two subtypes based on their projection patterns: striatonigral and striatopallidal neurons. Striatonigral neurons contain the peptide dynorphin and express predominantly adenosine A1 and dopamine D1 receptors, while striatopallidal neurons contain the peptide enkephalin and express adenosine A2A and dopamine D2 receptors (Ferre, 2008). A1 receptor expression has also been demonstrated on striatal acetylcholine interneurons (Alexander and Reddington, 1989). The affinity of the A1 receptors is high (Kd in low nM range) and therefore a tonic inhibitory tone may exist, as basal adenosine levels are estimated to be 40–200 nM in the striatum (Ballarin et al., 1991; Pazzagli et al., 1995).
A1 receptors have functional interactions with other receptors. For example, A1 receptors can form heteromers with dopamine D1 receptors or A2A receptors. Most studies have found an antagonistic interaction between A1 and D1 receptors in heteromers; that is, binding by an A1 agonist decreases the affinity of D1 receptors. This interaction involves intramembrane receptor-receptor interactions as well as interactions at the second messenger level (Franco et al., 2007). However, one study found the opposite effect, a synergistic interaction whereby A1 inhibition of stimulated dopamine release is partially dependent on activation of D1 receptors (O’Neill et al., 2007). This effect did not involve an intramembrane interaction and is likely an indirect, downstream action. Therefore, A1 regulation of neurotransmission can be mediated by other receptors.
While A1 receptors are known to modulate neurotransmitter release, it is not known if A1 receptors also self-regulate adenosine release. In this study, we examine evoked dopamine and adenosine release in the caudate-putamen in vivo and determine that A1 receptors regulate both dopamine and adenosine release. These studies were performed using fast-scan cyclic voltammetry with carbon-fiber microelectrodes, which allows rapid, simultaneous measurements of adenosine and dopamine release (Cechova and Venton, 2008; Swamy and Venton, 2007). The effect of D1 receptors in modulating adenosine release was also tested because of the known interactions between A1 and D1 receptors. A D1 antagonist can partially block the effects of an A1 agonist. This study shows that A1 receptors regulate evoked adenosine release as well as dopamine release and exhibit some characteristics of an autoreceptor.
2. Experimental procedures
2.1 Chemicals and drugs
Electrodes were calibrated in a Tris buffer that mimicked cerebral spinal fluid containing 15 mM Tris-base, 140 mM NaCl, 3.25 mM KCl, 1.2 mM CaCl2, 1.25 mM NaH2PO4, 1.2 mM MgCl2 and 2.0 mM Na2SO4, pH=7.4 (all from Fisher, Fair Lawn, NJ). Adenosine, dopamine, and all drugs were acquired from Sigma-Aldrich (Milwaukee, WI, USA). All aqueous solutions were made using deionized water (Milli-Q Biocel, Millipore, Billerica, MA). Stock solutions (10 mM) of adenosine and dopamine were made in 0.1 M perchloric acid (Fisher) and kept refrigerated for no longer than 1 month.
All drugs were administered intraperitoneally (i.p.) and dissolved in 1 mL of saline unless otherwise noted. The following drugs were used: 8-Cyclopentyl-1,3-dipropylxanthine, an adenosine A1 receptor antagonist (DPCPX, 6 mg/kg, 150 μL of DMSO added to 1 mL saline to dissolve); N6-cyclopentyladenosine, an adenosine A1 receptor agonist (CPA, 1 mg/kg); SCH-23390, a dopamine D1 receptor antagonist (0.1 mg/kg); α-methyl-p-tyrosine methyl ester hydrochloride, a tyrosine hydroxylase inhibitor (AMPT, 250 mg/kg in 2 mL saline); and haloperidol, a D2 receptor antagonist (0.5 mg/kg, 150 μl acetic acid added to 1 mL saline to dissolve). When drugs were administered together, the same doses were used as when they were administered individually.
2.2 Electrodes and electrochemistry
Fast-scan cyclic voltammetry at a carbon-fiber microelectrode was used to detect dopamine and adenosine simultaneously (Cechova and Venton, 2008). Cylindrical microelectrodes were made using 7 μm diameter T-650 carbon fibers (Cytec Engineering Materials, West Patterson, NJ, USA) pulled in a glass capillary (1.2 × 0.68 mm, A-M Systems, Inc., Carlsborg, WA, USA). The extended fiber was cut with a scalpel under a microscope to a length of approximately 50 μm. The electrode was linearly ramped from −0.4 V to 1.5 V and back at 400 V/s every 100 ms. All cyclic voltammograms were acquired with a Chem-Clamp voltammeter (Dagan Corporation, Minneapolis, MN, USA) and background subtracted to remove the large electrode charging current. Hardware and software for data collection were the same as described previously (Heien et al., 2003).
Before each experiment, the microelectrode was soaked for 10 minutes in isopropanol and calibrated using 1 μM dopamine and 5 μM adenosine solutions, prepared fresh daily by diluting 10 mM stock solutions with Tris buffer. Calibrations were performed in a flow injection analysis system (Strand and Venton, 2008). Three second long injections of neurochemicals were made to mimic fast concentration changes that occur in the brain and at least 6 injections were averaged for each compound. The peak oxidative current is at 0.6 V for dopamine and 1.5 V for adenosine, and these currents are proportional to concentration. The peak oxidative current for the known calibration concentration was used to convert the current measured during the in vivo experiment to concentration using a simple ratio calculation. For some experiments, electrodes were also calibrated with 0.5 μM dopamine and 1 μM adenosine, and the estimated concentration values were not significantly different than when the higher concentrations were used. The limit of detection for adenosine with fast-scan cyclic voltammetry is around 15 nM and the limit of detection for dopamine is 5 nM.
Microelectrodes were also calibrated in the presence of the pharmacological agents to ensure that the drugs did not interfere with dopamine and adenosine detection. Saline and quinpirole are not electroactive and therefore they do not interfere with dopamine and adenosine detection in the brain. When tested at concentrations of 1 μM in the Tris buffer, AMPT and haloperidol had no effect and CPA and DPCPX slightly decreased the sensitivity of microelectrodes to adenosine and dopamine (in the range of 5–7%). SCH-23390 slightly increased the sensitivity of microelectrode to dopamine. The combination of SCH-23390 and CPA had no effect on the microelectrode sensitivity and CPA +DPCPX decreased sensitivity by about 15 %. These interferences are small and would not affect the interpretation of results.
2.3 Animals and surgery
Male, Sprague-Dawley rats (250–300g, Charles River, Wilmington, MA, USA) were housed in a vivarium with a 12-h light/dark cycle, with food and water provided ad libitum. All animal experiments were approved by the Animal Care and Use Committee of the University of Virginia. On the day of the experiment, the rat was anesthetized with urethane (Sigma-Aldrich, Milwaukee, WI, USA) at a dose of 1.5 g/kg, i.p. (urethane dissolved in a 50 % wt solution of sterile saline). The rat was placed into a stereotaxic frame and the surgical site was locally infiltrated with 0.2 mL (s.c.) of 0.25 % of bupivacaine to provide analgesia. The skin was cut to expose the skull and holes were drilled for the placement of electrodes. The carbon-fiber microelectrode was inserted in the caudate-putamen, with coordinates (in mm from bregma): AP +1.2, ML +2.1, and DV −4.5 (Paxinos and Watson, 1998). The bipolar stimulating electrode (metal, MS 303/2, Plastics One Inc., Roanoke, VA, USA) was lowered into the substantia nigra (AP −5.4, ML +1.2, and DV −8.0). The DV placement of the stimulating electrode was adjusted downward until a robust dopamine signal was measured. A reference electrode, a silver/silver chloride wire, was inserted in the contralateral side of the brain. The carbon-fiber electrode was allowed to equilibrate (to obtain a stable background charging current at the microelectrode) in the brain for 30–40 minutes after implantation before the experiment was performed. The body temperature of the rat was maintained at 37°C with a heating pad and thermistor probe connected to a temperature controller (FHC, Bowdoin, ME, USA). Electrical stimulations were applied using a BSI-950 Biphasic Stimulus Isolator (Dagan Corporation). A stimulation train of 60 biphasic square pulses at 60 Hz (2 ms wide and 300 μA per phase) was used.
2.4 Data collection and analysis
Data collection began 30–40 minutes after microelectrode implantation. The voltage waveform (−0.4 V to +1.5 V, scan rate 400 V/s, repetition rate 10 Hz) was continually applied to the microelectrode. To estimate concentrations, the peak current for adenosine at 1.5 V and for dopamine at 0.6 V was converted to concentration using the calibration values obtained before the experiment. For pharmacology experiments, stimulation trains were repeated every 3 minutes. Six baseline stimulation files were recorded before a drug was administered to the rat. For each rat, an average predrug stimulated current was measured and then all stimulated release values were normalized by dividing by the predrug value and multiplying by 100. These normalized data were then averaged across multiple animals per group (n= 5–7 animals).
2.5 Statistics
Statistical analyses were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA). All data are presented as mean ± standard error of the mean (SEM) for n numbers of animals. Data were considered statistically significant at the 95 % confidence level. To compare the effect of drugs and time on concentrations of dopamine and adenosine, a two-way ANOVA was applied with a Bonferroni post-test.
3. Results
Fast-scan cyclic voltammetry (FSCV) is useful for monitoring fast changes in neurotransmitters, and compounds with different oxidation potentials can be discriminated electrochemically (Swamy and Venton, 2007). Using FSCV, adenosine and dopamine can be detected simultaneously at carbon-fiber microelectrodes because the oxidation potential for adenosine is 1.5 V while the oxidation potential for dopamine is 0.6 V. Fig. 1 shows a false-color plot of all the data collected for a single stimulation. A 60 pulse, 60 Hz stimulation was applied at 5 s (marked by red line underneath) to the substantia nigra region, which contains dopamine cell bodies. Efflux was measured at a carbon-fiber microelectrode implanted in the caudate-putamen. The small green circle on the bottom half of the color plot at five seconds is due to dopamine oxidation. Adenosine appears in the color plot as a green area at the center, 1.5 V. When the current at the peak oxidation potential is plotted, concentration changes can be monitored over time, as shown in the traces above the color plot. The concentration vs time trace for dopamine increases when the stimulation is applied and decreases afterwards due to rapid uptake. The plot for adenosine shows two phases of release, one that peaks approximately 5 s after the stimulation and another that peaks 15 s after stimulation. The first adenosine peak is more consistent between stimulations in an animal while the second adenosine peak varied more widely in magnitude between stimulations. Uptake for adenosine is slower than for dopamine and the adenosine signal lasts longer than the dopamine signal. For all experiments, the peak stimulated efflux was 0.15 ± 0.03 μM (mean ± SEM) for dopamine, 0.62 ± 0.11 μM for the first adenosine and 0.34 ± 0.05 μM for the second adenosine peak (n= 39).
Figure 1.
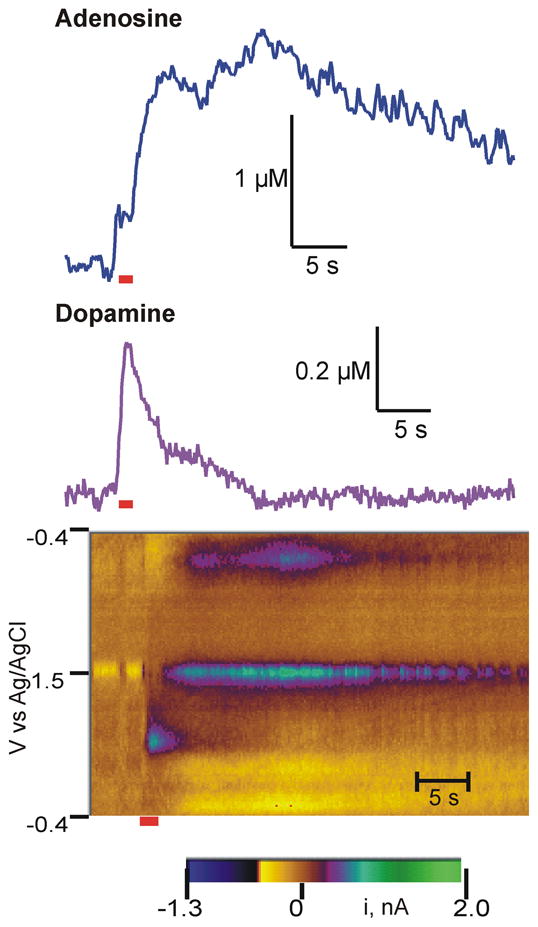
Example stimulated adenosine and dopamine release data. An electrical stimulation was applied to the SN/VTA region and adenosine and dopamine measured in the caudate-putamen. The stimulation was applied at 5 s and was 60 pulses at 60 Hz (2 ms, 300 μA each phase, duration marked by red line). The electrode was scanned from −0.4 V to 1.5 V at 400 V/s and back at 10 Hz. The green/purple area on the lower half the color plot is due to dopamine and the area in the middle of the plot is due to adenosine oxidation. Above the color plot, current vs time traces show changes over time. While dopamine rises quickly during the stimulation, adenosine peaks later and stays elevated longer.
Our lab has previously characterized stimulated adenosine release measured with fast-scan cyclic voltammetry using pharmacology. The adenosine uptake inhibitor propentofylline decreased evoked adenosine release while the adenosine metabolic inhibitor ABT-702 increased release, showing that the measured signal responded as expected to adenosine pharmacological agents (Cechova and Venton, 2008). Other compounds could possibly be interferents, appearing at similar oxidation peaks to adenosine. Oxidation of H2O2 has been observed at carbon-fiber microelectrodes (Sanford et al., 2010), but its oxidation peak occurs after adenosine and the electrode is 10 times more sensitive to adenosine than H2O2. Histamine has a similar CV but was pharmacologically ruled out as an interferent, as a histamine synthesis precursor had no effect on the signal (Cechova and Venton, 2008). These studies confirmed that the signal measured with fast-scan cyclic voltammetry was adenosine.
3.1 A1 regulation of dopamine and adenosine
To determine if the A1 receptor regulates adenosine and dopamine concentrations, an A1 agonist and an A1 antagonist were administered. CPA, a selective A1 receptor agonist, is known to cause strong cardiovascular responses including rapid decreases in blood pressure and heart rate (Schindler et al., 2005). Therefore, we selected a moderate dose of 1 mg/kg that would have a neurochemical effect without compromising circulation (Karcz-Kubicha et al., 2003). A large dose of DPCPX (6 mg/kg, i.p.), a selective A1 antagonist, was chosen to demonstrate an effect. High doses in the 5–6 mg/kg range have previously been shown to cause anxiogenic effects in mice (Prediger et al., 2004) and increased locomotion (Kuzmin et al., 2006).
Figure 2 shows example stimulated release data collected in an animal before (black) and 60 min after (dashed grey) i.p. administration of the drugs. CPA administration decreased evoked dopamine but DPCPX administration did not change dopamine. After CPA injection, the first adenosine peak is nearly eliminated and the second adenosine peak is sharply reduced. In contrast, both adenosine peaks increased after DPCPX administration, particularly the second peak. Therefore, the A1 agonist decreased both evoked adenosine and dopamine release while the A1 antagonist increased evoked adenosine.
Figure 2.

Example data of effect of A1 drugs on stimulated dopamine and adenosine release. A. Stimulated dopamine release before (dark line) and in the same animal 60 min. after (dashed grey line) 1 mg/kg (i.p.) CPA, an A1 agonist. B. Stimulated adenosine release in the same animal as A, before and 60 min after CPA. C. Stimulated dopamine release in one animal before (dark line) and 60 min after (dashed grey line) 6 mg/kg (i.p.) DPCPX, an A1 antagonist. D. Evoked adenosine release in the same animal as C, before and 60 min after DPCPX. Stimulation parameters are the same as Fig. 1 (1 s stimulation) and the stimulation duration is marked by a line underneath the traces.
To determine the effect of pharmacological agents on stimulated release, the peak heights from repeated stimulations were examined. Stimulations were repeated every 3 min. Several baseline stimulations were performed and an average predrug release value was calculated from these data. Then a drug was administered and stimulations repeated for at least 60 minutes after administration. Each stimulated release data point for an animal was normalized and expressed as percentage of the average predrug value. Then, data from several rats (5–7 rats) were averaged and plotted (as in Fig. 3).
Figure 3.

Time course of A1 drug effects on stimulated release. Stimulations were repeated every 3 min (60 pulse, 60 Hz, 300 μA). A. Dopamine, B. 1st adenosine peak, and C. 2nd peak adenosine peak. Drugs were administered i.p. at time 0 (1 mg/kg CPA, 6 mg/kg DPCPX, or 1 mL saline). For each animal, the peak currents were averaged for 6 stimulations before drug to obtain a predrug value. Each stimulated current was divided by that predrug value (and multiplied by 100) to obtain the normalized data, expressed as % of predrug levels. Data from multiple animals were then averaged for n=5–7 animals per group. Error bars are standard error of the mean (SEM) and are shown in one direction only for clarity. Error bars for saline are shown as a dashed line.
Fig. 3 shows stimulated dopamine and adenosine efflux for each time point after CPA, DPCPX, or saline injection. For the saline control group, stimulated dopamine release slightly decreased with time, likely due to long term depression of exocytotic dopamine release (Montague et al., 2004). A two-way ANOVA for the dopamine data (Fig. 3A) shows a significant interaction between drug and time (F50=1.76, p<0.0001) as well as a significant effect of time (F25=6.12, p<0.001). Bonferonni post-tests for dopamine showed significant differences at 57 and 60 min between the CPA and saline control group (p<0.05). DPCPX had no significant effect on dopamine efflux at any time point.
For adenosine, the effect of the A1 agonist CPA was rapid, with adenosine efflux being severely diminished for the first stimulation after drug, at 3 minutes (Fig. 3B and C). This contrasts with the longer time required, nearly an hour, for CPA to affect evoked dopamine release. DPCPX administration also quickly increased the first peak of adenosine efflux, with a large increase evidenced in the first stimulation after drug. However, the effect of DPCPX on the first adenosine peak was not as sustained as the effect of CPA (Fig. 3B). The second adenosine peak also doubled after DPCPX administration and these effects lasted for an hour (Fig. 3C). A two-way ANOVA for the first adenosine peak shows a significant interaction between drug and time (F50=4.67, p<0.0001) as well as significant effects of drug (F2=23.83, p<0.0001) and time (F25=3.68, p<0.0001). Bonferonni post-tests demonstrate that the CPA group is significantly different than the saline group at 3 min, 9–15 min, 21–30 min and 45–57 min (p<0.05). The DPCPX group was significantly different from the saline group at 3 and 6 min (p<0.05). For the second adenosine peak, a two-way ANOVA showed a significant interaction between time and drug (F50=3.52, p<0.001) and a significant main effect of drug (F2=58.2, p<0.0001). Because the second peak was more variable, the standard deviation of the measurements is large and significant differences were more difficult to detect. CPA was significantly different than saline at 57 min and DPCPX was significantly different than saline at 3–15, 21, 30, 39–51, and 57–60 min with Bonferonni post-tests (p<0.05). For all time points after drug, DPCPX was significantly greater than CPA (p<0.05).
If the effects on adenosine release are mediated by the A1 receptor, then administration of an A1 antagonist should prevent the effect of the A1 agonist. To test this, CPA was injected 18 min after DPCPX was administered (Fig. 4). After DPCPX + CPA, the evoked dopamine and adenosine levels are about the same as predrug levels, indicating that DPCPX blocks the effect of CPA.
Figure 4.

Effect of combined administration of DPCPX and CPA. Stimulated release was evoked every 3 min and normalized release is plotted for A. dopamine, B. 1st adenosine peak and C. 2nd adenosine peak. Stimulation and normalization parameters are the same as Fig. 3. DPCPX (6 mg/kg, i.p.) was administered at time 0 and then CPA (1 mg/kg, i.p.) was administered at 18 min. Data are mean ± SEM for 5 animals.
3.2 A1–D1 receptor interactions
SCH-23390 (SCH) is a selective D1 receptor antagonist that crosses the blood-brain barrier (Bourne, 2001) and has been used in vivo with typical doses between 0.05 and 0.1 mg/kg (Shaham and Stewart, 1996; Xu et al., 2005). SCH-23390 (0.1 mg/kg) had no significant effect on stimulated dopamine release (Fig. 5). Dopamine release did not change after administration of the combination of CPA and SCH-23390, in contrast with CPA administration alone, which caused a significant decrease in dopamine at later time points (Fig. 5). A two-way ANOVA of the dopamine data (Fig. 5A) shows a significant interaction between drug and time (F72=2.04, p<0.01), a significant main effect of time (F24=6.88, p < 0.001), and a significant effect of drug (F3=3.66, p<0.05). Using Bonferonni post-tests, SCH was different from the CPA group at 57 min and SCH + CPA was different from CPA at 36–39 min (p<0.05).
Figure 5.

A1–D1 effects on stimulated A. dopamine, B. 1st adenosine peak, and C.2nd adenosine peak. CPA (1 mg/kg, i.p.) and saline data is replotted (without error bars for clarity) from Fig. 3 for comparison. SCH 23390 (0.1 mg/kg, i.p.) a D1 antagonist and the combination of CPA (1 mg/kg) and SCH 23390 (0.1 mg/kg) are also shown (n=5–7 per group, error bars are SEM). Drugs were administered at time 0. Stimulation and normalization parameters are the same as Fig. 3
For the first stimulated adenosine peak, evoked release after SCH-23390 administration was not different from saline controls (Fig. 5B). Combining SCH-23390 with CPA attenuated the decrease in adenosine efflux caused by the A1 agonist (Fig. 5B). For the first adenosine peak, there was a significant interaction between drug and time (two-way ANOVA, F72=3.65, p <0.0001) as well as significant main effects of drug (F3=21.01, p <0.0001) and time (F24=4.44, p <0.0001). The Bonferroni post-test shows a significant difference for SCH-23390 + CPA on adenosine concentrations at all times post-drug (p < 0.05). SCH + CPA was significantly different than SCH at 12–15 and 30 min and SCH + CPA was significantly different from the CPA group at 30–36 min. The trends for the second adenosine peak were similar, but the variance is larger, so the differences are not statistically significant (Fig. 5C). While CPA administration decreases the second adenosine peak, the addition of SCH23390 attenuates that effect.
3.3 Is adenosine release dependent on dopamine release?
Because both dopamine and adenosine release decreased after the A1 receptor agonist, we tested the effect of dopamine release on adenosine to investigate if the trends in adenosine release follow dopamine release. AMPT decreases dopamine synthesis and lowers dopamine release (Ewing et al., 1983). The dose of 250 mg/kg previously has been shown to decrease dopamine release by two-thirds when long, 10-s stimulation trains are applied (Venton et al., 2006). Fig. 6 shows that stimulated dopamine release decreased by about 50 % with our shorter, 1-s stimulations (60 pulses at 60 Hz), but stimulated adenosine levels did not change. Therefore, adenosine levels do not decrease simply because dopamine levels decrease.
Figure 6.

Effect of modulating dopamine release on adenosine release. AMPT (250 mg/kg, i.p.), a synthesis inhibitor, or haloperidol (0.5 mg/kg), a D2 antagonist, were administered at time 0. Drug effects on stimulated A. dopamine and B. 1st adenosine peak and C. 2nd adenosine peak were measured (n=4–6 per group, error bars, SEM). Stimulation and normalization parameters are the same as Fig. 3.
Haloperidol is a D2 receptor antagonist that has previously been shown to increase stimulated dopamine release with a 0.5 mg/kg dose (Garris et al., 2003). Fig. 6 shows dopamine release doubled after haloperidol (0.5 mg/kg i.p.) but adenosine release did not change. This demonstrates that more dopamine release does not lead to higher adenosine concentrations. For dopamine, a two-way ANOVA comparing AMPT, haloperidol, and saline data showed a significant interaction between drug and time (F50=9.77, p <0.001) and significant main effects of drug (F2=30.7, p<0.001) and time (F25=1.83, p<0.05). Haloperidol was significantly different from saline for all time points after 15 min and AMPT was significantly different from saline from 30 min on (p<0.05). A two-way ANOVA of the first adenosine peak data (Fig. 6B) showed no significant main effects or significant differences between the drugs and saline (p>0.05). Similarly, there were no significant effects for the second adenosine peak (p>0.05, Fig. 6C). Thus, adenosine concentrations do not simply change with changing dopamine concentrations.
4. Discussion
Adenosine is a metabolic byproduct and neuromodulator that links cellular metabolism with neurotransmission. In this paper, we demonstrate that A1 receptors modulate not only other neurotransmitters, such as dopamine, but also release of adenosine itself. This is the first demonstration of an adenosine receptor that self-regulates its own concentration, indicating that A1 receptors have some properties of an autoreceptor. In addition, A1-D1 receptor interactions play a role in modulating both adenosine and dopamine concentrations. Therefore, stimulated adenosine efflux is regulated by its own receptor and receptor interactions can vary the concentration available to act as a modulator.
4.1 A1 receptor regulates adenosine and dopamine release
Adenosine’s actions as a presynaptic heteroreceptor have been widely reported. A1 receptors can inhibit glutamate, acetylcholine, dopamine, and serotonin release (Borycz et al., 2007; Christofi, 2008; Cunha et al., 1994; Flagmeyer et al., 1997). For example, A1 receptors modulate both basal (Okada et al., 1996; Wood et al., 1989) and evoked dopamine release (O’Neill et al., 2007). This action is due to inhibition of presynaptic calcium channels and may also be related to downstream effects of second messengers (Hamilton and Smith, 1991). A1 receptors are inhibitory, coupled to G-proteins that downregulate adenyl cyclase activity. In the striatum, A1 receptors are localized in dopaminergic and glutamatergic terminals (Ferre et al., 1997). Fig. 7 shows the location of adenosine and dopamine receptors in the striatum.
Figure 7.

Schematic of striatal processes and location of dopamine and adenosine receptors. Possible sources of ATP or adenosine release are marked with arrows. Abbreviations: acetylcholine (ach), glutamate (glu), enkephalin (enk), and dynorphin (dyn).
In our experiments, the A1 receptor agonist CPA decreased both stimulated adenosine and dopamine release while the adenosine receptor antagonist DPCPX increased stimulated adenosine release. In addition, DPCPX administration blocked the effect of CPA, further demonstrating that the dramatic effect of CPA administration on stimulated adenosine release is due to A1 receptor activation. This is the first report that an adenosine receptor can modulate its own release. Both the first and second peaks of adenosine release were affected by the A1 drugs, although the antagonist caused a more sustained increase in the second adenosine peak than the first. This may indicate a different modulation of the two peaks and that the second peak may be regulated more strongly by endogenous adenosine. The results of dopamine release regulation agree with previous studies in striatal slices that showed that the A1 agonist CPA decreased stimulated dopamine release and DPCPX had no effect (O’Neill et al., 2007). Therefore, activating A1 receptors inhibits both adenosine and dopamine release.
Stimulated adenosine release decreased in the first stimulation after CPA administration and adenosine release increased in the first stimulation after DPCPX administration, as well. This suggests a direct, fast effect of A1 receptors in modulating adenosine release. In contrast, the decrease in dopamine release was not immediate after CPA administration, but stimulated release decreased gradually over an hour. The previous brain slice studies also measured the effects of CPA after an hour, suggesting a delayed effect (O’Neill et al., 2007). This delayed action likely indicates that the inhibition is not a direct effect of presynaptic A1 receptors but a slower, indirect effect. Borycz et al. (2007) found that only ~25 % of dopamine terminals expressed presynaptic A1 receptors, so the modulation of dopamine release could come from suppression of glutamate or other neurotransmitters. Indeed, Quarta et al. (2004) have found that modulation of dopamine release by adenosine receptors in the nucleus accumbens depends on glutamate neurotransmission and NMDA receptor activation.
4.2 A1 regulation of adenosine and dopamine release is mediated by D1 receptors
Many studies have reported that the effects of A1 receptors can be modulated by D1 receptors (Franco et al., 2007). D1 receptors are postsynaptic dopamine receptors that regulate dopaminergic signaling but not its release. A1 and D1 receptors can form heteromers and have intramembrane interactions (Ferre et al., 1997). Most of these reported interactions are antagonistic, where activation of the A1 receptor decreases the affinity of dopamine for D1 receptor binding. However, O’Neill et al. (2007) found a synergistic effect of D1 and A1 receptors on stimulated dopamine release. Unlike the antagonistic findings, the synergistic effect was not due to intramembrane interactions, so it is not known whether the effect is due to heteromers. Similarly, our studies also found a synergistic effect of A1 and D1 receptors on both dopamine and adenosine release. By itself, the D1 antagonist SCH 23390 had no significant effect on either dopamine or adenosine release, which agrees with dopamine studies that find that D1 receptors do not regulate dopamine release.(Missale et al., 1998) When administered concurrently with CPA, SCH 23390 blocked the reduction in adenosine release by the A1 agonist. These are the first results to show that A1-D1 receptor interactions can modulate adenosine as well as dopamine release.
The previous study by O’Neill et al. (2007) that found a synergistic interaction between D1 and A1 receptors on dopamine release also found that intramembrane interactions were not required for the effect. Thus, the A1 and D1 interaction might not be due to a heteromer or coexpression on dopamine neurons but instead be due to downstream interactions. A1 and D1 receptors have opposite effects on adenylyl cyclase so the interaction is unlikely to be due to adenyl cyclase, although it could involve another intracellular pathway. The downstream effect might also be modulation of another neurotransmitter, which then has an effect through the neuronal network. For example, both A1 (Alexander and Reddington, 1989) and D1 (Acquas and Di, 2001) receptors are expressed on cholinergic interneurons, so acetylcholine could be a downstream neurotransmitter involved in the receptor interaction. Alternatively, A1 and D1 receptors are coexpressed on dynorphinergic GABA neurons, and GABA modulation could play a role in the interaction. Future studies could determine the role of these neurons in the modulation of adenosine and dopamine release.
4.3 Adenosine release is not a downstream action of dopamine release
Because CPA decreased both dopamine and adenosine release and a dopamine receptor mediated the effects of adenosine release, we tested if the decrease in adenosine release is a downstream effect of dopamine release. The effects of CPA on adenosine release were much faster than for inhibition of dopamine release which suggested that the modulation of adenosine release is not due to dopamine signaling. To confirm this, the synthesis inhibitor AMPT was administered to decrease dopamine release. Although dopamine decreased by 50%, the adenosine signal did not change significantly. Similarly, increasing stimulated dopamine using a D2 receptor antagonist, haloperidol, did not increase adenosine levels. Therefore, changes in dopamine levels do not regulate adenosine release and the effects of the A1 agonist CPA are not just due to modulation of dopamine.
4.4 Is the A1 receptor an autoreceptor?
A standard definition of an autoreceptor is a presynaptically-located receptor that regulates the release of its own ligand from the cell on which it is located. Does the A1 receptor meet this definition? The first requirement is that the receptor act presynaptically. The phrase “adenosine autoreceptor” is found in the literature in the early 1990’s. Although these studies did not show that A1 receptors regulated adenosine release, they did identify the presynaptic effects of adenosine (Hamilton and Smith, 1991; Yawo and Chuhma, 1993). They established that adenosine acts through A1 receptors and G-protein coupled mechanisms to inhibit Ca2+ influx into presynaptic neurons (Hamilton and Smith, 1991). This inhibition can decrease calcium-dependent exocytosis and neurotransmitter release (Kimura et al., 2003). Therefore, these studies demonstrated adenosine can act presynaptically but did not actually identify the A1 receptor as an autoreceptor.
The second requirement for an autoreceptor is that it regulates the release of its own ligand. Our work shows that activation of A1 receptors dramatically decreases the evoked release of adenosine. In addition, blocking adenosine receptors with an antagonist increases adenosine efflux. Therefore, endogenous concentrations of adenosine are able to activate the A1 receptor. Thus, the A1 receptor regulates the concentration of its own ligand and meets this important test for an autoreceptor.
The third requirement is that release be from the neuron on which the A1 receptor is located. The cellular source of electrically-stimulated adenosine is complicated because adenosine is present in all neurons and glia. A1 receptors could be regulating release of adenosine from neurons where they are located presynaptically. For example, adenosine or ATP may be directly released from dopamine or glutamate neurons expressing presynaptic A1 receptors. In glutamatergic neurons, ATP is co-released with glutamate and activation of A1 receptors in glutamatergic terminals inhibits glutamate release, which could also inhibit ATP release (Calabresi et al., 1997; Flagmeyer et al., 1997). Additionally, lower activity in glutamatergic or dopaminergic terminals could lead to less intracellularly produced adenosine that would be effluxed from the neuron by nucleoside transporters. If adenosine release was primarily from dopaminergic or glutamatergic terminals, the presynaptic A1 receptors could be considered autoreceptors. However, adenosine may also be released from postsynaptic cells. Activation of postsynaptic adenosine A1 receptors can also suppress activity in GABAergic neurons. Lower activity of GABAergic cells could lead to less intracellular adenosine formation that would be available for transport to the extracellular space. In addition, acetylcholine interneurons might also be responsible for synthesizing and releasing adenosine (Wojcik and Neff, 1983). Astrocytes are known to release ATP so the source of adenosine might not be neuronal (Halassa et al., 2009). Fig. 7 summarizes possible sources of ATP or adenosine release. Because the source of adenosine is not known, we cannot determine if adenosine is released from a neuron with presynaptic A1 receptors. Thus, A1 receptors have some properties of an autoreceptor, including modulating their own release and acting presynaptically, but it is not known if the modulation occurs presynaptically to a neuron releasing adenosine.
The A1 receptor can regulate evoked adenosine release, changing the amount of adenosine available for signaling presynaptically or postsynaptically. By responding to large concentrations of adenosine in the extracellular space, the A1 receptor may prevent adenosine from accumulating and causing sustained inhibition. Future research to determine the cellular source of adenosine will reveal whether this regulation of adenosine release occurs at specific neurons where adenosine is released or on a more global scale.
5. Conclusions
In this study, we show that A1 receptors can modulate evoked adenosine release; the first reports of an adenosine receptor self-regulating its own concentration. These effects are mediated in part by D1 receptors but are not a downstream action of dopamine release. Therefore, the regulation of neurotransmission by adenosine is complex and A1 receptors may regulate neurotransmission both through direct presynaptic effects and indirect effects by modulating adenosine levels as well.
Acknowledgments
This work was supported by grants to BJV from the American Heart Association (0765318U) and the NIH (R21-EB007830).
Abbreviations used
- AMPT
α-methyl-p-tyrosine
- CPA
N6-cyclopentyladenosine
- DPCPX
8-Cyclopentyl-1,3-dipropylxanthine
- DA
dopamine
- DYN
dynorphinergic
- ENK
enkephalinergic
- FSCV
fast-scan cyclic voltammetry
- i.p
intraperitoneal
- SCH
SCH-23390
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acquas E, Di CG. Role of dopamine D1 receptors in the control of striatal acetylcholine release by endogenous dopamine. Neurol Sci. 2001;22:41–42. doi: 10.1007/s100720170037. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Reddington M. The cellular localization of adenosine receptors in rat neostriatum. Neuroscience. 1989;28:645–651. doi: 10.1016/0306-4522(89)90011-0. [DOI] [PubMed] [Google Scholar]
- Ballarin M, Fredholm BB, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand. 1991;142:97–103. doi: 10.1111/j.1748-1716.1991.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Borycz J, Pereira MF, Melani A, Rodrigues RJ, Kofalvi A, Panlilio L, Pedata F, Goldberg SR, Cunha RA, Ferre S. Differential glutamate-dependent and glutamate-independent adenosine A1 receptor-mediated modulation of dopamine release in different striatal compartments. J Neurochem. 2007;101:355–363. doi: 10.1111/j.1471-4159.2006.04386.x. [DOI] [PubMed] [Google Scholar]
- Bourne JA. SCH 23390: the first selective dopamine D1-like receptor antagonist. CNS Drug Rev. 2001;7:399–414. doi: 10.1111/j.1527-3458.2001.tb00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Bernardi G. Endogenous adenosine mediates the presynaptic inhibition induced by aglycemia at corticostriatal synapses. J Neurosci. 1997;17:4509–4516. doi: 10.1523/JNEUROSCI.17-12-04509.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cechova S, Venton BJ. Transient adenosine efflux in the rat caudate-putamen. J Neurochem. 2008;105:1253–1263. doi: 10.1111/j.1471-4159.2008.05223.x. [DOI] [PubMed] [Google Scholar]
- Christofi FL. Purinergic receptors and gastrointestinal secretomotor function. Purinergic Signalling. 2008;4:213–236. doi: 10.1007/s11302-008-9104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Milusheva E, Vizi ES, Ribeiro JA, Sebastiao AM. Excitatory and inhibitory effects of A1 and A2A adenosine receptor activation on the electrically evoked [3H]acetylcholine release from different areas of the rat hippocampus. J Neurochem. 1994;63:207–214. doi: 10.1046/j.1471-4159.1994.63010207.x. [DOI] [PubMed] [Google Scholar]
- Ewing AG, Bigelow JC, Wightman RM. Direct in vivo monitoring of dopamine released from two striatal compartments in the rat. Science. 1983;221:169–171. doi: 10.1126/science.6857277. [DOI] [PubMed] [Google Scholar]
- Ferre S. An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem. 2008;105:1067–1079. doi: 10.1111/j.1471-4159.2007.05196.x. [DOI] [PubMed] [Google Scholar]
- Ferre S, Fredholm BB, Morelli M, Popoli P, Fuxe K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997;20:482–487. doi: 10.1016/s0166-2236(97)01096-5. [DOI] [PubMed] [Google Scholar]
- Flagmeyer I, Haas HL, Stevens DR. Adenosine A1 receptor-mediated depression of corticostriatal and thalamostriatal glutamatergic synaptic potentials in vitro. Brain Res. 1997;778:178–185. doi: 10.1016/s0006-8993(97)01060-3. [DOI] [PubMed] [Google Scholar]
- Franco R, Lluis C, Canela EI, Mallol J, Agnati L, Casado V, Ciruela F, Ferre S, Fuxe K. Receptor-receptor interactions involving adenosine A1 or dopamine D1 receptors and accessory proteins. J Neural Transm. 2007;114:93–104. doi: 10.1007/s00702-006-0566-7. [DOI] [PubMed] [Google Scholar]
- Garris PA, Budygin EA, Phillips PEM, Venton BJ, Robinson DL, Bergstrom BP, Rebec GV, Wightman RM. A role for presynaptic mechanisms in the actions of nomifensine and haloperidol. Neuroscience. 2003;118:819–829. doi: 10.1016/s0306-4522(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology. 2009;57:343–346. doi: 10.1016/j.neuropharm.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BR, Smith DO. Autoreceptor-mediated purinergic and cholinergic inhibition of motor nerve terminal calcium currents in the rat. J Physiol. 1991;432:327–341. doi: 10.1113/jphysiol.1991.sp018387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien MLAV, Phillips PEM, Stuber GD, Seipel AT, Wightman RM. Overoxidation of carbon-fiber microelectrodes enhances dopamine adsorption and increases sensitivity. Analyst. 2003;128:1413–1419. doi: 10.1039/b307024g. [DOI] [PubMed] [Google Scholar]
- Karcz-Kubicha M, Antoniou K, Terasmaa A, Quarta D, Solinas M, Justinova Z, Pezzola A, Reggio R, Muller CE, Fuxe K, Goldberg SR, Popoli P, Ferre S. Involvement of adenosine A1 and A2A receptors in the motor effects of caffeine after its acute and chronic administration. Neuropsychopharmacology. 2003;28:1281–1291. doi: 10.1038/sj.npp.1300167. [DOI] [PubMed] [Google Scholar]
- Kimura M, Saitoh N, Takahashi T. Adenosine A(1) receptor-mediated presynaptic inhibition at the calyx of Held of immature rats. J Physiol. 2003;553:415–426. doi: 10.1113/jphysiol.2003.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin A, Johansson B, Gimenez L, Ogren SO, Fredholm BB. Combination of adenosine A(1) and A(2A) receptor blocking agents induces caffeine-like locomotor stimulation in mice. Eur Neuropsychopharm. 2006;16:129–136. doi: 10.1016/j.euroneuro.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Masino SA, Dulla CG. Adenosine, glutamate and pH: interactions and implications. Neurol Res. 2005;27:149–152. doi: 10.1179/016164105X21850. [DOI] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber MJ, Caron MG. Dopamine Receptors: From Structure to Function. Physiolog Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Montague PR, McClure SM, Baldwin PR, Phillips PE, Budygin EA, Stuber GD, Kilpatrick MR, Wightman RM. Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci. 2004;24:1754–1759. doi: 10.1523/JNEUROSCI.4279-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill C, Nolan BJ, Macari A, O’Boyle KM, O’Connor JJ. Adenosine A1 receptor-mediated inhibition of dopamine release from rat striatal slices is modulated by D1 dopamine receptors. Eur J Neurosci. 2007;26:3421–3428. doi: 10.1111/j.1460-9568.2007.05953.x. [DOI] [PubMed] [Google Scholar]
- Okada M, Mizuno K, Kaneko S. Adenosine A1 and A2 receptors modulate extracellular dopamine levels in the rat striatum. Neurosci Lett. 1996;212:53–56. doi: 10.1016/0304-3940(96)12780-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pazzagli M, Corsi C, Fratti S, Pedata F, Pepeu G. Regulation of extracellular adenosine levels in the striatum of aging rats. Brain Res. 1995;684:103–106. doi: 10.1016/0006-8993(95)00471-2. [DOI] [PubMed] [Google Scholar]
- Prediger RDS, Batista LC, Takahashi RN. Adenosine A(1) receptors modulate the anxiolytic-like effect of ethanol in the elevated plus-maze in mice. Eur J Pharmacol. 2004;499:147–154. doi: 10.1016/j.ejphar.2004.07.106. [DOI] [PubMed] [Google Scholar]
- Quarta D, Borycz J, Solinas M, Patkar K, Hockemeyer J, Ciruela F, Lluis C, Franco R, Woods AS, Goldberg SR, Ferre S. Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-D-aspartate receptor stimulation. J Neurochem. 2004;91:873–880. doi: 10.1111/j.1471-4159.2004.02761.x. [DOI] [PubMed] [Google Scholar]
- Sanford AL, Morton SW, Whitehouse KL, Oara HM, Lugo-Morales LZ, Roberts JG, Sombers LA. Voltammetric detection of hydrogen peroxide at carbon-fiber microelectrodes. Analytical Chemistry. 2010;82:5205–5210. doi: 10.1021/ac100536s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CW, Karcz-Kubicha M, Thorndike EB, Muller CE, Tella SR, Ferre S, Goldberg SR. Role of central and peripheral adenosine receptors in the cardiovascular responses to intraperitoneal injections of adenosine A1 and A2A subtype receptor agonists. Br J Pharmacol. 2005;144:642–650. doi: 10.1038/sj.bjp.0706043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Stewart J. Effects of opioid and dopamine receptor antagonists on relapse induced by stress and re-exposure to heroin in rats. Psychopharmacology (Berl) 1996;125:385–391. doi: 10.1007/BF02246022. [DOI] [PubMed] [Google Scholar]
- Strand AM, Venton BJ. Flame etching enhances the sensitivity of carbon-fiber microelectrodes. Anal Chem. 2008;80:3708–3715. doi: 10.1021/ac8001275. [DOI] [PubMed] [Google Scholar]
- Swamy BEK, Venton BJ. Susbsecond detection of physiological adenosine concentrations using fast-scan cyclic voltammetry. Anal Chem. 2007;79:744–750. doi: 10.1021/ac061820i. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Seipel AT, Phillips PE, Wetsel WC, Gitler D, Greengard P, Augustine GJ, Wightman RM. Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J Neurosci. 2006;26:3206–3209. doi: 10.1523/JNEUROSCI.4901-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik WJ, Neff NH. Location of adenosine release and adenosine-A2 receptors to rat striatal neurons. Life Sciences. 1983;33:755–763. doi: 10.1016/0024-3205(83)90781-6. [DOI] [PubMed] [Google Scholar]
- Wood PL, Kim HS, Boyar WC, Hutchison A. Inhibition of nigrostriatal release of dopamine in the rat by adenosine receptor agonists: A1 receptor mediation. Neuropharmacology. 1989;28:21–25. doi: 10.1016/0028-3908(89)90062-2. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhu JP, Angulo JA. Induction of striatal pre- and postsynaptic damage by methamphetamine requires the dopamine receptors. Synapse. 2005;58:110–121. doi: 10.1002/syn.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. Preferential inhibition of omega-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature. 1993;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]